

Abstract
The regulation of gene expression through alternative pre-mRNA splicing is common in metazoans and is often controlled by intracellular signaling pathways that are important in cell physiology. We have shown that the alternative splicing of a number of genes is controlled by membrane depolarization and Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) through CaMKIV-responsive RNA elements (CaRRE1 and CaRRE2); however, the trans-acting factors remain unknown. Here we show that the heterogeneous nuclear ribonucleoprotein (hnRNP) L is a CaRRE1 binding factor in nuclear extracts. An hnRNP L high affinity CA (cytidine-adenosine) repeat element is sufficient to mediate CaMKIV and hnRNP L repression of splicing in a location (3′-splice site proximity)-dependent way. Depletion of hnRNP L by RNA interference followed by rescue with coexpressed exogenous hnRNP L demonstrates that hnRNP L mediates the CaMKIV-regulated splicing through CA repeats in heterologous contexts. Depletion of hnRNP L also led to increased inclusion of the stress axis-regulated exon and a CA repeat-harboring exon under depolarization or with activated CaMKIV. Moreover, hnRNP L binding to CaRRE1 was increased by CaMKIV and, conversely, was reduced by pretreatments with protein phosphatases. Therefore, hnRNP L is an essential component of CaMKIV-regulated alternative splicing through CA repeats, with its phosphorylation likely playing a critical role.
Alternative pre-mRNA splicing allows the generation of multiple protein isoforms from a single transcript, contributing greatly to proteomic complexity (1–6). It is essential for normal cell function, and aberrant splicing is implicated in the development of human genetic diseases (7, 8). Many alternative splicing events can be regulated by cell signals and may play important roles in cell physiology (9–11). However, little is known of the essential molecular components between cell signals and the splicing machinery.
Alternative splicing in mammalian systems is often controlled by multiple cis-acting regulatory elements in introns and exons (1, 12). These elements are mostly bound by transacting protein factors and in some cases by small nucleolar RNA (13) or form RNA secondary structures (14). The transacting factors are thought to control the assembly of constitutive splicing components to the target splice sites. For cell signal control of alternative splicing, RNA elements and trans-acting factors have been identified in several systems (9, 10, 15–23).
CA repeats are the most abundant and highly polymorphic dinucleotide repeats in the human genome. They are widely used in genetic linkage analyses (24), and their instability is characteristic of the mutator phenotype of cells defective in DNA repair genes (25). Recent studies indicate that long (≥19 repeats) or short clustered CA repeats in downstream introns are constitutive enhancers or repressors depending on their proximity to the 5′-splice site (26, 27). These effects are mediated by the heterogeneous nuclear ribonucleoprotein (hnRNP)2 L and/or its homologue hnRNP LL (hnRNP L-like), splicing factors implicated in the regulation of a number of alternative exons (22, 23, 26–33).
Membrane depolarization and activated Ca2+/calmodulin-dependent protein kinase IV (CaMKIV) control the alternative inclusion of many exons (11, 34), including stress axis-regulated exon (STREX) of the Ca2+/voltage-sensitive Slo potassium channel gene (35, 36). This control is through distinct CaMKIV-responsive RNA elements (CaRRE1 and CaRRE2) or the UAGG motif (11, 16–19). Particularly, the CaRRE1 contains CA dinucleotide repeats as well as other nucleotides that are critical for the CaMKIV effect on splicing (18, 19), but the CaRRE1-binding factor(s) is unknown. In this report we identified hnRNP L as a CaRRE1 binding factor and examined its role in the CaMKIV-regulated splicing.
EXPERIMENTAL PROCEDURES
Plasmid Construction
Splicing reporter or mutant plasmids were made using PCR with Pfu DNA polymerase and inserts confirmed by sequencing. DUP175, DUP175ST, CaMKIV-dCT (IV), CaMKIV-dCTK75E (IVm), and protein kinase A plasmids are as described previously (16, 37–40). The hnRNP L-FLAG expression plasmid was provided by Drs. Peter Stoilov and Ste-fan Stamm, and His-YB-1 was from Dr. Thomas Cooper.
The oligonucleotides used for the hnRNP L shRNA are: forward strand (hnRNP L sense sequence underlined) 5′-tccctcccaagattaaccttcacttcaagagagtgaaggttaatcttggga-3′; 5′-aaaatccc-aagattaaccttcactctcttgaagtgaaggttaatcttggga-3′ (reverse strand). To make shRNA expression plasmid, the two complementary oligos were annealed in a heating block after denaturation, phosphorylated at 37 °C, and inserted into the BbsI sites of the plasmid siRNA [H1.4]-R vector (provided by Dr. Owen N. Witte) (41). The shRNA expression cassette, driven by the human H1 promoter, was then cut off by XbaI and cloned into FG12 (42). The resulting construct was confirmed by XhoI digestion and DNA sequencing. FG12 also carries a green fluorescent protein reporter gene driven by an internal ubiquitin-C promoter (42).
Cell Culture and Transfection
HEK293T cells were cultured in Dulbecco’s modified Eagle’s medium with 10% of new-born calf serum and rat GH3 pituitary cells in F10 nutrient mixture (Invitrogen) with 10% of horse serum plus 2.5% of fetal bovine serum.
Transfections of overnight HEK293T cultures were performed with Lipofectamine 2000 (Invitrogen) according to the supplier’s procedure. For the HEK293T cells in 24-well plates, 0.1 μg of splicing reporter minigene plasmid and 0.3 μg of another plasmid including CaMKIV-dCT (IV), CaMKIV-dCTK75E (IVm), or hnRNP L-FLAG were used.
RNA Interference
Vesicular Stomatitis Virus Glycoprotein-pseudotyped lentiviral vectors were produced by calcium phosphate-mediated transfection of HEK293T cells as described previously (43). In brief, 2 × 107 HEK293T cells were transfected with 5 μg of pHCMVG, 12.5 μg of pCMVΔR8.2DVPR, and 12.5 μg of the lentiviral plasmids. Culture supernatant was collected on days 2 and 3 post-transfection, pooled, and filtered through a 0.22-μm filter. Viral supernatant was further concentrated 100 times by ultracentrifugation at 17,000 rpm for 90 min. Titers of the concentrated virus were determined by enhanced green fluorescent protein expression in transduced HEK293T cells. For transduction, HEK293T cells were plated at a density of 6 × 104 cells per well in a 24-well plate (Falcon) on day 1. On day 2, 25 μl of lentivirus stock (titer ~1 × 108 units) was mixed with 225 μl of Dulbecco’s modified Eagle’s medium (Invitrogen) then with 2.0 μl of Polybrene (1 μg/μl), gently vortexed, allowed to sit at room temperature for 5 min, incubated at 37 °C for 10 min, and then pipetted onto the cells. The cells were then incubated with the virus supernatant at 37 °C, 5% CO2 for 2.5–3.0 h before fresh media were supplied. On day 3 the cells were infected again as on day 2 and transferred into a 6-well plate. On day 5, the cells were cotransfected with 0.5 μg of splicing reporter minigene plasmids and 1.5 μg of CaMKIV-dCT or CaMKIV-dCTK75E plasmid. On day 6, the cells were harvested for Western blots (with anti-hnRNP L 4D11, Santa Cruz Biotechnology) and semi-quantitative reverse transcription (RT)-PCR assay. For transduction of GH3 cells, similar procedures were followed.
