

Abstract
Activated protein C (APC), a protease with anticoagulant and cytoprotective activities, protects neurons and cerebrovascular endothelium from ischemic injury. A recombinant APC, drotrecogin alfa (activated) (DrotAA) (Xigris®), was approved by the Food and Drug Administration for the treatment of sepsis; however, serious bleeding was a dose-limiting side effect. A modified APC, containing 405 amino acid residues, 3K3A-APC, was designed to possess significantly reduced anticoagulant activity (< 10 %) while maintaining full cytoprotective properties. The preclinical safety assessment of 3K3A-APC was conducted to support initiation of ischemic stroke clinical trials. The safety and toxicokinetics of 3K3A-APC were studied in CD-1 mice and cynomolgus monkeys. Multiple-dose (14-day), intravenous GLP toxicology assessed toxicity, histopathology, immunogenicity, and toxicokinetics. Dose-related increases in plasma total 3K3A-APC were observed in mice and monkeys with no evidence of accumulation over 14 days. The elimination T1/2 in monkeys was 1 hour. 3K3A-APC was well tolerated in mice and monkeys, and no signs of 3K3A-APC toxicity were identified in mice or monkeys at any time. Additionally, wild-type APC (DrotAA) was studied to obtain comparative anticoagulant data using clotting assays. Anticoagulant activity of 3K3A-APC was observed in monkeys at doses of 1 and 5 mg/kg/day. In contrast, DrotAA showed prolongation of clotting assays in monkeys at doses 1/10th of those showing effects with 3K3A-APC. Based upon the anticoagulant profiles, the risk for APC-induced bleeding in clinical trials of 3K3A-APC is greatly reduced relative to wild type APC which makes this new drug a feasible therapy for ischemic stroke patients.
Keywords: neuroprotection, anticoagulant activity, toxicology, pharmacokinetics
INTRODUCTION
Activated protein C (APC) is a serine protease with systemic anticoagulant, anti-inflammatory and anti-apoptotic activities [1]. APC is generated from its zymogen protein C that is activated by thrombin on the surface of the endothelial cells which requires two membrane receptors, thrombomodulin and endothelial protein C receptor (EPCR) (Figure 1).
Figure 1.
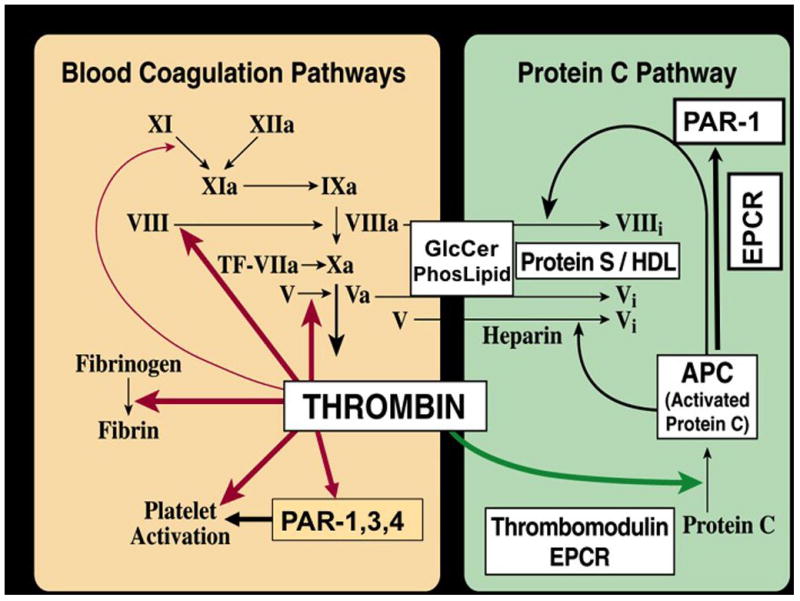
APC’s anticoagulant activity is independent of its direct cellular effects (Figure 2). APC’s anticoagulant activity is mediated by irreversible proteolytic degradation of factors Va and VIIIa with contributions by various cofactors, whereas its cytoprotective activities are mediated by proteolytic activation of protease activated receptor 1 (PAR1) [1]. APC’s cellular signaling results in cytoprotective alterations of gene expression profiles, anti-inflammatory activities and anti-apoptotic activity [2–6].
Figure 2.

APC protects neurons and brain endothelial cells from ischemic injury and divergent inducers of apoptosis by inhibiting the mitochondria-mediated and death receptors-mediated apoptotic pathways[4,7–9]. Early post-ischemic administration of APC within 4 hours of an ischemic insult is neuroprotective in rodent models of transient ischemia [4,10] and embolic stroke [11]. APC also protects against diabetic endothelial and glomerular injury [12], and is also protective in animal models of sepsis, ischemia-reperfusion injury of kidney and lung, and thrombosis models [13]. Regarding central nervous system disorders, APC is protective in rodent models of spinal cord injury [14], amyotrophic lateral sclerosis[15], and multiple sclerosis [16].
