

Abstract
We demonstrated a unique optofluidic lab-on-a-chip device that can measure optically encoded forward scattering signals. From the design of the spatial pattern, we can measure the position and velocity of each cell in the flow and generate a 2-D cell distribution plot over the cross section of the channel. Moreover, we have demonstrated that the cell distribution is highly sensitive to its size and stiffness. The latter is an important biomarker for cell classification and our method offers a simple and unequivocal method to classify cells by their size and stiffness. We have proved the concept using live and fixed HeLa cells. Due to the stiffness and size difference of neutrophils compared to other types of white blood cells, we have demonstrated detection of neutrophils from other blood cells. Finally, we have performed the test using 5 μL of human blood. In a greatly simplified blood preparation process, skipping the usual steps of anticoagulation, centrifuge, antibody labelling or staining, filtering, etc., we have demonstrated that our device and detection principle can count neutrophils in whole human blood. Our system is compact, inexpensive and simple to fabricate and operate, having a commodity laser diode and a Si PIN photoreceiver as the main pieces of hardware. Although the results are still preliminary, the studies indicate that this optofluidic device holds promise to be a point-of-care and home care device to measure neutrophil concentration, which is the key indicator of the immune functions for cancer patients undergoing chemotherapy.
Introduction
Detection and classification of cells using sophisticated tools, such as flow cytometers and fluorescence-activated-cell-sorters (FACS) are often performed clinically for disease diagnosis and prognosis. To reduce cost, time from test to outcome, as well as chances of hospital infection, performing such tests in point-of-care clinics or the patient’s residence is highly attractive, provided that the devices are able to generate reliable results, are easy to operate, compact and affordable.1–3
Today most sophisticated equipment for cell-based assays is located in major medical centers. The high cost of equipment acquisition, maintenance and medical personnel, as well as the support of the massive outfit and infrastructure, contributes to the high and rising health care cost in most developed countries, particularly the United States. The lack of such sophisticated medical equipment in resource-limited countries and territories, on the other hand, presents major challenges in delivering quality health care services to populations living in these areas. To address this barrier in health care, development of a new class of medical devices that are suitable for point-of-care or personal use has become a global focus of biomedical device research.
To meet this challenge, lab-on-a-chip devices that combine microfluidics with other technologies have attracted significant attention. Since blood and bodily fluids are the easiest samples to acquire with minimal invasiveness, and such samples, particularly blood, contain rich health and disease information, a large family of lab-on-a-chip devices are microfluidic devices that handle blood and bodily fluids. In general terms, they can be considered as different types of lab-on-a-chip flow cytometers.4,5 Various effects, including acoustic,6 magnetic,7 optical 8,9 and electrical 10,11 mechanisms have been incorporated into the microfluidic devices to manipulate and detect cells, beads and biomolecules in lab-on-a-chip flow cytometers. However, most existing methods require labelling to detect or isolate specific subpopulations of cells from the sample mixture,12,13 and require sample preparation procedures, such as anticoagulation, antibody labelling, staining, centrifugation, filtering, etc., that are too complicated to be handled by people without medical training. To address this issue, we suggest that instead of developing general purpose systems, such as full-bloomed flow cytometers, our chance of success in moving the lab-on-a-chip technology to clinics will be significantly better if we develop devices for specific applications, targeting specific biomarkers without labelling. To meet this objective, three issues need to be addressed: (1) identify an effective biomarker or markers for unequivocal determination of the condition of a certain disease, (2) develop an accurate and low-cost method to read the biomarker signals, and (3) develop straightforward and minimally invasive sample extraction and processing procedures. In this paper, in addition to cell size, which has been used to classify cells since the invention of the Coulter counter, we use cell stiffness as an additional biomarker, because the stiffness of cells is cell type specific 14 and also gives information about the health and life cycle of cells.15 To detect the cell effects in microfluidics attributed to cell stiffness, we invented a unique method of optical space-time coding 16 that enables us to unambiguously measure the position of each cell travelling in the microfluidic channel. Using the principle that the stable positions of cells in a microfluidic channel depend upon cell size and stiffness, we can classify cells by their stiffness in the flow channel at very high throughput. Since the detection method needs only a semiconductor diode laser, and a Si PIN photoreceiver requires no sheath flow for flow confinement, the system is simple and compact and can be fabricated at very low cost. Finally, the test draws only a minimum amount of peripheral blood (~5 μL) similar to the blood draw for a glucose test and requires minimum sample processing.