RT-PCR
HEK293T cells were harvested 18–22 h after transfection. Cytoplasmic RNA of HEK293T or total RNA of GH3 cells was extracted using GenElute Mammalian Total RNA Miniprep Kit (Sigma-Aldrich). RT-PCR was performed as previously described (16), except that about 400 ng of HEK293T cytoplasmic RNA or GH3 total RNA was used for 10 μl of reverse transcription reaction. PCR reaction of splicing reporters was performed with forward primer DUP8a (5′-CTCAAACAGACACCATGCATGG-3′) and reverse primer DUP1 (5′-GCAGCTCACTCAGTGTGGCA-3′) for 25 cycles. Products were resolved in 2.5% agarose gels with ethidium bromide, visualized, and documented with a digital camera under UV. For hot PCR of endogenous genes of GH3 cells with a 32P-labeled primer, 1 μl of the reverse transcription product was used for 25 cycles. The PCR products were resolved in 5% polyacrylamide, 7.5 M urea gels, which were dried, exposed, and scanned by phosphorimaging (Bio-Rad). The percentage of the middle exon-included product is expressed as the percent band intensities relative to the total of exon-included and -excluded products. The band intensities of the agarose gels were not calibrated for product lengths.
Nuclear Extract Preparation
HeLa nuclear extracts were prepared on a 5-liter scale (44). Nuclear extracts of HEK293T or GH3 cells were made in a smaller scale (45). Briefly, for the small scale preparation, 3-day cultures in 2–3 dishes of 150 mm in diameter were phosphate-buffered saline-washed three times, resuspended in 5× cell pellet volume of ice-cold buffer A (10 mM HEPES, pH 7.9, 10 mM KCl, 0.4% IGEPAL, 0.2 mM EDTA, 1.5 mM MgCl2, 0.5 mM dithiothreitol, 1.0 mM phenylmethylsulfonyl fluoride, 1/1000 protease inhibitor mixture), and left on ice for 10 min. Then the nuclei were pelleted at 10,000 rpm for 1 min, resuspended in buffer B (20 mM HEPES, pH 7.9, 25% glycerol, 0.2 mM EDTA, 1.5 mM MgCl2, 0.42 M NaCl, 1.0 mM dithiothreitol, 1.0 mM phenylmethylsulfonyl fluoride, 1/1000 protease inhibitor mixture) with shaking for 2 h and centrifuged at 13000 rpm for 5 min. The resulting supernatant was saved as nuclear extracts and diluted 2–3 times before use in DG buffer (20 mM HEPES-KOH, pH 7.9, 20% glycerol, 80 mM potassium glutamate, 0.2 mM EDTA, 0.2 mM PSMF, 1.0 mM DTT) (44). For HEK293T nuclear extracts expressing kinases or together with hnRNP L-FLAG, 5.0 μg of CaMKIVm or CaMKIV or 1.5 μg of hnRNP L-FLAG plus 3.0 μg of CaMKIV plasmids were transfected into a well of cells grown in 6-well plates using Lipofectamine 2000.
UV Cross-linking and Immunoprecipitation
DNA templates of RNA transcripts were made from splicing reporter plasmids by PCR with T7 promoter-tagged primers. RNA probes were made with T7 RNA polymerase in in vitro transcription (46). The wild type RNA probe sequences (CA repeat elements underlined) are: CaRRE1 (DUP175ST upstream 3′-splice site), gaagacucuuggguuucugauagggcccuugccauuaaccgcgcucuuccucuccucccauccaaccacaugguuauagGCUGCUGGUGGUCUACCCUUGGACAGCUUGCUUACAUUUGCUUCUGACACAACUGUGUUCACUAGCAACCUCAAACAGACACCAUG; L3, gagacucuuggguuucugauagggcccacugacucucucugccuauuggucuauuuucccacccuuagGCUGCUGGUGAUACAUGACACACACACGCA; GALNT11, cuauuuucccacccuuagACACAGCCCACACACGCAUUGGACAGCUUGCUUACAUUUGCUUCUGACACAACUGUGUUCACUAGCAACCUCAAACAGACACCAUG; PTEN2, cuauuuucccacccuuagCCCACACACAAUCUACCCUUGGACAGCUUGCUUACAUUUGCUCUGACACAACUGUGUUCACUAGCAACCUCAAACAGACACCAUG.
UV cross-linking was performed similarly as described (47). Briefly, HeLa (~60 μg), HEK, or GH3 (~40 μg) nuclear extract was incubated with 3 × 105 cpm of [α-32P]CTP-labeled RNA transcripts in cross-linking buffer (12 mM Hepes-KOH, pH7.9, 12% glycerol, 48 mM potassium glutamate, 0.12 mM EDTA, 0.12 mM phenylmethylsulfonyl fluoride, 0.6 mM dithiothreitol, 0.4 mM ATP, 2.2 mM MgCl2, 20 mM phosphocreatine, 0.2 μg/μl yeast tRNA) in 13-μl reaction at 30 °C for 20 min, irradiated with UV light (254 nm) for 25 min on ice, and digested with 8 units of RNase T1 plus 4.0 μg of RNase A for 30 min at 37 °C. For immunoprecipitation, 10 μl of settled resin of immobilized protein G (Pierce) in 200 μl of RIPA buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1% Triton X-100, 0.5% deoxycholic acid, 0.1% SDS) were incubated with 1.0 μg of anti-hnRNP L (4D11, Santa Cruz), anti-hnRNP LL (Aviva Systems Biology, San Diego, CA), polypyrimidine tract-binding protein (PTB) (BB7) (48), or control antibodies (U2AF65, Sigma-Aldrich, or hnRNP F/H, Santa Cruz) at 4 °C for 4 h with rotation, and washed 5 times with 1 ml of RIPA buffer. Then 10 μl of the beads in 200 μl of RIPA buffer was incubated with 3–8 volumes of UV cross-linking reactions with rotation at 4 °C overnight. The beads were then washed 5 times with 1 ml of RIPA buffer (containing 150 mM NaCl). The resulting proteins were denatured in SDS loading buffer at 95 °C for 5 min and resolved in 4–20% or 8% SDS-polyacrylamide gels, which were dried, exposed, and scanned by phosphorimaging (Bio-Rad).
Pretreatment of nuclear extracts (about 40 μg) with phosphatases were carried out in λ-protein phosphatase (λ-PPase) buffer lacking the detergent Brij 35 (λ-PPase 1× reaction buffer, New England Biolabs) or calf intestinal alkaline phosphatase (CIAP) buffer (1× dephosphorylation buffer, Invitrogen) with 600 units of λ-PPase or 10 units of CIAP at 30 °C for 30 min or 37 °C for 20 min, respectively. Under this condition, we found that the phosphatases efficiently reduced the 32P-labeled nuclear extract proteins without disrupting subsequent UV cross-linking. In vivo labeling of proteins with [32P]orthophosphoric acid was carried out as described previously (40).