A form of human recombinant wild type APC with slightly increased anticoagulant activity, drotrecogin-alfa activated (DrotAA) (Xigris®), was approved by the FDA for use in patients with severe sepsis [17]. DrotAA was approved for studies in patients with ischemic stroke [18]. The Recombinant Human Activated Protein C Worldwide Evaluation in Severe Sepsis (PROWESS) trial for APC infusion in patients with severe sepsis reported low, but significant risk for serious bleeding, including increased risk for intracerebral hemorrhage with DrotAA treatment [18]. Moreover, at higher pharmacological doses, wild type APC is toxic to non-human primates and produces disseminated bleeding [19].
ZZ Biotech LLC is developing 3K3A-APC, a newly designed, modified, recombinant activated protein C (APC) pharmaceutical for the treatment of acute ischemic stroke. 3K3A-APC is derived from a 461-residue protein C zymogen precursor expressed via recombinant technology in Chinese Hamster Ovary (CHO) cells. Its amino acid sequence differs from that of the wild-type human Activated Protein C (APC) and of the commercial product, DrotAA, which is approved by the Food and Drug Administration (FDA) for treatment of sepsis, in that 3 sequential lysine residues have been replaced with 3 sequential alanine residues; the amino acid substitutions are K191A-K192A-K193A [20]. This change in pharmaceutical design retains the cytoprotective effects of native (wild-type) APC while significantly reducing its anticoagulant effects by > 90 %. Bleeding is a dose limiting side effect of Xigris in clinical use (Xigris package insert, Eli Lilly, 2008). Thus, the development of APC variants with reduced anticoagulant activity has the potential to provide effective therapies with reduced risk.
The three amino acid changes in APC, K191A-K192A-K193A, alter a factor Va binding exosite in APC resulting in greatly reduced anticoagulant activity, i.e., by > 90%[21], but do not affect exosites that recognize EPCR or PAR1 which are important for APC’s cellular activity. Thus, 3K3A-APC retains normal anti-apoptotic activity in a model of staurosporine-induced apoptosis in human umbilical vein cells [22, 23]. Furthermore, 3K3A-APC exerts enhanced neuroprotective activity and eliminates risk for intracerebral bleeding that has been seen with DrotAA in a mouse model of ischemic stroke [22].
The preclinical safety of 3K3-APC in a rodent and non-rodent species prior to initiating human clinical trials in stroke is a critical consideration in advancing this new chemical entity into human clinical development. Based upon 3K3A-APC’s activity in a mouse model of ischemic stroke [22,23], the mouse was selected as the rodent species for the toxicology assessment. For the non-rodent species, the cynomolgus monkey was selected based upon the historical use of this species for assessing the toxicology of recombinant wildtype human APC, drotrecogin alfa (activated) (DrotAA), Xigris [24,25]. The intravenous route of exposure in these studies parallels the intended route of administration of 3K3A-APC in humans. The duration of 14 days of daily dosing supports up to 14 days of dosing in humans.
The pharmacokinetics of 3K3A-APC has been studied as part of the pivotal GLP toxicology studies in CD-1 mice and cynomolgus monkeys. The primary assay for 3K3A-APC exposure in the toxicology studies was a validated total-antigen ELISA which measured total drug that includes both free (active) enzyme and the inhibited enzyme that is inactive due to binding to plasma protease inhibitors (inactive). Active drug anticoagulant activity was measured in monkey plasma samples using standard clotting assays. Serum anti-3K3A-APC antibodies were measured in mice and monkeys in the toxicology studies using validated methods.
METHODS
Reagents
Human 3K3A-Protein C (3K3A-PC) zymogen was stably transfected into Chinese hamster ovary (CHO) cells that were grown in suspension in CD OptiCHO medium (Invitrogen) supplemented with 2 mM CaCl2, 10 μg/ml vitamin K, and 2 mM GlutaMAX (Invitrogen). After the required number of serial expansions of culture volumes using shake flask passages and WAVE culturing, the final stirred-tank bioreactor volume was either 200 l or 2500 l in which culture was performed for ten days at 35 degrees C. After 0.2-μm filtration, the clarified culture supernatant was subjected to purification using chromatography on a Q Sepharose fast flow ion exchange column and hydrophobic affinity column. The purified 3K3A-PC zymogen was then activated with recombinant human thrombin (Recothrom, Zymogenetics) to generate 3K3A-Activated Protein C (3K3A-APC). Thrombin was removed from the reaction mixture using ion exchange chromatography with a UnoSphere S flow through resin (BioRad). The concentration of 3K3A-APC was adjusted using tangential flow filtration (TFF). Following the concentration step, Uno Sphere Q Ion Exchange flow through was used with buffer exchange and concentration adjustment to polish the preparation, prior to Planova filtration and addition of polysorbate-80. 3K3A-APC was characterized by SDS-PAGE under reduced and non-reduced conditions, SE-FPLC, amidolytic activity by a chromogenic assay, and optical density at 280 nm. There was no detectable thrombin in the purified 3K3A-APC based on a thrombin-time clotting assay using purified fibrinogen. Xigris® was purchased from Eli Lilly and Co. (Indianapolis, IN) [24,25]. This biologic compound is recombinant wildtype APC with the normal amino acid sequence that was made in a different cell line (HEK293 cells) than 3K3A-APC (CHO cells) such that XIGRIS (also known as DrotAA) differs from 3K3A-APC by 3 amino acid residues and also in its carbohydrate structures. The names XIGRIS, Drot AA and recombinant wildtype APC used herein refer to the same Eli Lilly commercially purchased recombinant biologic product.