To demonstrate point-of-care and home care functions, we have applied the device to measure the neutrophil concentration in the blood because neutrophil concentration is an important indicator for the functions of the immune system.17 For a healthy person, the total concentration of white blood cells is between 4000 and 11 000 μL−1 of blood, with 53–62% of white blood cells being neutrophils and the majority of the rest being lymphocytes. A neutrophil is a granulocyte named by the shape of its nucleus and is softer than monocytes and lymphocytes,18,19 a property that facilitates its migration to the sites of infection from blood vessels. Clinically, a patient is considered to be in mild neutropenia if the neutrophil count is between 1000 and 1500 μL−1, in moderate neutropenia when the neutrophil density drops to 500–1000 μL−1, and in severe neutropenia when the neutrophil density falls below 500 μL−1. Severe neutropenia leads to a very high risk of infection, which could be life threatening, particularly for cancer patients undergoing chemotherapy.20 Each year over 90 000 patient deaths result from hospital infection in the United States.21 Cancer patients undergoing chemotherapy typically make 12 to 24 hospital visits, often only for neutrophil counts. This represents a particularly high risk group and therefore a device that can easily and accurately measure the immune function of cancer patients is tremendously valuable. Therefore, we have chosen such a device for demonstration of clinical applications of lab-on-a-chip technologies.
We have designed and fabricated a simple optofluidic lab-on-a-chip device consisting of a straight microfluidic channel with spatially coded patterns that modulate the excitation light intensity experienced by the particle passing the patterns. The technique effectively converts a spatial pattern (code) into an optical intensity-modulated time-domain signal that can be readily processed by digital signal processing (DSP) algorithms.
The method of converting a spatial pattern into a temporal signal has been explored by a few groups.8,22,23 However, the previous methods detect only fluorescent signals for labelled cells and cell labelling is a dedicated and expensive process that cannot be done correctly and reliably by persons without medical training and access to proper facilities. Above all, these earlier methods are elegant ways to detect the same information as a conventional flow cytometer does, thus producing no new information about cell properties. Recently, our group reported a specially designed optical space–time coding method that can detect new information, namely the position of the cell in the channel and its travel speed, which is not detectable by the previous methods.24 With the designed pattern, the waveform of the optical forward scattering (FS) signal directly corresponds to the cell position inside the microfluidic channel. After applying the DSP technique to decipher the FS waveform, we can measure the cell position very precisely. The equilibrium position of cells inside a microfluidic channel is highly useful because it directly results from cell size and stiffness.25–28 Thus, one can use cell position to distinguish softer and larger neutrophils from red blood cells and other types of white blood cells.
Furthermore, since the space–time coded signal is carried in both forward and side scattering signals, we have the option to only detect the forward scattering (FS) signal from which all of the needed information, such as cell position and velocity, can be obtained. This makes the system simple, robust, compact and inexpensive since the forward scattering signal is orders of magnitude stronger than the side scattering signal, thus requiring no sophisticated optics or photomultiplier tubes (PMTs) to perform the measurements.