RESULTS
hnRNP L Is a CaRRE1 Binding Factor
To identify the protein factor(s) binding to the CaRRE1 element of the STREX exon, we carried out a UV cross-linking assay with the upstream 3′-splice site of the middle exon of the splicing reporter DUP175ST (Fig. 1), which harbors the CaRRE1 (16). In HeLa nuclear extracts the wild type RNA transcript was cross-linked mainly to three protein bands of about 55, 65, and 110 kDa, respectively (Fig. 1A, right, lane 1). Mutation of the CaRRE1 element mainly reduced the 65- and 110-kDa bands (lane 2). Similarly cross-linked bands were observed in HEK293T nuclear extracts (lanes 3 and 4).
FIGURE 1. Binding of hnRNP L to CaRRE1.

A, left, diagram of the CaRRE1 RNA probe (wild type (WT)) and its mutant (Mut). The line represents the intron, and gray open box represents the downstream exon of DUP175ST. The CA repeat and upstream A/C-rich region is boxed, and the mutated nucleotides are underlined. Right, phosphorimages of SDS-PAGE gels of HeLa (lanes 1–2) or HEK293T (HEK, lanes 3–7) nuclear extract (NE) proteins UV-cross-linked to the wild type or mutant CaRRE1 RNA probes, with the protein kDa size markers indicated to the left. In lanes 5–7, 4, 8, or 4 times of UV-cross-linked nuclear extracts were used for immunoprecipitation (IP) with antibodies (Ab) against hnRNP L (L), hnRNP LL (LL), or PTB (PTB), respectively. B, left, UV cross-linking of the CaRRE1 wild type RNA probe in nuclear extracts of GH3 cells non-treated (NT) or treated with depolarizing concentrations of KCl (50 mM) for 12 h, similarly as in A. M, molecular size marker (kDa). Immunoprecipitation antibodies against hnRNP L (L), hnRNP F/H (F/H), or hnRNP LL (LL) are indicated above the lanes. Three (lanes 3–5) or eight times (lanes 6 and 7) of the cross-linked nuclear extracts were used for immunoprecipitation. Right, a bar graph of the ratio of hnRNP L/p55′ (mean ± S.E.) in GH3 nuclear extracts from non-treated or KCl-treated cells, with the p value and number of samples indicated (Student’s t test, paired non-treated and KCl-treated samples). C, specificity of the antibodies used against hnRNP L or LL in the experiments. The same blot of a GH3 nuclear extract was probed with hnRNP LL and hnRNP L antibodies, respectively, and aligned side by side with molecular markers. The hnRNP LL band is clearly lower than hnRNP L. *, a nonspecific band common for the anti-L, -LL, and -PTB antibodies (as seen in lanes 5–7 in A).
Immunoprecipitation of the cross-linked proteins in the HEK293T nuclear extracts with antibodies against hnRNP L (lane 5), hnRNP LL (lane 6), or PTB (lane 7) indicates that the 65-kDa band contains hnRNP L, and the 55-kDa band contains PTB. hnRNP LL is not observed in the precipitates even with twice the nuclear extract input (lane 6).
To examine the CaRRE1-binding proteins in depolarized cells, we carried out a similar UV cross-linking assay with nuclear extracts prepared from non-treated or KCl (50 mM)-depolarized GH3 cells (Fig. 1B). The pattern of the cross-linked major bands was similar to that in HeLa and HEK293T nuclear extracts. Interestingly, the ratio of the 65- and 55-kDa bands appears increased by about 10% upon depolarization (compare lanes 1 and 2 and the graph on the right). The cross-linked 65-kDa products was immunoprecipitated by anti-hnRNP L as well (lanes 3 and 4) but not by anti-hnRNP F/H (lane 5) or anti-hnRNP LL (lanes 6 and 7). Taken together with the previous finding that hnRNP L selectively binds CA repeat elements and controls alternative splicing (26, 27), these data suggest that hnRNP L is a CaRRE1 binding factor.
An hnRNP L High Affinity CA Repeat Element Is Sufficient to Mediate CaMKIV- and hnRNP L Repression of Splicing in a Location-dependent Way
To isolate the role of hnRNP L and CA repeats from the influence of non-CA nucleotides of CaRRE1, we tested the effect of known hnRNP L binding simple CA repeats in CaMKIV-regulated splicing. We first replaced 17 nt of the CaRRE1 with a high or low affinity sequence of hnRNP L (27) to make splicing reporters L1 and L1m (Fig. 2). These reporter plasmids were each cotransfected with constitutively active CaMKIV-dCT (IV) or kinase-dead mutant (CaMKIV-dCTK75E, IVm) into HEK293T cells, and inclusion of the middle exon was measured by semiquantitative RT-PCR (PCR for 25 cycles). Exon inclusion levels of both L1 and L1m reporters were similar to the DUP175ST when co-expressed with CaMKIVm (Fig. 2, lanes 1, 3, and 5), suggesting that the 17-nt L1 and L1m elements here are not constitutive splicing silencers. However, when co-expressed with CaMKIV, exon inclusion levels of the L1 reporter were reduced by 18% (Fig. 2, compare lanes 3 and 4), similarly as the CaRRE1 reporter DUP175ST (lanes 1 and 2). In contrast, the L1m reporter did not respond to CaMKIV (lanes 5 and 6). Thus, the hnRNP L high affinity CA repeat element can specifically replace CaRRE1 in response to CaMKIV.
FIGURE 2. Effect of CaRRE1 replacement by an hnRNP L-high affinity CA repeat element in CaMKIV-regulated splicing.

Upper, diagram of the exons (boxes) and introns (horizontal lines) of the splicing reporter mini-gene DUP175ST and its CaRRE1 in the upstream 3′-splice site of STREX (heavy line). In reporters L1 or L1m, the CaRRE1 was replaced with the hnRNP L high affinity CA repeats (L1) or low affinity sequence (L1m). Arrowheads, locations of PCR primers. Lower, an agarose gel of RT-PCR products from RNA samples of HEK293T cells co-transfected with each of the above splicing reporters and kinase-dead mutant (IVm) or constitutively active CaMKIV (IV), with average percentages (±S.D.) of exon inclusion under each lane, exon-included or -excluded products indicated to the right, and molecular size makers (M) to the indicated to the left. NC, PCR negative control. *, likely product from pre-mRNA. **, a cryptic 3′-splice product from 65 nt downstream in the middle exon.
To determine whether the L1 element is sufficient to mediate CaMKIV repression of exon inclusion outside of the 3′-splice site, we placed 20 nt of the original L1 sequence inside the middle exon or in the flanking introns of the CaMKIV-nonresponsive DUP175 (16, 37) and assayed its response to CaMKIV co-expression (Fig. 3A, reporters L2–L5). The middle exon was strongly repressed by CaMKIV only in the L3 reporter that contains the CA repeat in the exon nearby the upstream 3′-splice site (27% reduction, Fig. 3B, lanes 5 and 6) but not in any of the other reporters L2, L4, and L5 that contain CA repeats at other locations. Moreover, the repression was abolished by A to G mutations in the CA repeats (L3m, lane 8). In comparison to CaMKIV, coexpressed protein kinase A barely reduced the exon inclusion of the L3 reporter (lane 14). Therefore, the high affinity hnRNP L binding CA repeat element confers CaMKIV-specific repression of exon inclusion in a location-dependent way.