Animal Studies
Mice
Crl:CD1(ICR) mice (10/sex/group) were dosed daily by intravenous injection with 3K3A-APC at 0, 0.4, 2, or 5 mg/kg/day for 14 days. An additional 10/sex/group in the control and high-dose groups were dosed for 14 days followed by a 14-day treatment-free recovery period. Separate groups of animals (8/sex/group) were dosed at 0, 0.4, 1 or 5 mg/kg/day of 3K3A-APC for toxicokinetic (TK) evaluations. The highest dose represented a maximal feasible dose based upon the concentration of 3K3A-APC in the clinical formulation that was being used in the toxicology study and the maximum volume that could be injected safely into this species. Mice in the vehicle control group were dosed with the formulation buffer containing 20 mM sodium citrate, 300 mM NaCl, and 200 ppm Polysorbate-80 at pH 6.0 (± 0.3). 3K3A-APC was formulated at a concentration of 1.0 mg/mL in the formulation buffer. 3K3A-APC and vehicle were slowly injected over a period of 1 minute via the lateral tail vein.
Mice were monitored for changes in clinical signs, body weights, and food intake. Clinical pathology assessments included standard hematology, clinical chemistry, coagulation and urinalysis parameters in all animals on Days 15 and 29. The toxicokinetics of 3K3A-APC were monitored on Days 1, 14 and 28 by measuring 3K3A-APC plasma concentrations by ELISA at the following time points: prior to injection and 5 minutes, 30 minutes, and 2 hours postdose. On Days 15 or 29, animals were necropsied, gross clinical observations were made, and selected tissues were weighed. A full tissue list was harvested from mice at necropsy. The tissues were collected in 10% formalin for subsequent histopathologic analysis. Mice were tested for 3K3A-APC antibodies following dosing on Day 14 and on Day 28 in recovery animals following 14 days of a treatment-free period.
Monkeys
Cynomolgus monkeys (4/sex/group) were dosed daily by intravenous injection with 0, 0.2, 1 or 5 mg/kg/day of 3K3A-APC for 14 days. An additional 2 groups of monkeys (3/sex/group) were dosed with either vehicle or 5 mg/kg/day of 3K3A-APC followed by a 14-day treatment-free recovery period. The top dose represented a maximal feasible dose based upon the concentration of 3K3A-APC in the clinical formulation that was being used in the toxicology study and the maximum volume that could be injected safely into this species. Monkeys in the vehicle control group were dosed with the formulation buffer containing 20 mM sodium citrate, 300 mM NaCl and 200 ppm Polysorbate-80 at pH 6.0 (± 0.3). 3K3A-APC was formulated at a concentration of 1.0 mg/mL in the formulation buffer. Animals were dosed by slow, intravenous injection at an approximate rate of 5 mL/minute.
Monkeys were monitored for changes in clinical signs, body weights, and food intake. Clinical pathology assessments included standard hematology, clinical chemistry, and urinalysis parameters in all animals. Coagulation parameters (prothrombin time (PT), activated partial thromboplastin time (aPTT), fibrinogen) were monitored 15 minutes and 3 hours after dose administration on Days 1 and 14. In addition, coagulation parameters were measured at necropsy on Days 15 and 29. The TK of 3K3A-APC were monitored on Days 1 and 14 by collecting plasma samples for ELISA measurements at the following timepoints: prior to injection, immediately following injection (within 2 minutes), and 0.25, 0.5, 1, 3 and 8 hours post-dose. On Days 15 and 29, animals were necropsied and gross observations were recorded, and selected tissues were weighed. A full tissue list was harvested from monkeys at necropsy, and the tissues were collected in 10% formalin for subsequent histopathologic analysis.
The following safety pharmacology assessments were measured prior to the study and at 2 time points during the 14-day treatment period: functional observational battery for neurological function, EKG (electrocardiogram) including quantitative QT, blood pressure, respiratory rate, and pulse oximetry which measures the percent of hemoglobin that is saturated with oxygen. Monkeys were tested for anti-3K3A-APC antibodies prior to dosing on Days 1 and 14 and on Day 29 in recovery animals (following 14 days of a treatment-free period).
The effects of recombinant wildtype APC (DrotAA) on coagulation parameters in cynomolgus monkeys was examined to compare with the coagulation effects noted with 3K3A-APC in the monkey toxicology study described above.
