

Summary
To guide development of new drugs targeting methionyl-tRNA synthetase (MetRS) for treatment of human African trypanosomiasis, crystal structure determinations of Trypanosoma brucei MetRS in complex with its substrate methionine and its intermediate product methionyl-adenylate were followed by those of the enzyme in complex with high-affinity aminoquinolone inhibitors via soaking experiments. Drastic changes in conformation of one of the two enzymes in the asymmetric unit allowed these inhibitors to occupy an enlarged methionine pocket and a new so-called auxiliary pocket. Interestingly, a small low-affinity compound caused the same conformational changes, removed the methionine without occupying the methionine pocket, and occupied the previously not existing auxiliary pocket. Analysis of these structures indicates that the binding of the inhibitors is the result of conformational selection, not induced fit.
Introduction
Human African trypanosomiasis (HAT) affects 50,000 to 70,000 inhabitants of Sub-Saharan Africa (Brun et al., 2010), with 60 million people at risk of infection (Hotez et al., 2007). HAT is caused by the protozoan Trypanosoma brucei. Left untreated the disease is uniformly fatal. Current therapeutic options have many shortcomings (Brun et al., 2010). Consequently, we aim to develop effective, safe, easy to administer and affordable drugs for treating the disease by targeting aminoacyl-tRNA synthetases (aaRS) (Ibba and Soll, 2000). AaRSs are crucial for protein synthesis in all organisms. Blocking aaRSs functions inhibits the proper charging of tRNAs leading to disruption of translation. The essential role of aaRSs thus presents opportunities for the development of new antimicrobials (Critchley et al., 2009; Critchley and Ochsner, 2008; Ochsner et al., 2007; Pohlmann and Brotz-Oesterhelt, 2004; Vondenhoff and Van Aerschot, 2011).
Methionyl-tRNA synthetase of T. brucei (TbMetRS) is one of our top targets for anti-trypanosomal drug design because of substantial sequence differences with the mammalian orthologs in the active site. We have validated MetRS as a drug target by RNAi (Shibata et al., 2011) and obtained a series of inhibitors with an aminoquinolone scaffold similar to two bacterial MetRS inhibitors (Critchley et al., 2009; Critchley and Ochsner, 2008). These inhibitors selectively inhibit TbMetRS and the growth of bloodstream form T. brucei in culture with EC50 values as low as 4 nM but with minimal effects on mammalian cells. One inhibitor was shown to have anti-trypanosomal activity in a mouse model (Shibata et al., 2011). To provide a structural platform to assist further anti-TbMetRS drug development, eight structures of TbMetRS in complex with its substrate methionine (Met), with the intermediate product methionyl-adenylate (MAMP), with five inhibitors, and with a ‘fragment’ of the inhibitors were determined (Table 1). Diffusing inhibitors and fragment into substrate-bound TbMetRS crystals caused large conformational changes in one but not the other subunit in the asymmetric unit.
Table 1.
Data collection and refinement statistics
|
TbMetRS• Met |
TbMetRS• MAMP |
TbMetRS• 1289 |
TbMetRS• 1312 |
TbMetRS• 1320 |
TbMetRS• 1325 |
TbMetRS• 1331 |
TbMetRS• 89 |
|
|---|---|---|---|---|---|---|---|---|
| Data collection | ||||||||
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||||||
| a, b, c (Å) | 85.0, 105.9, 207.2 | 85.5, 105.9, 208.4 | 87.5, 105.9, 207.6 | 86.9, 106.1, 207.4 | 86.5, 105.9, 207.6 | 86.6, 105.6, 207.4 | 89.2, 105.7, 205.9 | 87.9, 105.7, 206.6 |
| Resolution (Å) | 50 – 2.90 (3.0 – 2.90)# | 50 – 2.95 (3.06 – 2.95) | 50 – 3.10 (3.15 – 3.10) | 50 – 3.10 (3.21 – 3.10) | 50 – 2.70 (2.75 – 2.70) | 50 – 2.90 (3.0 – 2.90) | 50 – 2.75 (2.80 – 2.75) | 50 – 2.60 (2.64 – 2.60) |
| Rmerge | 0.14 (0.73) | 0.21 (0.99) | 0.19 (0.72) | 0.24 (0.87) | 0.14 (0.82) | 0.17 (0.67) | 0.14 (0.76) | 0.15 (0.76) |
| Rpim | 0.06 (0.30) | 0.09 (0.39) | 0.10 (0.34) | 0.11 (0.37) | 0.06 (0.33) | 0.09 (0.34) | 0.07 (0.36) | 0.08 (0.34) |
| CC*## | 1.0 (0.96) | 1.0 (0.94) | 1.0 (0.93) | 0.99 (0.90) | 1.0 (0.93) | 1.0 (0.94) | 1.0 (0.91) | 1.0 (0.92) |
| I / σI | 12.4 (2.0) | 8.9 (1.9) | 9.1 (1.9) | 6.7 (1.6) | 11.6 (1.9) | 8.6 (2.1) | 10.1 (2.1) | 10.5 (2.0) |
| Completeness (%) | 99.9 (100) | 99.9 (100) | 99.7 (100) | 99.5 (98.9) | 99.6 (99.2) | 99.9 (100) | 97.8 (97) | 99.9 (99.6) |
| Redundancy | 6.1 (5.7) | 6.4 (6.4) | 4.5 (4.3) | 6.1 (5.7) | 6.2 (6.1) | 4.3 (4.3) | 3.9 (3.8) | 5.0 (5.0) |
| Refinement | ||||||||
| Resolution (Å) | 30 – 2.90 | 30 – 2.94 | 30 – 3.15 | 30 – 3.10 | 30 – 2.70 | 30 – 2.90 | 30 – 2.75 | 30 – 2.60 |
| No. reflections | 39,977 | 38,631 | 32,066 | 33,506 | 49,997 | 40,543 | 47,066 | 56,834 |
| Rwork / Rfree | 0.209/0.254 | 0.212/0.266 | 0.191/0.240 | 0.202/0.258 | 0.188/0.228 | 0.190/0.243 | 0.182/0.230 | 0.177/0.224 |
| No. atoms | ||||||||
| Protein | 8495 | 8435 | 8257 | 8221 | 8480 | 8368 | 8561 | 8542 |
| Met or MAMP | 18 | 62 | 9 | 9 | 9 | 9 | 9 | 9 |
| Inhibitor | - | - | 28 | 25 | 27 | 34 | 23 | 12 |
| Solvent ligand/ion | 72 | 66 | 149 | 137 | 135 | 115 | 130 | 140 |
| Water | 81 | 103 | 169 | 180 | 302 | 217 | 310 | 461 |
| B-factors | ||||||||
| Protein | 48.5 | 58.7 | 44.9 | 38.0 | 54.5 | 43.0 | 40.4 | 43.6 |
| Met or MAMP | 28.3 | 46.2 | 27.7 | 11.9 | 39.1 | 28.6 | 26.8 | 32.3 |
| Inhibitor | - | - | 44.6 | 32.2 | 49.9 | 43.5 | 30.2 | 31.2 |
| Solvent ligand/ion | 57.0 | 63.4 | 56.1 | 40.9 | 71.6 | 60.3 | 60.6 | 67.1 |
| Water | 31.4 | 34.0 | 27.1 | 20.4 | 47.0 | 26.3 | 29.8 | 38.0 |
| R.m.s. deviations | ||||||||
| Bond lengths (Å) | 0.008 | 0.007 | 0.008 | 0.008 | 0.008 | 0.007 | 0.008 | 0.008 |
| Bond angles (°) | 1.16 | 1.12 | 1.19 | 1.21 | 1.19 | 1.13 | 1.18 | 1.18 |
Values in parentheses are for highest-resolution shell.