Experimental methods
Device design and operation principle
To encode the position and velocity information into the FS signal from a travelling cell, we designed a mask pattern that contains four trapezoidal slits, as shown in Fig. 1(a). A semiconductor laser source was used to illuminate from the bottom of the microchannel. Through these slit openings, the forward scattering signal was detected by the Si photodiode on the opposite side of the semiconductor laser diode. The four-slit mask was formed by Ti/Au (100 nm/200 nm) metal layers deposited on a glass slide using standard E-beam evaporation and a metal lift-off process. Each trapezoidal slit has base lengths of 100 μm and 50 μm and the four slits were arranged in an alternating pattern with a separation of 50 μm to form a sensing area of a total pattern length of 450 μm, as shown in Fig. 1(a). Depending on the trajectory of moving samples, the forward scattering signals were encoded by the spatial mask and exhibited distinct waveforms the in time domain (Fig. 1(b)). We later used these waveforms to obtain the positions of these cells in the microfluidic channel. The microfluidic channel is 5 cm long and has its inlet and outlet at the ends. The channel has a rectangular cross section, being 100 μm wide and 45 μm high. The spatial mask was placed at 4.5 cm from the inlet. A PIN silicon photoreceiver was used to collect 5–10 degree FS signals. For convenience of discussion, we define the direction of channel width as the x-axis and the direction of channel height as the y-axis. The intensity-modulated FS signal by the trapezoidal slits displays 4 peaks (Fig. 1(b)). The ratio between the width of the first peak (W1) and the second peak (W2) yields information about the cell position in the x-axis. The same information can also be obtained from the ratio of W4 and W3. The redundancy helps reduce noise and improve measurement accuracy. The velocity of the cell is acquired by dividing the total pattern length (450 μm) by the duration of the signal. Knowing the position in the x-axis and the velocity, the cell position in the y-axis can be obtained using the property of laminar flow that gives rise to a parabolic velocity profile represented by the simple relation: υ(x, y) = υMax(x) [1 − (y/h)2], wherein υ is the velocity at position (x, y), υMax is the maximal velocity at position (x,0) (we define y = 0 to be the line across the middle of the channel height) and h represents the half-height of the channel.
Fig. 1.

(a) A schematic representation of the encoded microfluidic device and the system setup. (b) An encoded forward scattering signal detected by the silicon PIN photoreceiver.
The knowledge of both the position and speed of each cell gives us an insight into the property of the cells. Previous studies suggest that when the ratio between the particle diameter and the channel dimension is greater than 0.07,25 the inertial effect would focus the particles toward equilibrium positions inside the channel. In addition, if similar sized cells have different stiffness, the more deformable cells tend to migrate laterally toward the central area in the x-axis.26–28
Device fabrication and system setup
The device in Fig. 1(a) was fabricated using a polydimethylsiloxane (PDMS) (Sylgard 184, Dow Corning, MI) soft lithography process. After curing, the PDMS film was separated from the silicon mold master. Holes for inlet and outlet were punched through the PDMS replica. On a separate glass slide, a Ti/Au film (100/200 nm in thickness) was deposited and lithographically patterned to form a spatial mask. With oxygen plasma treatment of the PDMS surface, the PDMS film patterned with the microfluidic channel and inlet and outlet was bonded to the glass slide. An alignment apparatus was used to assure good alignment between the Ti/Au patterns and the microfluidic channel. A semiconductor diode laser was used as the light source and a silicon PIN photoreceiver was used to detect FS signals. A syringe pump (Harvard Apparatus, Pump Elite 11) was used to inject samples to the device at designed flow rates. After the measurements of forward scattering signals, a custom signal processing algorithm implemented in MATLAB was used to remove noise and extract events. To characterize the device and test the operation principle, we at first introduced only one cell type at a time before testing samples with mixed cell types.
Sample preparation
For device characterization with selected cell types, we have prepared samples with cancer cells (HeLa cells), red blood cells (RBCs) and white blood cells (WBCs). HeLa (human cervical epithelioid carcinoma) cells were cultured in the growth medium in a humidified incubator at 37 °C in 5% CO2. For fixed HeLa cells, paraformaldehyde was used prior to the experiment. Whole blood samples used to produce RBC and WBC samples were purchased from the blood bank. To prepare white blood cell samples, whole blood was lysed with a commercial lysing buffer (eBioscience, CA). To prepare RBC samples, 100 μL of whole blood was centrifuged at 10 000 rpm. 5 μL of red blood cells was carefully taken out from the bottom of canonical tubes (below the buffy coat) and then diluted 2 million times using a buffer solution. The buffer solution used for sample dilution consists of 10 mM ethylenediaminetetraacetic acid (EDTA), 1% bovine serum albumin (BSA) and 1 × phosphate buffered saline (PBS).
For neutrophil counting tests, 5 μL of whole blood was taken from healthy volunteers using a finger prick. The blood was first diluted with 1 mL of buffer solution. Then, 500 μL RBC lysing agent was added to 150 μL of the diluted blood to create a total dilution of ~ 870 × in the WBC concentration. After keeping the sample in the ambient environment for 10 min, the blood mixed with the lysing agent was introduced into the microfluidic device for measurement without further preparation.