A total of 6 non-naïve cynomolgus monkeys weighing approximately 2.5 to 3.5 kg were used in this study (3 males and 3 females). Vials of DrotAA containing 5 mg were reconstituted with 2.5 mL Sterile Water for Injection, USP. An additional 2.5 mL of Sterile Saline for Injection, USP, was added to the vials, resulting in 5 mL of 1.0 mg/mL DrotAA. DrotAA was administered intravenously via slow bolus injection (approximately 5 mL/minute) at doses of 0.02 to 1 mg/kg. Blood samples (approximately 1.2 mL/sample) were collected from the femoral vein/artery at pre-dose, and 15 minutes, 3 hours, and 24 hours after dosing for PT and aPTT assays.
Bioanalytical Methods
ELISA methods were used to determine the concentration of anti-3K3A-APC antibodies in mice and monkey serum samples in the pivotal toxicology studies. The sample analyses were performed by Charles River Laboratories, Reno, NV, according to validated ELISA methods for mice and monkeys. The presence of antibodies was monitored prior to dosing and after 14 days of dosing. Animals were also sampled at Day 28 following a 14-day treatment-free recovery period. Serum samples from mice and monkeys were initially screened for antibodies and samples above the plate specific negative cut off (PSNCO) were subsequently tested in an immunodepletion assay to confirm the presence of antibodies, if sufficient serum was available. In monkeys, all animals that screened positive were tested by immunodepletion. In the majority of mice that screened positive, the volume of serum was insufficient to conduct the immunodepletion assay and therefore confirmation of antibodies to 3K3A-APC was limited in this species.
Total plasma 3K3A-APC antigen [both free 3K3A-APC (active) and 3K3A-APC bound to protease inhibitors (inactive)] was measured in mice and monkeys using validated ELISA methods. The lower limits of quantification (LLOQ) for the mouse and monkey 3K3A-APC assays were 0.5 and 10 μg/mL, respectively.
Statistical analysis
Data were presented as mean ± SEM. Student’s t-test and ANOVA was used to determine statistically significant differences. P < 0.05 was considered statistically significant.
RESULTS
Animal Studies
Mice
No signs of toxicity including effects on body weights were observed in mice during the dosing or recovery phases of the 14-day toxicology study of this newly designed pharmaceutical, 3K3A-APC. No effects attributable to 3K3A-APC were observed in the clinical pathology assessments including coagulation parameters. Since the amount of blood needed to assess coagulation parameters in mice could only be taken at necropsy 24 hours after the last dose, it is possible that any effects on coagulation parameters had reversed by the time the samples were taken. The assessment of coagulation effects was studied in more detail in monkeys where it was possible to obtain multiple blood samples following dosing. No gross pathological findings or effects on organ weights were observed in 3K3A-APC-treated groups at necropsies on Days 15 and 29. No target organ toxicities were identified in histopathological analyses.
Monkeys
Toxicology Study
No signs of toxicity were observed in monkeys during the dosing or recovery phases of the 14-day toxicology study. No effects on neurological, respiratory (rate and blood oxygenation), or cardiovascular functions or ECG, including QTc values, were observed. No effects attributable to 3K3A-APC were observed in the clinical pathology parameters with the exception of aPTT and PT parameters. Dose-related increases in aPTT were observed on Days 1 and 14, with increases of approximately 1.5 and 4.5 times control values at doses of 1.0 and 5.0 mg/kg of 3K3A-APC, respectively (Table 1). The PT was increased only at the highest dose of 3K3A-APC tested. The effects of 3K3A-APC on aPTT and PT were reduced 3 hours after injection and clotting time values were normal 24 hours post injection. The aPTT effects were similar in magnitude on Days 1 and 14 of the study, indicating no cumulative effects of multiple dosing with 3K3A-APC. No clinical signs of bruising or bleeding were observed in monkeys.
Table 1.
Mean Coagulation Parameters In Monkeys Dosed With 3K3A-APC
| Dose (mg/kg) | PT | aPTT | ||
|---|---|---|---|---|
| Mean Value1 ± SD (seconds) | Fold Increase2 | Mean Value1 ± SD (seconds) | Fold Increase2 | |
| Day 1 | ||||
| 0 | 11.7 ± 0.5 | -- | 23.9 ± 1.7 | -- |
| 0.2 | 11.2 ± 0.3 | -- | 26.1 ± 2.2 | -- |
| 1 | 11.5 ± 0.6 | -- | 36.7 ± 3.1* | 1.54 |
| 5 | 16.0 ± 2.2* | 1.37 | 109.1 ± 42.6* | 4.56 |
| Day 14 | ||||
| 0 | 11.4 ± 0.5 | -- | 23.3 ± 1.8 | -- |
| 0.2 | 11.2 ± 0.3 | -- | 26.0 ± 2.4 | -- |
| 1 | 11.3 ± 0.4 | -- | 35.2 ± 5.0* | 1.51 |
| 5 | 15.2 ± 2.3* | 1.33 | 112.0 ± 59.5* | 4.81 |
mean ± SD of combined male and female values measured 15 minutes following dosing
relative to concurrent control group value
p < 0.05 relative to control values
Upon necropsy, no gross pathology observations attributed to 3K3A-APC treatment were observed. Organ weights were comparable between control and 3K3A-APC groups. No target organ toxicities were identified in histopathological analyses.