CC* should be close to 1.0 but as demonstrated by (Karplus and Diederichs, 2012), even CC* values as low as 0.4 indicate significantly more useful signal compared to noise.
The mechanism of conformational changes in proteins upon binding to large and small molecules is currently a hotly debated issue. For more than 50 years the ‘induced-fit’ model (Koshland, 1958), where the ligand binds and thereby induces changes in conformation has been widely accepted (Boehr and Wright, 2008; Csermely et al., 2010). Several more recent reports support, however, ‘conformational selection’, also called conformational sampling, population shift or fluctuation fit (Bakan and Bahar, 2009; Boehr et al., 2009; Changeux and Edelstein, 2011; Csermely et al., 2010; Gsponer et al., 2008; Koglin et al., 2008; Lange et al., 2008; Tang et al., 2007), as an alternative mechanism. In this paradigm, a protein is thought to be present in an ensemble of multiple conformations in the absence of ligand, and the ligand binds to one of these pre-existing ligand-free conformations. The eight new structures reported here, when combined with previously known ligand-free structures of related enzymes, provide a platform to compare the two mechanisms of ligand binding.
Results
The synthetase in complex with Met and MetAMP
The TbMetRS•Met crystals contain two copies of the enzyme in the asymmetric unit (AU) (Figure 1). TbMetRS is characterized by several key structural features: (i) a Rossmann fold catalytic core with an inserted connective peptide (CP) domain; (ii) a stem-contact fold (SCF) domain containing the conserved KMSKS signature (KISKS in trypanosomatids); and (iii) an anti-codon binding α-helix bundle. Of special interest is the CP domain which in TbMetRS harbors one “knuckle”, which defines TbMetRS as member of the MetRS1 subfamily (Gentry et al., 2003; Green et al., 2009). (MetRS with two knuckles belong to the MetRS2 subfamily). The structure of TbMetRS•Met is closest to that of Leishmania major MetRS in complex with two intermediate products (LmMetRS•MAMP•PPi•Mg2+) (Larson et al., 2011). However, in the TbMetRS•Met structure, the enzyme adopts a slightly more open active site conformation than in the LmMetRS•MAMP•PPi•Mg2+ complex (Figure S1).
Figure 1. The structure of TbMetRS•Met.

TbMetRS•Met has two subunits in one AU (separated by the dashed line). Each subunit has four domains typical of MetRS: the Rossmann fold (green), CP domain (cyan), SCF (red) and the anticodon binding helix-bundle. The first part of the CP domain is further divided into the CP base (olive) and CP knuckle (deep blue). Met bound in the active site is shown in CPK model. See also Figures S1 to S5 and Table S1.
There is no major difference between the structures of the two subunits in the AU of TbMetRS (RMSD of 0.40 Å) except for a slight displacement in part of the CP domain (Asp353 to Arg408) in subunit B. For the purpose of the discussion below, we further divide this first part of CP domain into two segments – the antiparallel β-stranded “base”, spanning Asp353 to Tyr363 and Thr398 to Arg408, and the knuckle, formed by residues Ser364 to Val397. Four residues at the tip of the knuckle in subunit B are disordered, while these residues are well-defined in subunit A (Figure S2).
Well-defined, and very similar, electron densities for Met are found in the active sites of both subunits in the AU (Figure S3). The Met binding pockets of TbMetRS in the two subunits are essentially the same and the protein adopts a conformation as observed in other MetRS structures in complex with Met (Ingvarsson and Unge, 2010; Serre et al., 2001) (Figure S4 and Table S1).
Upon soaking of Met,ATP and Mg2+ into the TbMetRS•Met crystals, the intermediate product MAMP is formed in the crystal and occupies the active site in each of the two subunits in the AU in the same manner (Figure S5). The overall structure of the TbMetRS•MAMP complex remains close to that of TbMetRS•Met, with an RMSD of 0.28 Å for the two subunits (Figure S1). The structure of TbMetRS•MAMP demonstrates access of ligands to both active sites in the crystal lattice, opening an avenue to study inhibitor binding through soaking experiments.
The synthetase in complex with inhibitors
Five potent inhibitors of TbMetRS, with low nanomolar EC50 values in T. brucei cell culture (Shibata et al., 2011), were soaked into TbMetRS•Met crystals. In an aminoacylation assay, the IC50 values of these inhibitors range from 8 to 65 nM, and correlate well with both the EC50 and the ability to change the melting temperature of the protein (ΔTm) (Table 2). All inhibitors contain a substituted benzyl group connected by a linker to an aminoquinolone (1289, 1312 and 1320) or to a benzimidazole (1331 and 1325) moiety.
Table 2.