Results and discussion
Live and fixed HeLa cell detection
Signals from live and fixed HeLa cells were first characterized using the space–time optically encoded signal from the lab-on-a-chip optofluidic device, since live and fixed HeLa cells are known to have different stiffness. HeLa cells were suspended in PBS buffer solution (ρ = 103 kg m−3, μ = 1.05 × 10−3 Pa·s). The live and fixed cells were tested separately at a flow rate of 50 μL min−1. The dimensions of our device produce a micro-channel hydraulic diameter, Dh = (2w × h)/(w + h), of 62.1 μm, where w and h are the width and height of the channel. Under the average flow velocity of 18 cm s−1 for 50 μL min−1, the Reynolds number (Re) is calculated to be 11.2. More than 1000 cells were detected within a measurement period of 90 s and the encoded FS signals were processed using custom developed algorithms implemented in MATLAB.24 The signals first pass a high pass filter and a low pass filter to remove noise and baseline drift. Then, a peak detection algorithm based on a predefined threshold was applied to register each event. With the aid of COMSOL simulation to retrieve the maximum flow velocity (ESI†Fig. 1), the spatial distribution of cells was obtained using the method discussed in the previous section. Fig. 2 shows the distribution of fixed and live HeLa cells over the cross section of the microfluidic channel. Detailed analyses on the HeLa cell distribution along the x-axis show a standard deviation (σx) of 12.89 μm for fixed cells and 8.66 μm for live cells at the Reynolds number of 11.2. Similar phenomena have been observed using a CCD camera and explained in the literature,29,30 although those measurements were made from a time averaged intensity profile of fluorescent beads instead of individual unlabelled cells.
Fig. 2.

Population distribution plots for fixed HeLa cells (left) and live HeLa cells (right) over the cross section (100 μm × 45 μm) of the microfluidic channel. The Reynolds number of the sample is 11.2.
A more prominent and useful feature for cell classification in Fig. 2(a) and (b) is that both live and fixed HeLa cells were tightly focused to narrow positions in the y-axis. At the Reynolds number of 11.2, the distribution of fixed HeLa cells was centered at 8.72 μm from the channel wall with a standard deviation (σy) of 1.03 μm. As a comparison, when live HeLa cells were introduced into the microfluidic device under the same conditions, their distribution was centered at 9.34 μm from the channel wall with a standard deviation (σy) of 0.73 μm. Given the ~20 μm average size of HeLa cells, the above represent an extremely tight cell distribution along the y-axis and manifest the high accuracy of our measurement technique.
Earlier reports showed that cell fixation by paraformaldehyde hardens cells due to the crosslink of proteins.31 As indicated in Fig. 2, the observation of live HeLa cells moving farther away from the channel wall compared to the stiffer fixed HeLa cells indicated that cells of the same size experienced a force influenced by their stiffness.32 In the microfluidic channel, cells generally experience a lift force, mainly determined by their size, to achieve their equilibrium position, which is about ~0.2h from the channel wall.33–35 The very tight population distribution represents steep energy minima. However, the most exciting discovery from the experiment is the first direct observation of the effect of cell stiffness on the cell distribution in a microfluidic channel. The flow between cells and the channel wall creates a repulsive lubrication force that results in a negative velocity and drives cells toward the channel wall. Because softer cells are more deformable to become thinner in the direction of force, the flow velocity difference across the cell is smaller than that of a stiffer cell.32 Therefore, softer cells have their equilibrium positions farther away from the channel wall and travel faster than the stiffer cells. Our results in Fig. 2 agree well with the theory and show the feasibility of using cell stiffness as a biomarker for cell distinction. Like most circulating cancer cells that migrate through the tissue barrier to enter the blood vessel to metastasize,36 live HeLa cells are more deformable than most normal cells. The equilibrium position of cells of different stiffness in a microfluidic channel might be used to identify cell phenotype or cell cycles.