Pharmacodynamic Study with DrotAA
Following single intravenous doses of DrotAA, the aPTT was elevated relative to pre-dose values at doses of 0.1, 0.2 or 1 mg/kg and the PT was elevated at doses of 0.2 and 1 mg/kg (Table 2).
Table 2.
Mean Coagulation Parameters in Monkeys 15 Minutes Following DrotAA Administration
| Dose (mg/kg) | PT | aPTT | ||
|---|---|---|---|---|
| Mean Value1 (seconds) | Fold Increase2 | Mean Value1 (seconds) | Fold Increase2 | |
| 0.02 | 12.0 ± 0.5 | -- | 28.9 ± 2.4 | -- |
| 0.1 | 12.7 ± 0.4 | -- | 36.8 ± 5.5 | 1.43 |
| 0.2 | 13.0 ± 0.3 | 1.14 | 46.2 ± 7.5 | 1.81 |
| 1 | 25.0 ± 3.0 | 2.06 | >200 * | >4.24 |
mean ± SD of combined male and female values
relative to pre-dose value
measurements were above the maximum readable by the equipment used (200 seconds)
The magnitude of the aPTT elevation at 0.1 mg/kg (~1.4-fold increase) was similar to what was observed with 1 mg/kg of 3K3A-APC, indicating an approximately 10-fold difference in anticoagulant activity between the two forms of APC. To compare further the aPTT elevation differences between DrotAA in this study and 3K3A-APC in the toxicology study discussed above, the changes in aPTT from baseline or pre-dose values are displayed in the Figure.
Similar to the observations in the 3K3A-APC toxicology study, prolonged aPTT values after administration of DrotAA returned to near normal values (< 30 seconds) 24 hours after dosing. However, the pre-dose values of aPTT prior to the 1 mg/kg dose (72 hours after dosing with 0.2 mg/kg) were elevated above normal (values ranged from 33.1 to 138.5 seconds). To confirm this observation, DrotAA was administered again at a dose of 1 mg/kg approximately 1 week following the first dosing of 1 mg/kg. The elevated pre-dose aPTT values were again observed prior to the second dose of 1 mg/kg (values ranged from 37.7 to 56.4 seconds). The mean aPTT values prior to and following dosing are summarized in Table 3. These findings are in contrast to what was observed with 3K3A-APC where pre-dose aPTT values on Day 14 of the study were normal following 13 consecutive doses of 3K3A-APC.
Table 3.
Mean Coagulation Parameters in Monkeys Dosed with DrotAA
| DrotAA Dose (mg/kg) | Day Dosed | Mean aPTT ± SD (seconds) | |||
|---|---|---|---|---|---|
| Pre-Dose | 15 min | 3 hr | 24 hr | ||
| 0.02 | 1 | 25.85 ± 2.8 | 28.90 ± 2.4 | 26.27 ± 2.2 | 26.10 ± 2.8 |
| 0.1 | 3 | 25.63 ± 2.8 | 36.75 ± 5.5 | 27.30 ± 2.1 | 26.38 ± 3.0 |
| 0.2 | 5 | 25.53± 2.3 | 46.18 ± 7.5 | 28.75 ± 2.2 | 26.33 ± 2.9 |
| 1.0 | 8 | 54.20 ± 41.5 | > 200* | 42.38 ± 4.1 | 28.92 ± 3.8 |
| 1.0 | 18 | 47.13 ± 8.3 | > 200 | 66.02 ± 43.0 | 29.07 ± 4.3 |
measurements were above the maximum readable by the equipment used (200 seconds)
Pharmacokinetics
Mice
The TK parameters of 3K3A-APC on Days 1 and 14 are summarized in Table 4.
Table 4.
Mean Total 3K3A-APC Toxicokinetic Values in Mice
| Dose (mg/kg) | Cmax (μg/mL) | AUC0-t (μg·hr/mL) | ||
|---|---|---|---|---|
| Males | Females | Males | Females | |
| Day 1 | ||||
| 0.4 | 1.18 | 2.84 | ND | 1.97 |
| 2 | 16.1 | 16.7 | 8.91 | 14.6 |
| 5 | 36.6 | 31.2 | 30.8 | 30.7 |
| Day 14 | ||||
| 0.4 | 1.97 | 2.41 | ND | ND |
| 2 | 12.0 | 13.6 | 6.45 | 8.26 |
| 5 | 16.7 | 36.2 | 24.2 | 31.1 |
There were no marked or consistent differences between male and female mice in the TK parameters calculated for Day 1 or Day 14.
There was no evidence for accumulation of 3K3A-APC. Mean accumulation factors (Day 14/Day 1) for AUC0-t were ≤ 1.0.
Monkeys
The plasma concentrations of 3K3A-APC were consistent between males and females on Days 1 and 14. The TK parameters of 3K3A-APC on Days 1 and 14 are summarized in Table 5. Additional TK parameters obtained in the high dose group (5 mg/kg/day) are shown in Table 6.