FIGURE 3. The hnRNP L high affinity CA repeat element in CaMKIV- and hnRNP L-regulated splicing and its location dependence.

A, the CA repeat element (or CG mutant as shown in sequence) is transferred to different positions of the middle exon or flanking introns in splicing reporters L2-L5 to replace the corresponding sequences of the vector DUP175 (except in reporter L2, the element was inserted upstream the branch point as indicated). Positions of the first nucleotide of the elements are indicated above the exon or introns, relative to the last nucleotide of upstream intron, first nucleotide of exon, or downstream intron, respectively. For comparison, the relative positions of L1 and L1m elements in DUP175ST are also indicated. B, an agarose gel of the RT-PCR products of the splicing reporters with co-expressed CaMKIVm, CaMKIV, or protein kinase A (PKA) in HEK293T cells, with average percentages (±S.D.) of exon inclusion under each lane and exon-included or -excluded products indicated to the right. NC, PCR negative control. *, same as in Fig. 2. C, effect of hnRNP L overexpression on splicing reporters L1-L5. Western blots (upper panel) of overexpressed hnRNP L-FLAG (anti-FLAG tag) and His-YB-1 (anti-His tag) from HEK293T cells cotransfected with reporters L1-L5 or mutants and His-YB-1 (Y) or hnRNP L-FLAG (L) plasmids. *, a nonspecific band recognized by the anti-His tag antibody. An agarose gel (lower panel) of RT-PCR products of the splicing reporters with the overexpressed hnRNP L or YB-1, similarly labeled as in B.
To determine whether hnRNP L is also able to repress exon inclusion through the CA repeats, we overexpressed hnRNP L-FLAG (L) with the vector DUP175 or the CA repeat-containing splicing reporters L1-L5 (Fig. 3C) with YB-1 (Y), another splicing factor known to interact with A/C-rich elements (49), as a control. Similar to CaMKIV, hnRNP L-FLAG coexpression strongly repressed inclusion of the middle exon of reporters L1 and L3 (lanes 4 and 10) but not of DUP175, L2, L4, and L5 (lanes 2, 8, 14, and 16), neither of the CA repeat mutants (lanes 6 and 12). In contrast, YB-1 coexpression did not change the splicing of any reporter (Y, odd-numbered lanes). Therefore, overexpressed hnRNP L specifically represses exon inclusion through the CA repeats, with the same location dependence as CaMKIV.
hnRNP L Mediates CaMKIV Repression of Splicing through the hnRNP L High Affinity CA Repeat Element Nearby the 3′-Splice Site
To determine whether hnRNP L is essential for the CaMKIV-regulated splicing, we examined the response of reporter L3 to CaMKIV without or with hnRNP L depletion by RNA interference (RNAi, Fig. 4). Exon inclusion of this L3 reporter was again reduced by CaMKIV in cells without hnRNP L depletion (Fig. 4A, lanes 1–4). In contrast, when the endogenous hnRNP L was effectively depleted by RNAi (11% protein left, lane 5, compared with controls), the L3 reporter lost its response to CaMKIV (96% ± 1.0 exon inclusion, n = 4, lane 5). Importantly, coexpression of the exogenous hnRNP L-FLAG protein in the RNAi cells restored the CaMKIV repression of the middle exon inclusion (lane 6). Thus, the loss of CaMKIV repression of exon inclusion by RNAi is indeed due to the specific depletion of hnRNP L.
FIGURE 4. Role of hnRNP L in CaMKIV-regulated splicing through the hnRNP L high affinity CA repeats.

A, effect of hnRNP L depletion by RNAi and rescue with exogenous hnRNP L on CaMKIV-regulated splicing. An agarose gel of RT-PCR products of the reporter L3 expressed in HEK293T cells infected with virus from lentiviral vector (Vec), shRNA against luciferase (shLuc) or hnRNP L (shL) and transfected with CaMKIV (IV), CaMKIV mutant (IVm), and hnRNP L-FLAG (+), with percentages of exon inclusion under each lane. Bottom, Western blots of hnRNP L and loading control hnRNP A1 (aligned with the agarose gel). Results are representative of 2–4 samples per lane. B, UV cross-linking of CA repeat or CG mutant (Mut) RNA probes with HeLa or HEK nuclear extracts (NE). WT, wild type. Upper, the probes containing the 3′-splice site of DUP175 and the CA repeat or CG mutant. Lower, a phosphorimage of HeLa (lanes 1–5) or HEK (lanes 6 –9) nuclear proteins cross-linked to the probes and resolved in a 4 –20% SDS-PAGE gel. Immunoprecipitating antibody (Ab) is against hnRNP L (L) or U2AF65 (C). A sixth of the cross-linking mix for immunoprecipitation (IP) was loaded in lanes 2 and 3 and a fourth in lanes 6 and 7.
To confirm hnRNP L binding to the CA repeats as to the CaRRE1, we cross-linked RNA transcripts containing the CA repeats or its mutant with HeLa or HEK nuclear extracts. The wild type transcript was strongly cross-linked to an ~65-kDa protein (Fig. 4B, lanes 2 and 6). Mutating A to G in the CA repeats almost abolished this protein band (lanes 3 and 7). Importantly, this protein was specifically immunoprecipitated by the antibody against hnRNP L (L, lanes 4 and 8) but barely by the control anti-U2AF65 antibody (C, lanes 5 and 9). Thus, hnRNP L interacts with the RNA probe in a CA repeat-dependent way. Taken together, the data from the RNAi/rescue and UV-cross-linking experiments (Fig. 4) strongly support hnRNP L as an essential factor mediating CaMKIV-regulated splicing through the hnRNP L highly affinity CA repeat element nearby the 3′-splice site.
hnRNP L Also Mediates CaMKIV-regulated Splicing through Short CA Repeats from Alternative Exons Nearby the 3′-Splice Site
To determine whether shorter CA repeats from alternative exons other than the artificially selected hnRNP L high affinity sequence are also sufficient to mediate CaMKIV repression of exon inclusion, we examined several exonic (CA)3–4 elements of human alternative exons from the ASAPII data base (50). Four elements with their flanking A/C-rich sequences were transferred to the same location of the DUP175 as in their endogenous exons and assayed for their responses to CaMKIV (Fig. 5A). Three of four tested sequences caused CaMKIV reduction of exon inclusion, as examplified in gels for the GALNT11 and PTEN2 elements (Fig. 5A, lanes 1 and 2 and lanes 5 and 6). Importantly, mutations of the nucleotide A to G in the CA repeats abolished the responses to CaMKIV (lanes 3 and 4 and lanes 7 and 8). Therefore, shorter CA repeat elements with adjacent A/C-rich sequences nearby the 3′-splice site of alternative exons are also sufficient to mediate CaMKIV-regulated splicing.