Classification of white blood cells and red blood cells
Next we performed an experiment to separate white blood cells (WBCs) and red blood cells (RBCs) where the two groups of cells have different sizes and stiffness. We first characterize the WBCs and RBCs separately. After dilution of the RBC-lysed whole blood to a WBC concentration of around 10 counts μL−1, WBCs were introduced into the device at the Reynolds number of 5.6. To test the RBCs, whole blood was diluted by 2 million times and introduced into the device at the Reynolds number of 5.6. A minimum of 1500 encoded FS signals from each cell type were detected and analysed to create the population distribution plots. Since the previous HeLa cell study shows a tight distribution along the y-axis and the cell velocity approximately follows the parabolic function of the y-position in a rectangular channel, we hypothesized that velocity could be used as a convenient parameter for cell separation. Fig. 3(a) shows the histogram for RBCs and WBCs using cell velocity as the parameter, and the results clearly support our hypothesis. The detailed distribution plots for RBCs and WBCs are shown in Fig. 3(b). As predicted, two distinctive regimes for WBCs and RBCs are present and the separation is primarily along the y-axis. From Fig. 3(b), RBCs are mostly populated at a distance of 5.09 μm from the channel wall in the y-axis, with σy being 0.94 μm. In contrast, larger WBCs are mostly populated at a distance of 9.93 μm from the channel wall, with σy being 1.80 μm. If we define “gating” in the x–y plane in a similar fashion to user-defined gating with a commercial flow cytometer, then the separation between RBCs and WBCs can be even more distinctive and accurate than the single parameter plot in Fig. 3(a). The experimental data implied that the inertial force induced by the size of cells dominates the distribution. Even though RBCs appear to be softer than WBCs, WBCs were farther away from the channel wall and travel faster because of their significantly larger size than RBCs. The results show that both cell size and cell stiffness contribute to the equilibrium position of cells in a microfluidic channel.
Fig. 3.
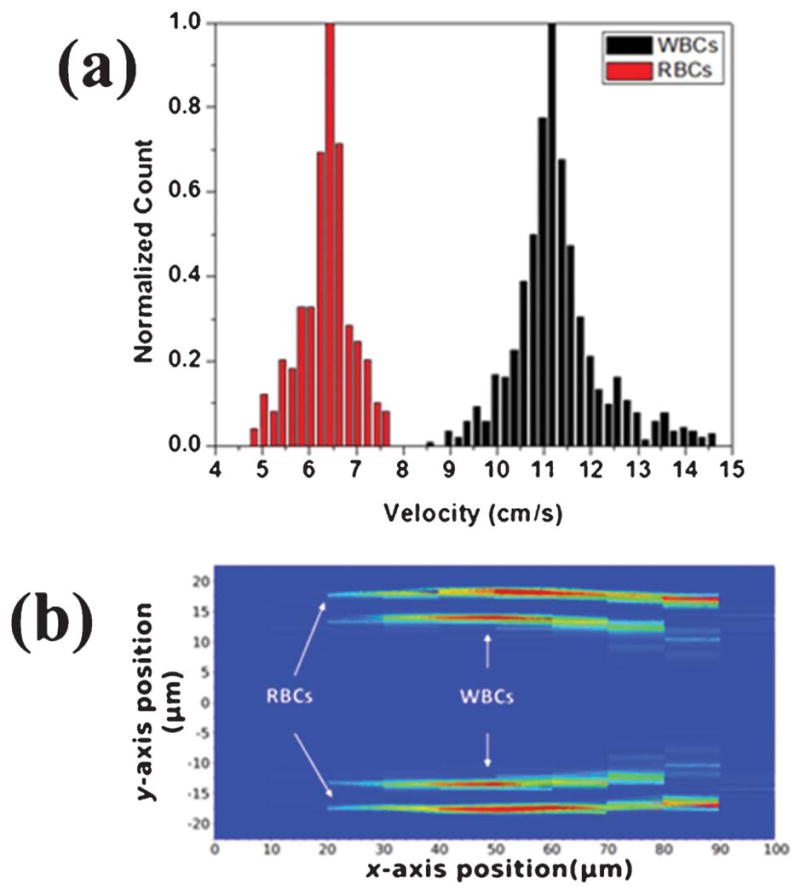
WBCs and RBCs tested separately at Re = 5.6. (a) A histogram of WBCs and RBCs using cell velocity as the parameter. (b) Superimposed population distributions for WBCs and RBCs within the microfluidic channel.