Table 5.
Total 3K3A-APC Toxicokinetic Values (Mean) in Monkeys
| Dose (mg/kg) | Cmax (μg/mL) | AUC0-t (μg·hr/mL) | ||
|---|---|---|---|---|
| Males | Females | Males | Females | |
| Day 1 | ||||
| 0.2 | ND | ND | ND | ND |
| 1 | 15.8 | 17.7 | ND | ND |
| 5 | 71.2 | 70.4 | 59.0 | 41.2 |
| Day 14 | ||||
| 0.2 | ND | ND | ND | ND |
| 1 | 15.9 | 14.3 | ND | ND |
| 5 | 65.1 | 69.5 | 72.1 | 60.8 |
Table 6.
Additional Total Toxicokinetic Parameters (Mean) of 3K3A-APC In Monkeys At 5 mg/kg
| Day | Sex | N | t1/2 (h) | Vd (mL/kg) | CL (mL/h/kg) | AUC0-inf (μg·h/mL) |
|---|---|---|---|---|---|---|
| 1 | M | 7 | 1.05 | 79.0 | 58.5 | 93.7 |
| F | 7 | 0.76 | 77.5 | 76.0 | 71.3 | |
| 14 | M | 7 | 1.25 | 89.4 | 57.9 | 103.0 |
| F | 7 | 1.06 | 78.3 | 57.4 | 95.6 |
There was no evidence for accumulation of 3K3A-APC. Mean accumulation factors (Day 14/Day 1) for AUC0-t ranged from approximately 0.9 – 1.5.
Antibody Assessments
Mice
In mice, a total of 21 of 80 (26%) of animals dosed with 3K3A-APC screened positive for anti-3K3A-APC antibodies. Of the 21 animals dosed with 3K3A-APC that screened positive, only 8 animals (38%) had sufficient sample volume to perform confirmatory immunodepletion analyses. Five of 8 samples confirmed positive for antibodies following immunodepletion; 3 of 20 animals in the 0.4 mg/kg group and 2 of 40 animals in the 5.0 mg/kg group were without evidence of a dose-dependent response.
Four of 40 mice (10%; 2 male and 2 female) in the control group screened positive for anti-3K3A-APC antibodies. The positive samples had absorbance values that were just above the PSNCO (0.002 to 0.008 A450). These screening results were likely false positives. A similar rate of false positives (16.7%) was observed in control serum during the assay validation process.
Monkeys
In monkeys, a total of 17 of 30 (57%) of animals dosed with 3K3A-APC screened positive for anti-3K3A-APC antibodies. Of the 17 animals dosed with 3K3A-APC that screened positive, 1 animal in each dose group (total of 3) was confirmed positive via immunodepletion. The percentage of animals treated with 3K3A-APC that confirmed positive for antibodies following immunodepletion was 3 of 30 animals (10%); these included 1 of 8 (12.5%), 1 of 8 (12.5%) and 1 of 14 (7.1%) for the 0.2, 1.0, and 5.0 mg/kg 3K3A-APC dose groups, respectively.
Four of 14 cynomolgus monkeys (28%) in the control group screened positive for anti-3K3A-APC antibodies. Additionally, 7 of 44 (16%) of the Day 1 (pre-dose) samples from monkeys dosed with 3K3A-APC in the study screened positive for anti-3K3A-APC antibodies. All samples from the control group monkeys and the Day 1 pre-dose samples that screened positive were confirmed negative via immunodepletion.
Anti-3K3A-APC antibodies were confirmed through immunodepletion in monkeys at all dose levels on Days 14 and 29, with no apparent dose-dependent response noted. These antibodies did not appear to affect plasma exposure to 3K3A-APC in that 3K3A-APC concentrations following dosing were not reduced in monkeys with antibodies to 3K3A-APC compared to those without.
DISCUSSION
3K3A-APC is a newly developed pharmaceutical to treat ischemic stroke patients. 3K3A-APC was well tolerated in mice and monkeys at intravenous doses up to 5 mg/kg/day for 14 days. The only effects attributed to 3K3A-APC were dose-related and reversible increases in the coagulation parameters, PT and aPTT, in monkeys at doses ≥ 1 mg/kg. Based on these findings, the no observed adverse effect level (NOAEL) of 3K3A-APC in mice and monkeys was 5 and 0.2 mg/kg/day, respectively. Antibodies to 3K3A-APC were observed in mice and monkeys. The generation of antibodies in these species was expected since 3K3A-APC is a protein of human origin. Dose-related increases in 3K3A-APC exposures (Cmax and AUC) were observed in mice and monkeys over the dose ranges tested. No significant gender differences in PK were observed in either species and no evidence of 3K3A-APC accumulation was observed. The elimination T1/2 of total 3K3A-APC in monkeys was approximately 1 hour, consistent with what has been reported in the literature with APC in humans [25].