FIGURE 5. Role of hnRNP L in CaMKIV-regulated splicing through short CA repeats from alternative exons.

A, CA repeat elements from GALNT11 and PTEN2 genes and mutants replace the corresponding sequences of the vector DUP175 (upper, aligned nucleotides starting from the first nt of the exon). The reporter and mutant responses to CaMKIV in HEK293T cells are shown in agarose gels (lower) of RT-PCR products, with average percentages (±S.D.) of exon inclusion under each lane and exon-included or -excluded products indicated to the right. NC, PCR negative control. * and **, same as in Fig. 2. B, a phosphorimage of an SDS-PAGE gel of UV-cross-linked products of the GALNT11 and PTEN2 element RNA probes (as diagrammed above) of reporters (wild type (WT)) or mutants (Mut) and immunoprecipitated (IP) with anti-hnRNP L (L) or anti-U2AF65 antibody (as a control (C)). The hnRNP L band is indicated to the right. M, molecular weight markers. C, effect of hnRNP L depletion on the CaMKIV-regulated splicing of the PTEN2 reporter. Experiment and gel labeling are similar to that for reporter L3 in Fig. 4A.
Similar to that for the hnRNP L high affinity CA repeat element (Fig. 4B), UV cross-linking and immunoprecipitation experiments indicate that hnRNP L binds the reporter RNA probes, and the CA repeats are critical (Fig. 5B). Moreover, similar tests with the PTEN2 reporter in RNAi/rescue experiments also support hnRNP L as an essential factor in the CaMKIV-regulated splicing (Fig. 5C). Therefore, hnRNP L also mediates CaMKIV-regulated splicing through the shorter CA repeats from endogenous alternative exons nearby the 3′-splice site. Taken together, the tests with hnRNP L high affinity CA repeats and the shorter ones from alternative exons strongly support that, at or in close proximity to the 3′-splice site, the simple CA repeats and hnRNP L are sufficient to confer CaMKIV response on an otherwise non-responsive heterologous gene.
RNA Interference of hnRNP L Expression Alters Endogenous Exon Inclusion and Response to Membrane Depolarization or CaMKIV
To determine whether hnRNP L is essential for depolarization regulation of endogenous exons, we examined endogenous splice variants in GH3 cells where we efficiently knocked down the hnRNP L protein with the specific shRNA as used in Figs. 4 and 5 (Fig. 6A, upper panel). The rat alternative exons of the GALNT11 and PTEN2 genes do not contain the same CA repeats as their human homologues, and their alternative splicing was not detectable in this rat cell line; however, we were able to analyze the STREX exon of the Slo gene as in the previous report (16). STREX inclusion was reduced from 33 to 18% by depolarization in cells without hnRNP L depletion (Fig. 6A, middle panel, lanes 1 and 2), similarly as reported (16). In cells depleted of hnRNP L (lane 3), STREX inclusion was increased to 47%, indicating that hnRNP L is a repressor of STREX exon inclusion, consistent with its cross-linking to the CaRRE1 (Fig. 1). In these hnRNP L-depleted cells, depolarization still repressed the STREX exon inclusion (compare lanes 3 and 4). However, the relative reduction of exon inclusion by depolarization in these hnRNP L-depleted cells (32%, with 15% of net reduction divided by 47%, lanes 3 and 4) is significantly less than that without hnRNP L depletion (45%, with 15% of net reduction divided by 33%, lanes 1 and 2). We also analyzed the human endogenous exons of the GALNT11 and PTEN2 genes in HEK293T cells and found that the GALNT11 exon is alternatively spliced in this cell line and regulated by hnRNP L and CaMKIV (Fig. 6B).
FIGURE 6. Role of hnRNP L in depolarization- or CaMKIV-regulated endogenous exons.
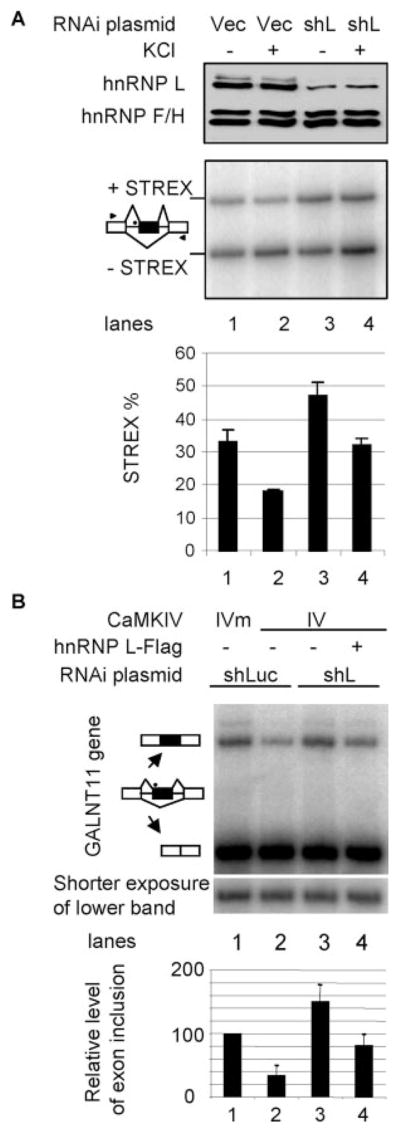
A, effect of hnRNP L depletion by RNAi on STREX exon inclusion and its regulation by depolarization in GH3 cells. Shown are a Western blot (upper panel) and a denaturing PAGE gel of RT-PCR products (middle panel, aligned with the blot) of rat GH3 cells infected with virus made from lentiviral vector (Vec) or plasmid expressing the specific shRNA against hnRNP L (shL) as in Figs. 4A and 5C and not treated (−) or treated (+) with KCl (50 mM) for 12 h. hnRNP F/H is a protein-loading control for the Western blots. The pre-mRNA splicing patterns for the products are indicated to the left, and treatments are indicated above the gels. The lower panel is a bar graph of the exon inclusion levels of the samples aligned with the above gels. The location of the CA repeat in the pre-mRNA intron (horizontal lines) is indicated as a black dot. Arrowheads, locations of PCR primers. B, effect of RNAi depletion of hnRNP L on the CaMKIV regulation of the GALNT11 exon in HEK293T cells as treated in Figs. 4A and 5C. Shown is a denaturing gel of RT-PCR products with the treatments indicated above and splicing patterns to the left of the gel. A shorter exposure of the lower band is shown below. The bar graph at the bottom is the level of exon inclusion (average ± S.D., n = 3) relative to the first lane (taken as 100%). This normalization reduces the effect from varying transfection efficiencies.
The GALNT11 exon is included in about 25% of transcripts in HEK293T cells when CaMKIVm is expressed (Fig. 6B, lane 1). This level was reduced more than half by CaMKIV (lane 2). Interestingly, the reduction was abolished upon knockdown of hnRNP L with the shL plasmid (lane 3). Importantly, the CaMKIV effect was rescued by coexpressed hnRNP L-FLAG (lane 4). Therefore, hnRNP L indeed mediates CaMKIV regulation of the endogenous GALNT11 exon in HEK293T cells.