Compound ID, structures & activity
| Compound | Chemical structure | ΔTm (°C) | IC50 (nM) | EC50 (nM) | Resolution of structure |
|---|---|---|---|---|---|
| Aminoquinolones | |||||
| 1289 | 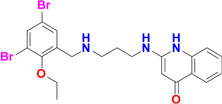 |
12 | 13 | 30 | 3.1 Å |
| 1312 | 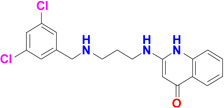 |
12 | 8 | 4 | 3.1 Å |
| 1320 | 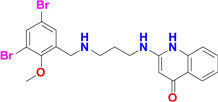 |
8.6* | 65 | 100 | 2.7 Å |
| Benzimidazoles | |||||
| 1325 | 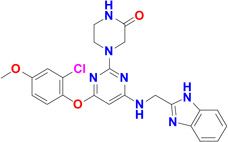 |
10.5 | 43 | 77 | 2.95 Å |
| 1331 | 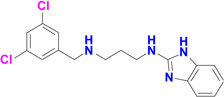 |
9.8 | 46 | 22 | 2.75 Å |
| Low molecular weight compound | |||||
| 89 | 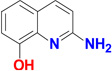 |
1.8** | ND | ND | 2.6 Å |
Concentration used for ΔTm determination is 50 µM in the presence of 10 mM Met, ATP and Mg2+ instead of 100 µM used for the other inhibitors
Concentration used for ΔTm determination is 800 µM against TbMetRS in the absence of Met, ATP and Mg2+
The results of the inhibitor soaks were a complete surprise, since the two MetRS subunits per AU behaved very differently towards the molecules introduced. While Met was retained in subunit A in all cases, each of the inhibitors replaced Met in subunit B and adopted a common binding mode (Figure S6). Typically the benzyl/phenyl group occupies and enlarges the size of the Met pocket forming an ‘enlarged Met pocket’ (EMP) (Figure 2A and 2B). Extending through the linker, the aminoquinolone or benzimidazole moiety of the inhibitors is inserted into a nearby pocket that was previously neither present in the TbMetRS•Met nor in the TbMetRS•MAMP structure. For ease of discussion, we call this the ‘auxiliary pocket’ (AP) (Figure 2C and 2D). Accompanying the enlargement of the Met pocket and the formation of the AP are extensive positional changes, some of these changes move residues to more than 30 Å away from the active site center, with over 80 residues shifting by up to 13 Å. These changes are manifold and include: (i) side chain rearrangements of residues in the Met pocket and the AP; (ii) displacements of at least 35 residues in helices α-2, α-3, α-7 and between strands β-10 and β-11 in the Rossmann fold to form the AP; and (iii) a large movement of the CP domain which creates access to the AP (Figure 2E and Table S2 to S4).
Figure 2. Structural changes in TbMetRS upon binding of inhibitors.


(A) Protein surface of TbMetRS•Met subunit B (pale cyan). Surface that will form the EMP upon binding of inhibitors is colored blue.
(B) Identical view as (A) showing the protein surface of TbMetRS•1320 subunit B (white). Note the enlargement of the narrow Met pocket seen in (A), to the EMP (blue) as well as formation of the AP, which is non-existent in (A).
(C) A 30° rotation view of (A) about the indicated axis, to better show the absence of the AP (red). Residues that would be within a 4.5 Å radius of 1320 upon binding of the inhibitor are shown in stick. The boundary of the CP knuckle is indicated by a dashed line.
(D) A 30° rotation view of (B) about the indicated axis, to better show the formation of the AP (red). Note the rearrangement of many residues in the formation of the AP, including the displacement of the CP knuckle (dashed line) away from the active site.
(E) The extensive conformational changes in TbMetRS upon binding of inhibitor. TbMetRS•Met subunit B (green) is superimposed with TbMetRS•1320 subunit B (sand); Parts of the backbone that undergo significant displacement are traced as red on TbMetRS•1320. Movements are highlighted with arrows.
See also Tables S2 to S4.
The substituted benzyl group and the linker part of the inhibitors hydrophobic occupy the enlarged Met binding pocket. Several hydrophobic side chains (Tyr250, Val473, Trp474 and Phe522) that delineate the smaller Met pocket have moved outwards by 2.4 Å to 6.7 Å to form the larger EMP (Figure 2A, 2B, 3A and Table S2). Two sub-pockets within the EMP can be discerned. The first is originally filled by the sulfur atom of Met in the TbMetRS•Met structure. Either a bromine (1289 and 1320), chlorine (1312 and 1331) or oxygen (1325) atom occupies, in the TbMetRS•inhibitor structures, essentially the same position as the sulfur atom in the TbMetRS•Met structure (Figure 3B and S7A). Another substitution on the ring, typically a halogen atom, fills the second, new, hydrophobic sub-pocket in the EMP created by the shifts of Val473, Trp474 and Phe522. The side chain hydroxyl oxygen of Tyr250 moves by not less than 6.5 Å, resulting in a stacking of its phenol ring against the amino-propyl linker of the inhibitors (Figure 3B and 3C). Otherwise no specific interactions can be observed for the largely solvent exposed linker, explaining the relatively weak density for this part of the molecule (Figure S6).
Figure 3. Stereo views of interactions between TbMetRS and inhibitors.




(A) Side chain rearrangements in TbMetRS active site. TbMetRS•Met subunit B (green) is superimposed with TbMetRS•1320 subunit B (sand). Met (magenta) and 1320 (light pink) are shown in ball and stick. Residues within a 4.5 Å radius of 1320 are shown in stick. Significant rearrangements are indicated with arrows, and side chains moving are labeled.
(B) to (D) Superpositions of all TbMetRS•inhibitor structures show a common binding mode. All inhibitors are depicted in ball and stick; residues within a 4.5 Å radius of inhibitors are shown in stick. The TbMetRS•inhibitor pairs are colored as follows: TbMetRS•1289 – cyan, orange; TbMetRS•1312 – deep blue, pale yellow; TbMetRS•1320 – sand, light pink; TbMetRS•1331 – deep purple, wheat; TbMetRS•1325 – limon, light blue; Met (magenta) is shown as reference.
(B) The substituted benzyl provides a hydrophobic moiety that is inserted into the hydrophobic EMP. The Br, Cl or O atom occupies a position very close to that of the sulfur atom of the substrate Met (shaded).
(C) The planar ring systems in the aminoquinolone or benzimidazole inhibitors, play an important role in stacking interactions, filling the deep auxiliary pocket (AP) (shaded).
(D) The two critical hydrogen bonds between Asp287 carboxylate and the nitrogens on the aminoquinolone and benzimidazole ring systems (2.6 to 3.3 Å) of the inhibitors.