Neutrophil counting from whole blood
Using cell size and stiffness as biomarkers that can be faithfully represented in the population distribution plot, we exploited the feasibility of counting neutrophil population from a very small amount (5 μL) of whole blood with minimum sample processing steps. Neutrophil count reveals the condition of the immune function and has been used as a key parameter for drug dosage and cytotoxicity in chemotherapy. To reduce the chance of hospital infection and cost, it is desirable to perform a neutrophil test at point-of-care clinics or even more preferably, at the patient’s residence, in a self-administered fashion. Therefore, in this experiment we have limited the blood sample to 5 μL. Without adding any anticogulation agent, the blood sample was diluted in a buffer solution to a total volume of 1 mL with RBC lysing agent. The RBC-lysed diluted blood was directly introduced to the device for testing without centrifugation and pipetting. Thus, the sample preparation procedure and the equipment requirement has been reduced to a minimum level, suitable for a self-administered test.
Within the WBC family, there exists a diverse size distribution among different subpopulations of WBCs. Essentially WBCs form a continuum in their size distribution, making cell classification by size alone very difficult without expensive flow cytometers that can give out very low intrinsic values of coefficients of variation (CVs). Since we discovered a method to measure cell positions within the channel and the cell positions depend strongly on cell stiffness, we have found an approach to count neutrophils from other types of WBCs, such as lymphocytes and monocytes, because of the high deformability of neutrophils. For example, being of similar size, neutrophils are more than twice as deformable as monocytes. Neutrophils are slightly softer than lymphocytes, 18 but also appreciably larger. All these are in favor of separating neutrophils from other WBCs (i.e. lymphocyes and monocytes) for an accurate neutrophil count.18
Taking 5 μL whole blood from purchased blood and from healthy volunteers, we prepared samples for neutrophil counts after RBC lysing and blood dilution. At Re = 16.8, 112.5 μL of diluted blood sample passed through the sensing area of the microfluidic device within 90 s, and the histogram of WBC velocity is shown in Fig. 4(a). The histogram shows a subpopulation of WBCs at a higher speed (i.e. velocity > 30 cm s−1) than the rest of population, which is believed to be from lymphocytes, partially monocytes and RBC residues since the samples were not centrifuged after lysing. The data became more revealing when presented in the cell distribution plot, as illustrated in Fig. 4(b), where the cells with the velocity over 30 cm s−1 are marked as red and considered as neutrophils. The population of neutrophils is clearly separated from that of other cells. To further verify the results from the microfluidic devices, we performed more blood tests from healthy donors and compared our results with the results from a commercial flow cytometer. In each run we passed 112.5 μL of diluted blood through our device in 90 s and 200 μL of diluted blood through a commercial flow cytometer in 15 min. The flow cytometer results in Fig. 4(c) show three populations, representing the major groups of WBCs. Table 1 summarizes the results of the neutrophil count from our device and the flow cytometer. Except for run 1, the results from our device agree with the flow cytometer results within 10%.
Fig. 4.

(a) A velocity histogram of white blood cells from 5 μL RBC-lysed whole blood sample (870 × dilution) at Re = 16.8. The histogram shows a distinguishable population of neutrophils due to their high deformability. The peak on the left of the neutrophil (i.e. V < 30 cm s−1) is the signal from other WBC types plus RBC residues, since the sample did not go through a centrifuge. (b) The distribution of RBC-lysed whole blood sample converted from (a) shows a separate band for neutrophils (red) from other cells (green). (c) A scatter plot of 5 μL RBC-lysed diluted whole blood sample (870 × vdilution) measured with a commercial flow cytometer (Accuri C6). Three populations clearly exhibit major WBC groups as the gating indicated.
Table 1.