The intravenous administration of recombinant wildtype human APC, DrotAA, to cynomolgus monkeys resulted in dose-related increases in aPTT over the dose range of 0.1 to 1 mg/kg. The magnitude of clotting time prolongations over pretreatment values at 0.1 mg/kg (~1.4-fold) for this version of wildtype APC was similar to what was observed with a dose of 1.0 mg/kg 3K3A-APC, indicating an approximately 10-fold difference in anticoagulant activity between the two proteins following their infusion. Dosing with Xigris also was associated with elevations in pre-dose aPTT values after multiple dosing which may be reflective of depletion of factors V and VIII. APC’s anticoagulant activity involves limited highly specific cleavages of factors V/Va and VIII/VIIIa, especially a rapid cleavage at Arg506 in factor Va [1,21]. Since APC inactivates factors V and VIII via irreversible limited proteolysis [1], the elevated pre-dose values observed in this study may be due to depletion of these factors and the inability to recover with repeated dosing of highly anticoagulant DrotAA. 3K3A-APC, on the other hand, has a greatly reduced ability to cleave Arg506 in factor Va [1,21] which may explain why multiple dosing with 3K3A-APC had no residual effect on aPTT values because it might not deplete endogenous factors V and VIII.
Conclusions
Intravenous administration of 3K3A-APC to mice and monkeys for 14 consecutive days at doses up to 5 mg/kg was well tolerated and did not result in any clinical signs of toxicity, effects on hematology or clinical chemistry parameters, or target organ toxicities following histopathological examination. No effects on neurological, respiratory, or cardiovascular functions were observed in monkeys. The only effect observed in monkeys was a dose-related and reversible prolongation of PT and aPTT clotting assays at doses ≥ 1 mg/kg of 3K3A-APC. Prolongation of coagulation assays is an expected pharmacological activity of APC, however, 3K3A-APC has approximately one tenth the anticoagulant effects of wild-type plasma-derived APC or of recombinant human APC, DrotAA (Xigris). These preclinical findings support the proposal that 3K3A-APC may have a reduced risk of bleeding in humans compared to the known bleeding side effect of DrotAA (Xigris package insert, Eli Lilly, 2008).
The no observed adverse effect levels (NOAEL) of 3K3A-APC in the mouse and monkey toxicology studies were 5.0 and 0.2 mg/kg/day, respectively, which represent human equivalent doses (HEDs) of 0.4 and 0.06 mg/kg, respectively (FDA Center for Drug Evaluation and Research, July 2005). Both mice and monkeys produced antibodies to human 3K3A-APC. The generation of antibodies in these species was expected since 3K3A-APC is a recombinant protein of human origin. Dose-related systemic exposure to 3K3A-APC measured by plasma ELISA was observed in both toxicology studies. No evidence of its accumulation over the 14 days of dosing was observed. Pharmacokinetics of the drug in monkeys revealed a relatively short terminal T1/2 of approximately 1 hour for all immunologic forms of the drug, i.e., both active enzyme and inactive complexes of plasma protease inhibitor-enzyme complexes, which is consistent with what has been reported for recombinant wildtype human APC (Drot AA) and its complexes with protease inhibitors in humans. These data provide positive evidence that the newly developed pharmaceutical, 3K3A-APC, may indeed make a promising stroke therapeutic.
Figure 3.

Clotting assays were used to compare the anticoagulant activity of 3K3A-APC versus XIGRIS (wildtype recombinant APC) following their infusions in cynomolgus monkey at indicated doses (x-axis). Plasma samples were assayed as described in Methods section using aPTT assays and the aPTT prolongation data are shown in sec on y-axis; this prolongation reflects the dose-dependent anticoagulant activity of each form of APC.
Acknowledgments
This work was supported by Socratech Biotechnology LLC, Rochester, NY. The authors thank Mike Ultee and Doug Rea, Laureate BioPharma, Princeton, New Jersey, for their valuable help in manufacturing of this drug, Jose A. Fernandez, The Scripps Research Institute, for technical advice, and Nienwen Chow, Sarah Lawrence, Frieder Hoffmann and Michael Sperber for their support in the characterization of the 3K3A-APC used in these studies.