See also Figure S6 to S7.
The AP is non-existent in the TbMetRS•Met structure (Figure 2C) and therefore the aminoquinolone or benzimidazole moiety of the inhibitors would clash in many ways with multiple residues in the substrate-bound conformation of TbMetRS (Figure 3A). Two of the many conformational changes occurring are mainly responsible for creating the AP. First, a rotation of ~30° by the CP, with as hinges residues Tyr355 and Tyr405 near both ends of the antiparallel β-strand CP base, thereby moving away from the active site and providing access to the AP (Figure 2E). Second, multiple helices in the Rossmann fold shift to form the AP (Table S3). The amino-propyl linker adopts an extended conformation and the planar aminoquinolone/benzimidazole moiety of the inhibitors is inserted between the N-termini of helices α-2 and α-7 in the Rossmann fold (Figure 2E). The movements of Tyr250, Gly290, Glu368, Val473 are particularly crucial since their original position in the substrate-bound structures would severely clash with the aminoquinolone or benzimidazole moiety (Figure 2C and 2D). The displacement of Glu368 in the CP domain exposes the AP. Finally, the flip of the Tyr250 side chain creates a groove that connects the EMP and AP. The large displacement of the CP domain needed for access to and formation of the AP probably explains why inhibitors are only observed in subunit B in the AU, since such a repositioning of the CP domain of subunit A is precluded by packing contacts in the crystals (Figure S2).
Eleven residues, mainly hydrophobic, define the AP (Table S4). The insertion of an aminoquinolone/benzimidazole ring into the AP is stabilized by multiple stacking interactions (Figure 3C). The aminoquinolone/benzimidazole ring is sandwiched between the side chains of Tyr250 and Val473, as well as the Tyr472-Val473 and His289-Gly290 peptide units. In addition, the indole ring of Trp474 stacks on top of Val473. These largely planar features of the enzyme form the walls of the pocket. The depth of the pocket is mainly determined by His289, Leu456, Ala460, Tyr472 and Asp476. The phenol ring of Tyr472 swings towards the inhibitors forming the new boundary for the AP. Side chains of Tyr250, Val473 and Trp474 all shift by at least 3.2 Å compared to the substrate-bound conformation. Importantly, Asp287 moves towards to the inhibitor, forming critical hydrogen bonds with the two amines of all inhibitors (Figure 3D and Figure S7B).
The synthetase in complex with a “fragment”
A low molecular weight compound, 2-amino-8-hydroxy-quinoline (89) was also used successfully in a soaking experiment. Compound 89 is a poor inhibitor, as it was found to increase the melting point of TbMetRS by only 1.8 °C at 0.8 mM, which is a much smaller change in melting temperature than observed for the other inhibitors, despite being used at higher concentration (Table 2). Compound 89 binds exclusively to the AP of subunit B (Figure 4A). Quite unexpectedly, even though none of the atoms of 89 are placed in the EMP, the Met pocket has been enlarged and the Met bound initially has disappeared (Figure 4B). A glycerol molecule from the cryo-solution fills part of the EMP (Figure S8). In sharp contrast, in subunit A, Met is retained, the Met pocket is unchanged in size, and no compound 89 is bound. In spite of the much lower affinity of compound 89 for TbMetRS than the other inhibitors, the structure of TbMetRS subunit B binding 89 is very similar to the structures of subunit B bound to the larger inhibitors. The molecule, an aminoquinoline, binds to the AP in the same way as seen before by the aminoquinolone moiety of the inhibitors although it lacks, compared to the inhibitors, the linker and the substituted benzyl moiety which targets the EMP (Figure 4). All the movements of side chains, main chain and secondary structure elements involved in the formation of EMP and AP upon binding compound 89 are remarkably similar to movements observed upon binding of the inhibitors (Figure 4E and Table S2 to S4).
Figure 4. The binding of compound 89 to TbMetRS.


(A) Surface representation of TbMetRS•89 subunit B (yellow) reveals that compound 89 (forest) binds exclusively to the AP (red).
(B) An alternative view of the pockets in TbMetRS•89 subunit B (yellow) show that Met has been displaced and the methionine pocket is enlarged to form the EMP, although none of the atoms of compound 89 are present in the EMP.
(C) Identical view and surface coloring as panel (A): TbMetRS•89 subunit B with inhibitor 1320 from TbMetRS•1320, after superposition of subunits B from both complexes. Compound 89 as ball and stick; 1320 in light pink stick.
(D) Identical view and surface coloring as panel b: TbMetRS•89 subunit B with inhibitor 1320 from TbMetRS•1320, after superposition of subunits B from both complexes. Compound 89 as ball and stick; 1320 in light pink stick.
(E) Superposition of TbMetRS•89 subunit B (yellow) with TbMetRS•1320 subunit B (sand) in stereo. Compound 89 (forest) and inhibitor 1320 (light pink) are shown as ball and stick. Conformations of residues within 4.5 Å of 1320 are identical in both structures except for a slight displacement of E368 as it is located in the flexible CP domain. See also Figure S8.
Discussion
The structures reported here reveal a considerable structural plasticity of the TbMetRS enzyme even in the crystalline state. The enzyme can adopt dramatically different conformations depending on the type of ligand bound. The eight new crystal structures presented here correspond globally with three states:
the Met-bound M-state characterized by a small Met pocket, a closed CP domain, an open/flexible KISKS motif, and a non-existing AP;
the MAMP-product bound P-state, with a small Met pocket, closed CP domain and a closed KISKS motif, while the AP is non-existent.
the inhibitor-bound I-state, where the Met pocket is enlarged, forming the EMP, the CP domain is open, the KISKS motif is flexible, and the AP is formed. Drastic changes of the I-state compared to the M-state involve backbone shifts of domain and secondary structure elements, combined with small and large side chain movements (Figure 2, 3 and Table S2 to S4). The ability of inhibitors to trigger so many changes despite the enzyme being in the crystalline state is highly unusual. In our case, the set of structures elucidated provides in addition a unique opportunity to arrive at a mechanism for the conformational changes observed.