CaMKIV Enhances hnRNP L Phosphorylation and Its Binding to CaRRE1
To determine how hnRNP L is regulated by CaMKIV, we examined both the protein expression levels and phosphorylation of hnRNP L. No significant changes in the hnRNP L total protein level by depolarization or CaMKIV were observed in GH3 cells or HEK293T cells, respectively. However, a consistent change in hnRNP L binding to CaRRE1 and its phosphorylation with CaMKIV expression was observed (Fig. 7).
FIGURE 7. hnRNP L is a phosphoprotein with its CaRER1 binding activity regulated by CaMKIV and sensitive to treatment with phosphatases.

A, a phosphorimage (upper) of the CaRRE1 probe cross-linked to HEK nuclear extracts of cells transfected with CaMKIVm or CaMKIV, with the protein levels of hnRNP L in the same gel shown below in Western blots after exposure to the phosphorimaging plate, rehydrated, blotted to a polyvinylidene difluoride membrane and probed with anti-hnRNP L antibody. The nuclear extract was made from green cells cotransfected with CaMKIV and enhanced green fluorescent protein plasmids and collected in flow cytometer. M, molecular weight markers. B, a phosphorimage (upper gel) of immunoprecipitated hnRNP L-FLAG from HEK cells coexpressing CaMKIVm or CaMKIV and in vivo labeled with [32P]orthophosphoric acid. At the bottom is the hnRNP L-FLAG protein level in a Western blot of the same gel. C, pretreatment of the nuclear extracts with λ-protein phosphatase (λ-PPase, left panel) or calf intestine alkaline phosphatase (CIAP, right panel) reduces hnRNP L-FLAG binding to the CaRRE1 probe in UV-cross-linking assays. HEK nuclear extracts (NE) from cells cotransfected with hnRNP L-FLAG and CaMKIV was treated (+) or not (−) with the phosphatases and cross-linked with the CaRRE1 RNA probe (upper gels). Western blots of the hnRNP L-FLAG in the same above-corresponding gels are shown (lower gels). Endogenous PTB is also shown in the left panel. The calf intestinal alkaline phosphatase-treated samples were immunoprecipitated (IP) with anti-FLAG antibody after cross-linking to avoid interference from the 65-kDa calf intestinal alkaline phosphatase monomer in gels. The activities of the phosphatases were verified by their ability to reduce the 32P signal of [α-32P]ATP-labeled protein bands in nuclear extracts. Each gel is representative of two to three experiments except the calf intestinal alkaline phosphatase gel (once).
We first examined the binding of endogenous hnRNP L to CaRRE1 with expressed CaMKIVm or CaMKIV in UV cross-linking assays (Fig. 7A). Relative to the cross-linked PTB and the corresponding protein levels in the same gels, the hnRNP L binding to CaRRE1 in CaMKIV-expressed cells was increased about 30% without an apparent change in total protein level.
We next in vivo labeled hnRNP L-FLAG with [32P]-orthophosphoric acid in HEK293T cells and immunoprecipitated it to examine its phosphorylation levels in response to CaMKIV expression (Fig. 7B). The total hnRNP L-FLAG protein signal in the Western blot was increased about four times by CaMKIV, likely due to CaMKIV enhancement of this exogenous gene expression through its CMV promoter in the vector; however, the 32P phosphorylation signal was increased about eight times in the same gel. Therefore, relative to its total protein signal, the level of hnRNP L-FLAG phosphorylation was doubled by the expressed CaMKIV.
To determine whether the hnRNP L phosphorylation in CaMKIV-expressed cells is critical for its binding to the CaRRE1, the HEK293T cell nuclear extract containing coexpressed hnRNP L-FLAG was pretreated with λ-protein phosphatase or calf intestinal alkaline phosphatase, and hnRNP L binding to CaRRE1 was examined with UV cross-linking and/or immunoprecipitation (Fig. 7C). Relative to the loaded protein levels of the same gels/lanes, hnRNP L-FLAG binding to CaRRE1 was reduced ~30% by either λ-protein phosphatase or calf intestinal alkaline phosphatase treatment. Therefore, the hnRNP L-FLAG binding to CaRRE1 was sensitive to dephosphorylation by phosphatases. Collectively these data support that hnRNP L binding to the CaRRE1 can be regulated by CaMKIV through phosphorylation. Taken together with the above evidence, we conclude that hnRNP L is an essential component in CaMKIV-regulated alternative splicing through CA repeats.
DISCUSSION
There have been many examples of cell signal regulation of alternative splicing, but the signal-responsive RNA elements and trans-acting factors are not known in most cases. In this work, our data demonstrate that hnRNP L mediates CaMKIV repression of splicing through CA repeats in a location-dependent way, likely through phosphorylation.
CA Repeats and hnRNP L in CaMKIV-regulated Splicing
The hnRNP L binding simple (CA)≥3 repeats with flanking A/C-rich nucleotides are sufficient to mediate the alternative splicing regulated by CaMKIV (Figs. 2–5). These repeat sequences do not appear similar to the CaRRE2 (GUGGUAGA) or the UAGG motif (17, 18) but share with CaRRE1 the CA dinucleotides.
Compared with the simple CA repeats and the SELEX elements of hnRNP L (27), the CaRRE1 contains three copies of CA dinucleotides and is C/A-rich (Fig. 1A), but it also appears more complex than just CA repeats. Its consensus (CACAUN-RUUAU) contains two copies of CA dinucleotides at its 5′ end (19), but its 3′ half is U-rich and appears more constrained than the 5′ CA dinucleotides (18). Consistently, replacing CaRRE1 with the hnRNP L high affinity CA repeat element conferred only partial response to CaMKIV, in comparison to CaRRE1 (Fig. 2), suggesting that the non-CA nucleotides of CaRRE1 is also required for the full response. Moreover, RNAi knockdown of hnRNP L did not abolish, but potentiated, the CaRRE1-mediated CaMKIV repression of exon inclusion of the DUP175ST reporter,3 implying that more elements/factors are involved in the CaRRE1 function than just the CA repeats and its bound hnRNP L.
hnRNP L binds long intronic CA repeats or exonic CA dinucleotide-containing elements (23, 26–30, 51–54). Consistent with these observations, hnRNP L binds the CA repeat RNA probes of the splicing reporters in a CA-dependent way (Figs. 1, 4, and 5).
Interestingly, the hnRNP L high affinity sequence shows location dependence in response to both CaMKIV and hnRNP L (Figs. 2 and 3). It confers CaMKIV repression only between the polypyrimidine tract and 3′ AG or close to the 3′ AG in the DUP175 reporters (Fig. 3), similar to the location of CaRRE1 (16, 19). In other reporters that allow the detection of splicing enhancer effect, the hnRNP L high affinity sequence (as in Fig. 3A, L2, L4, and L5) is a splicing enhancer when placed in introns further away from the 3′-splice site,3 similarly as seen by others for CA repeat elements (27). The underlying molecular basis of the location dependence remains unknown. Interestingly, at the 3′-splice site, the hnRNP L/p55 (PTB) ratio increased upon depolarization (Fig. 1B) or CaMKIV expression (Fig. 7A), suggesting the rearrangements of hnRNP L and p55 binding during the regulation. At the other locations with different sequences flanking the CA repeats, different binding of hnRNP L and other proteins likely occurred which probably cause differential recruitment of constitutive splicing factors and, thus, a different effect on splicing.