A summary of test results from 870 × diluted RBC-lysed blood samples using our device and a commercial flow cytometer (Accuri C6)
| Trial | I | II | III | IV |
|---|---|---|---|---|
| Our device (neutrophil count μL−1) | 5.80 | 7.74 | 5.72 | 6.20 |
| Accuri C6 (neutrophil count μL−1) | 4.38 | 7.09 | 6.39 | 6.77 |
Conclusions
We demonstrated a unique optofluidic lab-on-a-chip device that can measure optically encoded forward scattering signals. From the design of the spatial pattern, we are able to acquire the position and velocity of each cell in the flow and generate 2-D cell distribution plots over the cross section of the channel. Moreover, we have demonstrated that the cell distribution within the microfluidic channel is highly sensitive to cell size and stiffness. The latter is an important biomarker for cell classification and our method offers a simple and unequivocal method to classify cells by their stiffness. We have proved this concept using live and fixed HeLa cells. Due to the softness of neutrophils compared to other types of white blood cells, we have demonstrated detection of neutrophils from other blood cells. Finally, we have performed the test using a very small amount of human whole blood. In a greatly simplified blood preparation process, skipping the usual steps of anticoagulation, centrifugation, antibody labelling or staining, filtering, etc., we have demonstrated that our device and detection principle can count neutrophils in whole human blood.
Our system is compact, inexpensive and simple to fabricate and operate, having a commodity laser diode and a Si PIN photoreceiver as the main hardware. Although the results are still preliminary, the studies indicate that this optofluidic device holds promise to be a point-of-care and home care device that measures the immune function of patients, which is of great value to cancer patients undergoing chemotherapy.
Supplementary Material
Acknowledgments
We acknowledge the technical support of the staff in the Nano3 (Nanoscience, Nanoengineering, Nanomedicine) Facility in Calit-2. We acknowledge Dr Jose Morachis for providing blood samples and HeLa cells for the experiment. One of the authors, Zhe Mei, was supported by Chinese Scholarship Council and National Natural Science Foundation of China No.61001063. The project described was supported by Grant Number NIH 1R43RR032225 and NIH 1R21RR024453. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
Footnotes
Published as part of a themed issue on optofluidics
Electronic Supplementary Information (ESI) available. See DOI: 10.1039/c2lc40560a
References
- 1.Toner M, Irimia D. Annu Rev Biomed Eng. 2000;7:77. doi: 10.1146/annurev.bioeng.7.011205.135108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yager P, Edwards T, Fu E, Helton K, Nelson K, Tam MR, Weigl BH. Nature. 2000;442:412. doi: 10.1038/nature05064. [DOI] [PubMed] [Google Scholar]
- 3.Whitesides GM. Nature. 2006;442:368. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 4.Cho SH, Chen CH, Tsai FS, Godin J, Lo YH. Lab Chip. 2010;10:1567. doi: 10.1039/c000136h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godin J, Chen CH, Cho SH, Qiao W, Tsai F, Lo YH. J Biophotonics. 2008;5:355. doi: 10.1002/jbio.200810018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nam J, Lim H, Kim D, Shin S. Lab Chip. 2011;11:3361. doi: 10.1039/c1lc20346k. [DOI] [PubMed] [Google Scholar]
- 7.Forbes TP, Forry SP. Lab Chip. 2012;12:1471. doi: 10.1039/c2lc40113d. [DOI] [PubMed] [Google Scholar]