Two global mechanisms have been proposed when binding of a ligand to a protein results in conformational changes of the protein upon forming a protein•ligand complex: induced fit and conformational selection (Boehr et al., 2009; Boehr and Wright, 2008; Changeux and Edelstein, 2011; Csermely et al., 2010). An induced fit mechanism explaining the conformational changes going from the M-state in the crystals to the I-state (Figure 5A and 5B), would require that the MetRS inhibitors or compound 89 somehow cause these conformational changes to take place, while the I-state would not exist prior to ligand binding. A conformational selection mechanism for inhibitor binding requires, instead, that a ligand-free I-state is one of the TbMetRS conformations in the ensemble of conformations existing prior to binding the ligand.
Figure 5. TbMetRS I-state is similar to F-state.

(A) The protein surface of the TbMetRS M-state (pale cyan). In the M-state the Met binding pocket is narrow and the AP is non-existent.
(B) to (D) are identical views as (A).
(B) The protein surface of the TbMetRS I-state (white). Inhibitor 1320 (light pink) is shown in ball and stick model.
(C) The protein surface of the TtMetRS F-state (pale blue). TtMetRS belongs to the MetRS1 subfamily.
(D) The protein surface of the AaMetRS•tRNA F-state (tRNA-bound but unoccupied active site, pale green). AaMetRS also belongs to the MetRS1 subfamily. In contrast to the M-state of TbMetRS (A), both the EMP and the AP are fully formed and accessible in the I-state of TbMetRS (A) and F-states of MetRS1 (C and D). See also Figures S9 to S13.
Among the eight structures of TbMetRS reported here, there is no ligand-free structure since we have been unable to obtain well-diffracting crystals of TbMetRS without methionine. However, crystal structures of ligand-free MetRS1 subfamily members have been reported: Thermus thermophilus MetRS (TtMetRS) (Sugiura et al., 2000) and the tRNA-bound but unoccupied active site structure of Aquifex aeolicus MetRS (AaMetRS) (Nakanishi et al., 2005). The ligand-free states (F-states) of these two MetRS1 enzymes have as hallmarks an unoccupied EMP, an unoccupied AP, an open CP domain and a flexible KISKS motif (Figure 5C and 5D). In other words, these ligand-free MetRS1 F-states are similar to the I-states observed in TbMetRS in complex with inhibitors and with compound 89 (Figure 5B to 5D). Given this similarity, it is highly likely that the F-state of TbMetRS also contains these characteristics, and hence its F-state is very similar in structure to its I-state. The combination of these data indicate that, even in the crystalline state, the conformation of subunit B adopts for a small fraction of the time the F-state in which the methionine is absent, the EMP and AP are present but unoccupied, the CP moves away, and the KISKS loop flexes. When these crystals are in a medium containing inhibitors or compound 89, these small molecules select the ligand-free F-state for forming the TbMetRS•ligand complex with the enzyme in an I-state very similar to that to the F-state (Figure 6). Hence, inhibitor and fragment binding to the TbMetRS•Met complex occurs largely via the mechanism of conformational selection, with possibly small structural adaptations after initial ligand binding.
Figure 6. A model for the binding of inhibitors through conformational selection.

The presence of Met (red) stabilizes the M-state as the major population in the crystals. The introduction of ATP and Mg2+ (green) by soaking stabilizes a partially closed active site and shifts the equilibrium towards the P-state. Further, due to the lack of crystal contacts in subunit B to stabilize the CP domain (especially the CP knuckle) and the KISKS motif (Figure S2), a minor population of F-state continues to exist in the crystal. The F-state differs from the M-state by the presence of the EMP and AP. Upon the introduction of inhibitors (blue) or compound 89 (dark blue), the F-state, which is in a conformation compatible with the binding due to the presence of the EMP and the AP, is stabilized and selected by the inhibitors. Hence the mechanism of binding is conformational selection. Abbreviations used in labels are: MP – Methionine pocket, EMP – enlarge Methionine pocket, AP – auxiliary pocket, Ad – adenine pocket. See also Figures S9 to S13.
It is of interest to contrast the properties of the MetRS1 and MetRS2 enzymes. In the F-state of MetRS2 enzymes, as seen in the structures of EcMetRS (Mechulam et al., 1999) and Pyrococcus abyssi MetRS (PaMetRS)(Crepin et al., 2004), the EMP is evident but the AP is occluded (Figure S9). Interestingly, aminoquinolone inhibitors are known to be ineffective against MetRS2 subfamily (Gentry et al., 2003; Green et al., 2009). This further supports the conformational selection mechanism for MetRS1 enzymes, since the conformation compatible with the inhibitor binding is not available in the F-state of MetRS2. Hence, there are globally two F-state conformations in MetRS enzymes dependent on subfamily. The F-state of MetRS1 is comparable and similar to the I-state of TbMetRS while these states are different for the MetRS2 enzymes (Figure 5 and Figure S9). (The basis for the separation of the two distinct F-states is further discussed in the Supplementary Text and Figure S10 to 13).
Summarizing, the eight new structures of TbMetRS, a validated drug target for African trypanosomiasis, in complex with a substrate, a product, five inhibitors and a low molecular weight compound, provide a precise picture of the mode of binding and the conformational changes occurring in this MetRS1 enzyme upon binding of a series of potent inhibitors and of a weak binding compound. These structures provide a precious platform for the further development of desperately needed anti-trypanosomal compounds. Completely unexpectedly, this series of structures indicates at the same time that conformational selection is the likely mechanism of the observed ligand binding by TbMetRS. These results support the idea that conformational selection plays a major role in ligand binding to proteins (Bakan and Bahar, 2009; Boehr et al., 2009; Changeux and Edelstein, 2011).
Experimental procedures
Protein expression and purification
Previous attempts to crystallize both the full-length and a catalytically active N-terminal truncation mutant of TbMetRS (Tb927.10.1500) within the Medical Structural Genomics of Parasitic Protozoa (MSGPP) consortium (Fan et al., 2008) were unsuccessful even though the same N-terminal truncation had been successful in the crystallization of the homologous MetRS from Leishmania major (LmMetRS) (Larson et al., 2011). In a next approach, this truncated form (residues 237 to 773) was chosen for site-directed mutagenesis of surface residues to promote crystallization through surface entropy reduction mutagenesis (SER) using suggestions from the SERP server (Goldschmidt et al., 2007). Instead of mutating residues to alanine as in ‘classical’ SER, some residues were also mutated to arginine. Several crystal forms were obtained after exploring multiple SER variants, all remote from the active site. Crystals used in the current study contain a triple mutation of 452KKE454 to ARA in TbMetRS.