Membrane depolarization increases the nuclear protein level of hnRNP A1 (17). We have not observed similar changes in the hnRNP L protein level in GH3 cells. Interestingly, however, hnRNP L is phosphorylated in the presence of CaMKIV (Fig. 7, B and C). In support of a critical role of its phosphorylation in the CaMKIV-regulated splicing, the hnRNP L binding to CaRRE1 is increased by CaMKIV and reduced by pretreatments with protein phosphatases (Fig. 7).
Recently an hnRNP L homologue hnRNP LL has also been shown to control alternative splicing of CA-containing exons (31–33). This protein was not immunoprecipitated from the nuclear extracts UV-cross-linked to the CaRRE1 probe in our experiments (Fig. 1). Whether hnRNP LL is also a component in the CaMKIV-regulated splicing remains to be determined. On the other hand, it is also interesting to note that the alternative exon targets have little overlap between even closely related hnRNP proteins (55).
Impact of the CaMKIV-regulated Splicing through CA Repeats and hnRNP L on Cell Physiology
Several possible implications are interesting for cell physiology, based on the regulation of splicing by depolarization/CaMKIV through different RNA elements including the CaRRE1 and CaRRE2 (16, 18, 19), the UAGG motif (17), and the CA repeats characterized here. First, CaMKIV could target different groups of alternative exons when these elements are in different groups of genes, making it possible to regulate a specific cellular function/pathway through a particular element. Second, CaMKIV could target different exons of the same gene, for example through CaRRE1 in the exon 5 and others in the exon 21 of the NMDAR1 gene (17, 18). Third, CaMKIV could also target multiple elements in the same exon, for example CaRRE1 and -2 and possibly also the UAGG motif in the NMDAR1 exon 21 (17, 18). Last, because Ca2+ signaling can be activated by different stimuli with distinct effects on gene expression (56), these different elements provide potential targets for specific responses to individual stimulus during splicing. Taken together with the spatial and temporal expression of trans-acting factors, the existence of multiple depolarization/CaMKIV-responsive elements provides diverse targets, likely for the specific and refined control of splicing in response to different stimuli.
At present, the exons containing these CaRRE elements from genome or alternative exon data base searches are mostly not overlapping. For the CA repeats, data base search identified exons of genes involved in cell survival/death; for example, the PTEN2 gene encoding a protein homologue of the tumor suppressor PTEN (57, 58). These are not among the CaRRE1 and -2 or the UAGG motif target genes identified previously (17–19). Taken together (the multiple depolarization/CaMKIV-responsive elements in NMDAR1 exons and the distinct target exons from genome searches with different elements) further work to obtain a complete set of the specific and common target exons of each element will allow a more accurate assessment of the physiological impact of the regulations.
In summary, these experiments identify hnRNP L as an essential factor mediating membrane depolarization/CaMKIV-regulated alternative splicing through short CA repeat elements at or in close proximity to the 3′-splice site. This provides a molecular target for dissecting the components between CaMKIV and the splicing factor as well as the molecular basis of location-dependence of protein kinase-regulated splicing.
Acknowledgments
We are grateful to Stefan Stamm, Peter Stoilov, Thomas Cooper, Owen Witte, and Doug Black for plasmids and antibodies and Steven Pind, Spencer Gibson, Say-Pham Hong, and Hongzhao Li for valuable help. We thank Doug Black for helpful comments.
Footnotes
The abbreviations used are: hnRNP, heterogeneous nuclear ribonucleoprotein; CaMKIV, Ca2+/calmodulin-dependent protein kinase IV; RIPA, radio-immune precipitation assay buffer; STREX, stress axis-regulated exon; shRNA, short hairpin RNA; RT, reverse transcription; RNAi, RNA-mediated interference; PTB, polypyrimidine tract-binding protein.
J. Yu and J. Xie, unpublished observation.
This work was supported by Canadian Institutes of Health Research Grant MOP68919 (to J. X.).
References
- 1.Black DL. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- 2.Black DL. Cell. 2000;103:367–370. doi: 10.1016/s0092-8674(00)00128-8. [DOI] [PubMed] [Google Scholar]
- 3.Modrek B, Resch A, Grasso C, Lee C. Nucleic Acids Res. 2001;29:2850–2859. doi: 10.1093/nar/29.13.2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson JM, Castle J, Garrett-Engele P, Kan Z, Loerch PM, Armour CD, Santos R, Schadt EE, Stoughton R, Shoemaker DD. Science. 2003;302:2141–2144. doi: 10.1126/science.1090100. [DOI] [PubMed] [Google Scholar]
- 5.Graveley BR. Trends Genet. 2001;17:100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 6.Maniatis T, Tasic B. Nature. 2002;418:236–243. doi: 10.1038/418236a. [DOI] [PubMed] [Google Scholar]
- 7.Faustino NA, Cooper TA. Genes Dev. 2003;17:419–437. doi: 10.1101/gad.1048803. [DOI] [PubMed] [Google Scholar]
- 8.Licatalosi DD, Darnell RB. Neuron. 2006;52:93–101. doi: 10.1016/j.neuron.2006.09.017. [DOI] [PubMed] [Google Scholar]
- 9.Shin C, Manley JL. Nat Rev Mol Cell Biol. 2004;5:727–738. doi: 10.1038/nrm1467. [DOI] [PubMed] [Google Scholar]
- 10.Stamm S. Hum Mol Genet. 2002;11:2409–2416. doi: 10.1093/hmg/11.20.2409. [DOI] [PubMed] [Google Scholar]
- 11.Xie J. Biochim Biophys Acta. 2008;1779:438–452. doi: 10.1016/j.bbagrm.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith CW, Valcarcel J. Trends Biochem Sci. 2000;25:381–388. doi: 10.1016/s0968-0004(00)01604-2. [DOI] [PubMed] [Google Scholar]
- 13.Kishore S, Stamm S. Science. 2006;311:230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 14.Buratti E, Baralle FE. Mol Cell Biol. 2004;24:10505–10514. doi: 10.1128/MCB.24.24.10505-10514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rothrock C, Cannon B, Hahm B, Lynch KW. Mol Cell. 2003;12:1317–1324. doi: 10.1016/s1097-2765(03)00434-9. [DOI] [PubMed] [Google Scholar]
- 16.Xie J, Black DL. Nature. 2001;410:936–939. doi: 10.1038/35073593. [DOI] [PubMed] [Google Scholar]