- 8.Cho SH, Qiao W, Tsai FS, Yamashita K, Lo YH. Appl Phys Lett. 2010;97:1. doi: 10.1063/1.3481695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dochow S, Krafft C, Neugebauer U, Bocklitz T, Henkel T, Mayer G, Albert J, Popp J. Lab Chip. 2011;11:1484. doi: 10.1039/c0lc00612b. [DOI] [PubMed] [Google Scholar]
- 10.Qiao Wen, Cho Gyoujin, Lo Y-H. Lab Chip. 2011;11:1074. doi: 10.1039/c0lc00457j. [DOI] [PubMed] [Google Scholar]
- 11.Barat D, Spencer D, Benazzi G, Mowlem MC, Morgan H. Lab Chip. 2012;12:118. doi: 10.1039/c1lc20785g. [DOI] [PubMed] [Google Scholar]
- 12.Holmes D, Morgan H. Anal Chem. 2010;82:1455. doi: 10.1021/ac902568p. [DOI] [PubMed] [Google Scholar]
- 13.Yun H, Bang H, Min J, Chung C, Chang JK, Han DC. Lab Chip. 2010;10:3243. doi: 10.1039/c0lc00041h. [DOI] [PubMed] [Google Scholar]
- 14.Hou HW, Bhagat AAS, Chong AGL, Mao P, Tan KSW, Han J, Lim CT. Lab Chip. 2010;10:2605. doi: 10.1039/c003873c. [DOI] [PubMed] [Google Scholar]
- 15.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Science. 1997;276:1425. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 16.Wu TF, Mei Z, Pion-Tonachini L, Zhao C, Qiao W, Arianpour A, Lo YH. AIP Adv. 2011;1:022155. doi: 10.1063/1.3609967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crawford J, Dale DC, Lyman GH. Cancer. 2004;100:228. doi: 10.1002/cncr.11882. [DOI] [PubMed] [Google Scholar]
- 18.Worthen GS, Downey GP, Henson P, Doherty D, Schwab B, Elson E, Henson P, Worthen G. Applied Physiology. 1990;69:1767. doi: 10.1152/jappl.1990.69.5.1767. [DOI] [PubMed] [Google Scholar]
- 19.Waugh RE, Hochmuth RM. CRC. Chapter 32: Mechanics and deformability of hematocytes. In: Bronzino JD, editor. Biomedical Engineering Fundamentals. 2. Boca Raton, FL: 2000. [Google Scholar]
- 20.Sipsas NV, Bodey GP, Kontoyiannis DP. Cancer. 2005;103:1103. doi: 10.1002/cncr.20890. [DOI] [PubMed] [Google Scholar]
- 21.Laxminarayan R, Smith DL, Real LA, Levin SA. Discov Med. 2005;5:303. [PubMed] [Google Scholar]
- 22.Kiesel P, Bassler M, Beck M, Johnson N. Appl Phys Lett. 2009;94:041107. [Google Scholar]
- 23.Kiesel P, Beck M, Johnson N. Cytometry A. 2011;79:317. doi: 10.1002/cyto.a.21042. [DOI] [PubMed] [Google Scholar]
- 24.Mei Z, Wu TF, Pion-Tonachini L, Qiao W, Zhao C, Liu Z, Lo YH. Biomicrofluidics. 2011;5:034116. doi: 10.1063/1.3624740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhagat AAS, Kuntaegowdanahalli SS, Papautsky I. Microfluid Nanofluid. 2009;7:217. [Google Scholar]
- 26.Hur SC, Nicole KH, Edward RBM, Di Carlo D. Lab Chip. 2011;11:912. doi: 10.1039/c0lc00595a. [DOI] [PubMed] [Google Scholar]
- 27.Kilimnik A, Mao W, Alexeev A. Phys Fluids. 2011;23:123302. [Google Scholar]
- 28.Beech JP, Holm SH, Adolfssona Karl, Tegenfeldt JO. Lab Chip. 2012;12:1048. doi: 10.1039/c2lc21083e. [DOI] [PubMed] [Google Scholar]
- 29.Hur SC, Tat Kwong Tse H, Di Carlo D. Lab Chip. 2010;10:274. doi: 10.1039/b919495a. [DOI] [PubMed] [Google Scholar]
- 30.Di Carlo D, Irimia D, Tompkins RG. Proc Natl Acad Sci U S A. 2007;104:18892. doi: 10.1073/pnas.0704958104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lulevich V, Zink T, Chen HY, Liu FT, Liu GY. Langmuir. 2006;22:8151. doi: 10.1021/la060561p. [DOI] [PubMed] [Google Scholar]
- 32.Mortazavi S, Tryggvason G. J Fluid Mech. 2000;411:325. [Google Scholar]
- 33.Chun B, Ladd A. Phys Fluids. 2006;18:031704. [Google Scholar]
- 34.Serge G, Silberberg A. J Fluid Mech. 1962;14:115. [Google Scholar]
- 35.Serge G, Silberberg A. J Fluid Mech. 1962;14:136. [Google Scholar]
- 36.Lekke M, Laidler P, Gil D, Lekki J, Stachura Z, Hrykiewicz AZ. Eur Biophys J. 1999;28:312. doi: 10.1007/s002490050213. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.