References
- 1.Mosnier LO, Zlokovic BV, Griffin JH. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 2.Joyce DE, Gelbert L, Ciaccia A, DeHoff B, Grinnell BW. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 3.Riewald M, Petrovan RJ, Donner A, Mueller BM, Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 4.Cheng T, Liu D, Griffin JH, Fernández JA, Castellino F, Rosen ED, Fukudome K, Zlokovic BV. Activated protein C blocks p53-mediated apoptosis in ischemic human brain endothelium and is neuroprotective. Nat Med. 2003;9:338–342. doi: 10.1038/nm826. [DOI] [PubMed] [Google Scholar]
- 5.Domotor E, Benzakour O, Griffin JH, Yule D, Fukudome K, Zlokovic BV. Activated protein C alters cytosolic calcium flux in human brain endothelium via binding to endothelial protein C receptor and activation of protease activated receptor-1. Blood. 2003;101:4797–4801. doi: 10.1182/blood-2002-12-3680. [DOI] [PubMed] [Google Scholar]
- 6.Mosnier LO, Griffin JH. Inhibition of staurosporine-induced apoptosis of endothelial cells by activated protein C requires protease-activated receptor-1 and endothelial cell protein C receptor. Biochem J. 2003;373:65–70. doi: 10.1042/BJ20030341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu D, Cheng T, Guo H, Fernández JA, Griffin JH, Song X, Zlokovic BV. Tissue plasminogen activator neurovascular toxicity is controlled by activated protein C. Nat Med. 2004;10:1379–1383. doi: 10.1038/nm1122. [DOI] [PubMed] [Google Scholar]
- 8.Guo H, Liu D, Gelbard H, Cheng T, Insalaco R, Fernández JA, Griffin JH, Zlokovic BV. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 9.Cheng T, Petraglia AL, Li Z, Thiyagarajan M, Zhong Z, Wu Z, Liu D, Maggirwar SB, Deane R, Fernández JA, LaRue B, Griffin JH, Chopp M, Zlokovic BV. Activated protein C inhibits tissue plasminogen activator-induced brain hemorrhage. Nat Med. 2006;12:1278–1285. doi: 10.1038/nm1498. [DOI] [PubMed] [Google Scholar]
- 10.Shibata M, Kumar SR, Amar A, Fernandez JA, Hofman F, Griffin JH, Zlokovic BV. Anti-inflammatory, antithrombotic, and neuroprotective effects of activated protein C in a murine model of focal ischemic stroke. Circulation. 2001;103:1799–1805. doi: 10.1161/01.cir.103.13.1799. [DOI] [PubMed] [Google Scholar]
- 11.Zlokovic BV, Zhang CL, Liu D, Fernandez J, Griffin JH, Chopp M. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. 2005;58:474–477. doi: 10.1002/ana.20602. [DOI] [PubMed] [Google Scholar]
- 12.Isermann B, Vinnikov IA, Madhusudhan T, Herzog S, Kashif M, Blautzik J, Corat MA, Zeier M, Blessing E, Oh J, Gerlitz B, Berg DT, Grinnell BW, Chavakis T, Esmon CT, Weiler H, Bierhaus A, Nawroth PP. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 13.Griffin JH, Zlokovic B, Fernandez JA. Activated protein C: potential therapy for severe sepsis, thrombosis, and stroke. Semin Hematol. 2002;39:197–205. doi: 10.1053/shem.2002.34093. [DOI] [PubMed] [Google Scholar]
- 14.Taoka Y, Schlag MG, Hopf R, et al. The long-term effects of pre-treatment with activated protein C in a rat model of compression-induced spinal cord injury. Spinal Cord. 2000;38:754–761. doi: 10.1038/sj.sc.3101096. [DOI] [PubMed] [Google Scholar]
- 15.Zhong Z, Ilieva H, Hallagan L, et al. Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest. 2009;119:3437–49. doi: 10.1172/JCI38476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, Grinnell BW, Raine CS, Sobel RA, Han DK, Steinman L. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. [DOI] [PubMed] [Google Scholar]
- 17.Bernard GR, Vincent JL, Laterre PF, LaRosa SP, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ., Jr Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 18.APCAST. The Activated Protein C in Acute Stroke Trial. http://clinicaltrials.gov/ct2/show/NCT00533546?term=apc&rank=25.
- 19.www.emea.europa.eu/humandocs/PDFs/EPAR/xigris/247102en6.pdf
- 20.Gale AJ, Tsavaler A, Griffin JH. Molecular characterization of an extended binding site for coagulation factor Va in the positive exosite of activated protein C. J Biol Chem. 2002;277:28836–28840. doi: 10.1074/jbc.M204363200. [DOI] [PubMed] [Google Scholar]
- 21.Mosnier LO, Gale AJ, Yegneswaran S, Griffin JH. Activated protein C variants with normal cytoprotective but reduced anticoagulant activity. Blood. 2004;104:1740–1744. doi: 10.1182/blood-2004-01-0110. [DOI] [PubMed] [Google Scholar]
- 22.Guo H, Singh I, Wang Y, Deane R, Barrett T, Fernández JA, Chow N, Griffin JH, Zlokovic BV. Neuroprotective activities of activated protein C mutant with reduced anticoagulant activity. Eur J Neurosci. 2009;29:1119–30. doi: 10.1111/j.1460-9568.2009.06664.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Guo H, Wang Y, Singh I, Liu D, Fernández JA, Griffin JH, Chow N, Zlokovic BV. Species-dependent neuroprotection by activated protein C mutants with reduced anticoagulant activity. J Neurochem. 2009;109:116–24. doi: 10.1111/j.1471-4159.2009.05921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xigris EMEA Scientific Discussion. 2004. [Google Scholar]
- 25.Short M, Schlichting D, Qualy R. From bench to bedside: a review of the clinical trial development plan of drotrecogin alfa (activated) Current Medical Research And Opinion. 2006;22(12):2525–2540. doi: 10.1185/030079906x154060. [DOI] [PubMed] [Google Scholar]