Plasmids carrying this triple mutation were transformed into Escherichia coli BL21(DE3) for expression. Protein was purified by a Ni-NTA affinity column followed by overnight cleavage of the N-terminal hexa-histidine tag using 3C protease at 4°C. Cleaved protein was purified by size-exclusion chromatography on a Superdex 75 column (Amersham Pharmacia Biotech) using a buffer containing 25 mM HEPES, 500 mM NaCl, 2 mM DTT, 5% glycerol, 0.025% NaN3 and 10 mM L-methionine at pH 7.0. Purified protein retained five residues of the 3C protease cleavage site (GPGSM) at the N-terminus.
Protein crystallization
The protein was screened for crystallization leads using a Phoenix crystallization robot (Art Robbins Instruments) using a variety of commercially available sparse matrix screens. One hit from the Wizard II screen (Emerald Biosystems) was optimized to yield diffracting quality crystals. The best crystals were obtained by vapor diffusion using sitting drops equilibrated at room temperature against a reservoir containing 2.0 to 2.3 M (NH4)2SO4, 0.2 M NaCl and 0.1 M sodium cacodylate pH 6.0 to 6.6. The drops consisted of 1µL protein at 10 mg/mL plus 1µL of the reservoir solution. An additional 1 mM TCEP and 10 mM L-Methionine were added to the protein solution immediately prior to setting up crystal trays. Crystals grew in 1–2 days at room temperature.
Compound synthesis
The synthesis and characterization of the compounds have been described recently (Shibata et al., 2011). All compounds are based on previously reported structures of bacterial MetRS inhibitors. Compounds 1289, 1312 and 1320 have a substituted benzyl group linked to an aminoquinolone moiety through an amino-propyl linker (Critchley et al., 2005; Jarvest et al., 2002). The aminoquinolone moiety is replaced by a benzimidazole moiety in compound 1331 (Jarvest et al., 2004). Compound 1325 is similar to another reported inhibitor (Li, 2008) where the substituted phenyl group and benzimidazole group is bridged by a pyrimidine moiety. Compound 89 is a low molecular weight ‘fragment’, similar to the aminoquinolone moiety of 1289, 1312 and 1320, found to increase the melting temperature of TbMetRS in a screen against the MSGPP fragment cocktail library (Verlinde et al., 2009).
Soaking of compounds
Attempts to co-crystalize the compounds with TbMetRS failed to yield high resolution diffraction data. Following an alternative avenue, TbMetRS•Met crystals were soaked in a cryo-solution containing the compounds. The ratio of compounds, reservoir and glycerol solutions was carefully adjusted to minimize precipitation. Typically, crystals were soaked in a 10 µL solution containing 1 µL of 20 mM inhibitor (or 500 mM 89) in 20% DMSO, 4 µL reservoir solution and 5 µL 60% glycerol in protein buffer. Crystals usually disintegrated upon soaking longer than several minutes and, typically, required to be flash frozen in liquid nitrogen within one minute.
Data collection and structure determination
Data were collected under cryogenic conditions at three different facilities: (1) home source facility using 1.54 Å wavelength for TbMetRS•Met, TbMetRS•MAMP, TbMetRS•1289, TbMetRS•1312, TbMetRS•1320 and TbMetRS•1325; (2) SSRL beam-line 11-1 using 0.98 Å wavelength for TbMetRS•1331; and (3) SSRL beam-line 9-2 using 0.98 Å for TbMetRS•1289. All data were integrated and scaled with the programs Denzo and Scalepack in HKL2000 (Otwinowski and Minor, 1997). Unmerged output from Scalepack was also scaled with the program Scala in the CCP4 suite (Collaborative Computational Project Number 4, 1994) to obtain Rpim (Weiss, 2001) and CC* (Karplus and Diederichs, 2012) values (Table 1). The initial structure of TbMetRS•Met was solved by molecular replacement using the coordinates of LmMetRS (3FKL) (Larson et al., 2011) as the search model with the program Phaser (McCoy et al., 2007). Iterated building/rebuilding and refinement of models were performed using Coot (Emsley et al., 2010) and REFMAC5 (Murshudov et al., 1997), respectively. In the final cycles of refinement, protein structures were refined with translational/libration/screw (TLS) groups identified by the TLS motion determination server (Painter and Merritt, 2006) before restrained refinement in REFMAC5. The refined structure of TbMetRS•Met was then used as search model for molecular replacement of the other structures. The PRODRG server (Schuttelkopf and van Aalten, 2004) was used to generate refinement restraints for all ligands. The structure validation server MolProbity (Chen et al., 2010) was used throughout the process to monitor the progress of structure determination. All refined structures showed good statistics with no outliers in Ramachandran plots based on MolProbity. The final crystallographic refinement statistics are given in the Table 1. Figures were created and rendered with Pymol (DeLano, 2002).
Aminoacylation assay
Enzyme activity was quantified by the attachment of [3H]methionine to tRNA in the presence of the T. brucei MetRS enzyme. Reactions were performed in 96-well filter plates with Durapore® membranes (MSHVN4B10; Millipore) in volumes of 75 µl. The reaction was performed with 25 mM HEPES (pH 7.9), 10 mM MgCl2, 50 mM KCl, 0.2 mM spermine, 0.1 mg/mL bovine serum albumin, 2.5 mM dithiothreitol, 1% DMSO, and 1 U/mL pyrophosphatase (I1643; Sigma). Recombinant enzyme (10 nM) and compound inhibitors (starting concentration varied depending on potency and included 12 serial two dilutions) were mixed with the buffer and preincubated for 15 min. To start the reaction, 400 µg/mL bulk Escherichia coli tRNA (R4251; Sigma), 0.1 mM ATP, and 250 nM [3H]methionine (80 Ci/mmol) were added. The plate was incubated without shaking at room temperature for 120 min. The reactions were stopped by the addition of 100 µL cold 10% trichloroacetic acid. The reaction components were separated from tRNA by filtration through a vacuum manifold and washed three times with cold 10% trichloroacetic acid. The filter plates were dried overnight, scintillation fluid was added, and the counts on the plates were determined in a scintillation plate counter. Samples were run in quadruplicate and percent inhibition was calculated using two different controls (no enzyme and no test compound) with the following formula:
Where Mnd is the average no drug control, Dtd is each test drug value, and Mne is the average no enzyme control. IC50 values were then calculated by non-linear regression (sigmoidal dose response) in Prism 3.0.