- 17.An P, Grabowski PJ. PLoS Biol. 2007;5:e36. doi: 10.1371/journal.pbio.0050036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JA, Xing Y, Nguyen D, Xie J, Lee CJ, Black DL. PLoS Biol. 2007;5:e40. doi: 10.1371/journal.pbio.0050040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie J, Jan C, Stoilov P, Park J, Black DL. RNA. 2005;11:1825–1834. doi: 10.1261/rna.2171205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matter N, Herrlich P, Konig H. Nature. 2002;420:691–695. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- 21.Hai Y, Cao W, Liu G, Hong SP, Elela SA, Klinck R, Chu J, Xie J. Nucleic Acids Res. 2008;36:3320–3331. doi: 10.1093/nar/gkn207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Melton AA, Jackson J, Wang J, Lynch KW. Mol Cell Biol. 2007;27:6972–6984. doi: 10.1128/MCB.00419-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rothrock CR, House AE, Lynch KW. EMBO J. 2005;24:2792–2802. doi: 10.1038/sj.emboj.7600745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dib C, Faure S, Fizames C, Samson D, Drouot N, Vignal A, Millasseau P, Marc S, Hazan J, Seboun E, Lathrop M, Gyapay G, Morissette J, Weissenbach J. Nature. 1996;380:152–154. doi: 10.1038/380152a0. [DOI] [PubMed] [Google Scholar]
- 25.Perucho M. Biol Chem. 1996;377:675–684. [PubMed] [Google Scholar]
- 26.Hui J, Stangl K, Lane WS, Bindereif A. Nat Struct Biol. 2003;10:33–37. doi: 10.1038/nsb875. [DOI] [PubMed] [Google Scholar]
- 27.Hui J, Hung LH, Heiner M, Schreiner S, Neumuller N, Reither G, Haas SA, Bindereif A. EMBO J. 2005;24:1988–1998. doi: 10.1038/sj.emboj.7600677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheli Y, Kunicki TJ. Blood. 2006;107:4391–4398. doi: 10.1182/blood-2005-12-4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffith BN, Walsh CM, Szeszel-Fedorowicz W, Timperman AT, Salati LM. Biochim Biophys Acta. 2006;1759:552–561. doi: 10.1016/j.bbaexp.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tong A, Nguyen J, Lynch KW. J Biol Chem. 2005;280:38297–38304. doi: 10.1074/jbc.M508123200. [DOI] [PubMed] [Google Scholar]
- 31.Hung LH, Heiner M, Hui J, Schreiner S, Benes V, Bindereif A. RNA. 2008;14:284–296. doi: 10.1261/rna.725208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Science. 2008;321:686–691. doi: 10.1126/science.1157610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Topp JD, Jackson J, Melton AA, Lynch KW. RNA. 2008;14:2038–2049. doi: 10.1261/rna.1212008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKee AE, Neretti N, Carvalho LE, Meyer CA, Fox EA, Brodsky AS, Silver PA. Genome Biology. 2007;8:R159. doi: 10.1186/gb-2007-8-8-r159. http://genomebiology.com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saito M, Nelson C, Salkoff L, Lingle CJ. J Biol Chem. 1997;272:11710–11717. doi: 10.1074/jbc.272.18.11710. [DOI] [PubMed] [Google Scholar]
- 36.Xie J, McCobb DP. Science. 1998;280:443–446. doi: 10.1126/science.280.5362.443. [DOI] [PubMed] [Google Scholar]
- 37.Dominski Z, Kole R. Mol Cell Biol. 1991;11:6075–6083. doi: 10.1128/mcb.11.12.6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miranti CK, Ginty DD, Huang G, Chatila T, Greenberg ME. Mol Cell Biol. 1995;15:3672–3684. doi: 10.1128/mcb.15.7.3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chatila T, Anderson KA, Ho N, Means AR. J Biol Chem. 1996;271:21542–21548. doi: 10.1074/jbc.271.35.21542. [DOI] [PubMed] [Google Scholar]
- 40.Xie J, Lee JA, Kress TL, Mowry KL, Black DL. Proc Natl Acad Sci U S A. 2003;100:8776–8781. doi: 10.1073/pnas.1432696100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Radu CG, Yang LV, Riedinger M, Au M, Witte ON. Proc Natl Acad Sci U S A. 2004;101:245–250. doi: 10.1073/pnas.2536801100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin XF, An DS, Chen IS, Baltimore D. Proc Natl Acad Sci U S A. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.An DS, Kung SK, Bonifacino A, Wersto RP, Metzger ME, Agricola BA, Mao SH, Chen IS, Donahue RE. J Virol. 2001;75:3547–3555. doi: 10.1128/JVI.75.8.3547-3555.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Black DL, Chan RC, Min H, Wang J, Bell L. In: RNA:Protein Interactions, A practical approach. Smith C, editor. Oxford University Press; New York: 1998. pp. 109–136. [Google Scholar]
- 45.Schreiber E, Matthias P, Muller MM, Schaffner W. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ma S, Liu G, Sun Y, Xie J. Biochim Biophys Acta. 2007;1773:912–923. doi: 10.1016/j.bbamcr.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 47.Chan RC, Black DL. Mol Cell Biol. 1997;17:4667–4676. doi: 10.1128/mcb.17.8.4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chou MY, Underwood JG, Nikolic J, Luu MH, Black DL. Mol Cell. 2000;5:949–957. doi: 10.1016/s1097-2765(00)80260-9. [DOI] [PubMed] [Google Scholar]
- 49.Stickeler E, Fraser SD, Honig A, Chen AL, Berget SM, Cooper TA. EMBO J. 2001;20:3821–3830. doi: 10.1093/emboj/20.14.3821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kim N, Alekseyenko AV, Roy M, Lee C. Nucleic Acids Res. 2007;35:93–98. doi: 10.1093/nar/gkl884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shih SC, Claffey KP. J Biol Chem. 1999;274:1359–1365. doi: 10.1074/jbc.274.3.1359. [DOI] [PubMed] [Google Scholar]
- 52.Hui J, Reither G, Bindereif A. RNA. 2003;9:931–936. doi: 10.1261/rna.5660803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liu X, Mertz JE. Genes Dev. 1995;9:1766–1780. doi: 10.1101/gad.9.14.1766. [DOI] [PubMed] [Google Scholar]
- 54.Guang S, Felthauser AM, Mertz JE. Mol Cell Biol. 2005;25:6303–6313. doi: 10.1128/MCB.25.15.6303-6313.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venables JP, Koh CS, Froehlich U, Lapointe E, Couture S, Inkel L, Bramard A, Paquet ER, Watier V, Durand M, Lucier JF, Gervais-Bird J, Tremblay K, Prinos P, Klinck R, Elela SA, Chabot B. Mol Cell Biol. 2008;28:6033–6043. doi: 10.1128/MCB.00726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bading H, Ginty DD, Greenberg ME. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- 57.Walker SM, Downes CP, Leslie NR. Biochem J. 2001;360:277–283. doi: 10.1042/0264-6021:3600277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forgacs E, Biesterveld EJ, Sekido Y, Fong K, Muneer S, Wistuba II, Milchgrub S, Brezinschek R, Virmani A, Gazdar AF, Minna JD. Oncogene. 1998;17:1557–1565. doi: 10.1038/sj.onc.1202070. [DOI] [PubMed] [Google Scholar]