Thermal shift assay
The thermal shift assay was performed as previously described (Shibata et al., 2011) with 0.42 mg/ml of TbMetRS, 100 µM of 1289, 1312, 1325, 1331 or 50 µM of 1320, 10 mM ATP magnesium salt, 10 mM L-methionine, and 5% dimethyl sulfoxide. The inclusion of 10 mM ATP and methionine in the assay buffer was to suppress the overall thermal shift signal so that only tight-binding inhibitors could cause a significant shift in the melting temperature (Tm) of MetRS. The assay for compound 89 was performed at 800 µM of 89 in the absence of Met, ATP and Mg2+. The assays were performed twice independently in triplicate in each experiment.
Supplementary Material
Highlights.
Complexes of MetRS from T. brucei with its substrate, intermediate and inhibitors
In crystals, substrate-bound MetRS undergoes extensive changes with inhibitor-binding
The inhibitor-bound conformation is surprisingly similar to the ligand-free state
This indicates inhibitor-binding occurs via conformational selection not by induced-fit
Acknowledgments
We thank Stewart Turley, Frank Zucker and Jonathan Kay for providing support for the X-ray data collection, database management and computing environment at the Biomolecular Structure Center of the University of Washington. We thank the Protein Expression and Purification Laboratory of Dr. Wesley Van Voorhis at the University of Washington for the initial cloning of wild-type TbMetRS. We also thank the staff of Stanford Synchrotron Radiation Lightsource (SSRL) for assistance during data collection. This work is funded by the National Institutes of Health (grants P01AI067921, (Medical Structural Genomics of Pathogenic Protozoa (MSGPP)), R56AI084004, and RO1AI084004.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Accession numbers
Coordinates and structure factors for TbMetRS•Met, TbMetRS•MAMP, TbMetRS•1289, TbMetRS•1312, TbMetRS•1320, TbMetRS•1325, TbMetRS•1331 and TbMetRS•89 are deposited in the Protein Data Bank under accession codes 4EG1, 4EG3, 4EG4, 4EG5, 4EGA, 4EG6, 4EG7 and 4EG8, respectively.
References
- Bakan A, Bahar I. The intrinsic dynamics of enzymes plays a dominant role in determining the structural changes induced upon inhibitor binding. Proc Natl Acad Sci U S A. 2009;106:14349–14354. doi: 10.1073/pnas.0904214106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, Nussinov R, Wright PE. The role of dynamic conformational ensembles in biomolecular recognition. Nat Chem Biol. 2009;5:789–796. doi: 10.1038/nchembio.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr DD, Wright PE. Biochemistry. How do proteins interact? Science. 2008;320:1429–1430. doi: 10.1126/science.1158818. [DOI] [PubMed] [Google Scholar]
- Brun R, Blum J, Chappuis F, Burri C. Human African trypanosomiasis. Lancet. 2010;375:148–159. doi: 10.1016/S0140-6736(09)60829-1. [DOI] [PubMed] [Google Scholar]
- Changeux JP, Edelstein S. Conformational selection or induced fit? 50 years of debate resolved. F1000 Biol Rep. 2011;3:19. doi: 10.3410/B3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr, Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Crepin T, Schmitt E, Blanquet S, Mechulam Y. Three-dimensional structure of methionyl-tRNA synthetase from Pyrococcus abyssi. Biochemistry. 2004;43:2635–2644. doi: 10.1021/bi0356247. [DOI] [PubMed] [Google Scholar]
- Critchley IA, Green LS, Young CL, Bullard JM, Evans RJ, Price M, Jarvis TC, Guiles JW, Janjic N, Ochsner UA. Spectrum of activity and mode of action of REP3123, a new antibiotic to treat Clostridium difficile infections. J Antimicrob Chemother. 2009;63:954–963. doi: 10.1093/jac/dkp041. [DOI] [PubMed] [Google Scholar]
- Critchley IA, Ochsner UA. Recent advances in the preclinical evaluation of the topical antibacterial agent REP8839. Curr Opin Chem Biol. 2008;12:409–417. doi: 10.1016/j.cbpa.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Critchley IA, Young CL, Stone KC, Ochsner UA, Guiles J, Tarasow T, Janjic N. Antibacterial activity of REP8839, a new antibiotic for topical use. Antimicrob Agents Chemother. 2005;49:4247–4252. doi: 10.1128/AAC.49.10.4247-4252.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P, Palotai R, Nussinov R. Induced fit, conformational selection and independent dynamic segments: an extended view of binding events. Trends Biochem Sci. 2010;35:539–546. doi: 10.1016/j.tibs.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. The PyMOL Molecular Graphics System. 2002 http://wwwpymolorg. [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallographica Section D. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan E, Baker D, Gelb MH, Buckner FS, Van Voorhis WC, Phizicky E, Dumont M, Mehlin C, Grayhack EJ, Sullivan M, et al. Structural Genomics of Pathogenic Protozoa: An Overview. In: Bostjan K, Mitchell G, Huber T, editors. Methods in Molecular Biology. 2008. pp. 497–513. [DOI] [PubMed] [Google Scholar]
- Gentry DR, Ingraham KA, Stanhope MJ, Rittenhouse S, Jarvest RL, O'Hanlon PJ, Brown JR, Holmes DJ. Variable sensitivity to bacterial methionyl-tRNA synthetase inhibitors reveals subpopulations of Streptococcus pneumoniae with two distinct methionyl-tRNA synthetase genes. Antimicrob Agents Chemother. 2003;47:1784–1789. doi: 10.1128/AAC.47.6.1784-1789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldschmidt L, Cooper DR, Derewenda ZS, Eisenberg D. Toward rational protein crystallization: A Web server for the design of crystallizable protein variants. Protein Sci. 2007;16:1569–1576. doi: 10.1110/ps.072914007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green LS, Bullard JM, Ribble W, Dean F, Ayers DF, Ochsner UA, Janjic N, Jarvis TC. Inhibition of methionyl-tRNA synthetase by REP8839 and effects of resistance mutations on enzyme activity. Antimicrob Agents Chemother. 2009;53:86–94. doi: 10.1128/AAC.00275-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gsponer J, Christodoulou J, Cavalli A, Bui JM, Richter B, Dobson CM, Vendruscolo M. A coupled equilibrium shift mechanism in calmodulin-mediated signal transduction. Structure. 2008;16:736–746. doi: 10.1016/j.str.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotez PJ, Molyneux DH, Fenwick A, Kumaresan J, Sachs SE, Sachs JD, Savioli L. Control of neglected tropical diseases. N Engl J Med. 2007;357:1018–1027. doi: 10.1056/NEJMra064142. [DOI] [PubMed] [Google Scholar]
- Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–650. doi: 10.1146/annurev.biochem.69.1.617. [DOI] [PubMed] [Google Scholar]
- Ingvarsson H, Unge T. Flexibility and communication within the structure of the Mycobacterium smegmatis methionyl-tRNA synthetase. FEBS J. 2010;277:3947–3962. doi: 10.1111/j.1742-4658.2010.07784.x. [DOI] [PubMed] [Google Scholar]
- Jarvest RL, Armstrong SA, Berge JM, Brown P, Elder JS, Brown MJ, Copley RC, Forrest AK, Hamprecht DW, O'Hanlon PJ, et al. Definition of the heterocyclic pharmacophore of bacterial methionyl tRNA synthetase inhibitors: potent antibacterially active non-quinolone analogues. Bioorg Med Chem Lett. 2004;14:3937–3941. doi: 10.1016/j.bmcl.2004.05.070. [DOI] [PubMed] [Google Scholar]
- Jarvest RL, Berge JM, Berry V, Boyd HF, Brown MJ, Elder JS, Forrest AK, Fosberry AP, Gentry DR, Hibbs MJ, et al. Nanomolar inhibitors of Staphylococcus aureus methionyl tRNA synthetase with potent antibacterial activity against gram-positive pathogens. J Med Chem. 2002;45:1959–1962. doi: 10.1021/jm025502x. [DOI] [PubMed] [Google Scholar]
- Karplus PA, Diederichs K. Linking crystallographic model and data quality. Science. 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koglin A, Lohr F, Bernhard F, Rogov VV, Frueh DP, Strieter ER, Mofid MR, Guntert P, Wagner G, Walsh CT, et al. Structural basis for the selectivity of the external thioesterase of the surfactin synthetase. Nature. 2008;454:907–911. doi: 10.1038/nature07161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshland DE. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc Natl Acad Sci U S A. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KF, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Larson ET, Kim JE, Zucker FH, Kelley A, Mueller N, Napuli AJ, Verlinde CL, Fan E, Buckner FS, Van Voorhis WC, et al. Structure of Leishmania major methionyl-tRNA synthetase in complex with intermediate products methionyladenylate and pyrophosphate. Biochimie. 2011;93:570–582. doi: 10.1016/j.biochi.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Hilgers H, Kedar GC, Stidham M, Brown-Driver V, Shaw KJ, Finn J. Discovery and SAR of a novel series of pyrimidine antibacterials targeting methionyl-tRNA synthetase; Interscience Conference on Antimicrobial Agents and Chemotherapy; 2008. [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechulam Y, Schmitt E, Maveyraud L, Zelwer C, Nureki O, Yokoyama S, Konno M, Blanquet S. Crystal structure of Escherichia coli methionyl-tRNA synthetase highlights species-specific features. Journal of Molecular Biology. 1999;294:1287–1297. doi: 10.1006/jmbi.1999.3339. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nakanishi K, Ogiso Y, Nakama T, Fukai S, Nureki O. Structural basis for anticodon recognition by methionyl-tRNA synthetase. Nat Struct Mol Biol. 2005;12:931–932. doi: 10.1038/nsmb988. [DOI] [PubMed] [Google Scholar]
- Ochsner UA, Sun X, Jarvis T, Critchley I, Janjic N. Aminoacyl-tRNA synthetases: essential and still promising targets for new anti-infective agents. Expert Opin Investig Drugs. 2007;16:573–593. doi: 10.1517/13543784.16.5.573. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter C, Sweet R, editors. Macromolecular Crystallography, Part A. Academic Press; 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- Painter J, Merritt EA. TLSMD web server for the generation of multi-group TLS models. J Appl Cryst. 2006;39:109–111. [Google Scholar]
- Pohlmann J, Brotz-Oesterhelt H. New aminoacyl-tRNA synthetase inhibitors as antibacterial agents. Curr Drug Targets Infect Disord. 2004;4:261–272. doi: 10.2174/1568005043340515. [DOI] [PubMed] [Google Scholar]
- Schuttelkopf AW, van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60:1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Serre L, Verdon G, Choinowski T, Hervouet N, Risler JL, Zelwer C. How methionyl-tRNA synthetase creates its amino acid recognition pocket upon L-methionine binding. J Mol Biol. 2001;306:863–876. doi: 10.1006/jmbi.2001.4408. [DOI] [PubMed] [Google Scholar]
- Shibata S, Gillespie JR, Kelley AM, Napuli AJ, Zhang Z, Kovzun KV, Pefley RM, Lam J, Zucker FH, Van Voorhis WC, et al. Selective inhibitors of methionyl-tRNA synthetase have potent activity against Trypanosoma brucei Infection in Mice. Antimicrob Agents Chemother. 2011;55:1982–1989. doi: 10.1128/AAC.01796-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura I, Nureki O, Ugaji-Yoshikawa Y, Kuwabara S, Shimada A, Tateno M, Lorber B, Giege R, Moras D, Yokoyama S, et al. The 2.0 A crystal structure of Thermus thermophilus methionyl-tRNA synthetase reveals two RNA-binding modules. Structure. 2000;8:197–208. doi: 10.1016/s0969-2126(00)00095-2. [DOI] [PubMed] [Google Scholar]
- Tang C, Schwieters CD, Clore GM. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449:1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- Verlinde CL, Fan E, Shibata S, Zhang Z, Sun Z, Deng W, Ross J, Kim J, Xiao L, Arakaki TL, et al. Fragment-based cocktail crystallography by the medical structural genomics of pathogenic protozoa consortium. Curr Top Med Chem. 2009;9:1678–1687. doi: 10.2174/156802609790102383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vondenhoff GH, Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur J Med Chem. 2011;46:5227–5236. doi: 10.1016/j.ejmech.2011.08.049. [DOI] [PubMed] [Google Scholar]
- Weiss MS. Global indicators of X-ray data quality. Journal of Applied Crystallography. 2001;34:130–135. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.