

Abstract
While neurodegenerative diseases are characterized by steady degeneration over relatively long timelines, it is widely believed that the early stages are the most promising for therapeutic intervention, before irreversible neuronal loss occurs. Developing a therapeutic response requires a precise measure of disease progression. However, since the early stages are for the most part asymptomatic, obtaining accurate measures of disease progression is difficult. Longitudinal databases of hundreds of subjects observed during several years with tens of validated biomarkers are becoming available, allowing the use of computational methods. We propose a widely applicable statistical methodology for creating a disease progression score (DPS), using multiple biomarkers, for subjects with a neurodegenerative disease. The proposed methodology was evaluated for Alzheimer’s disease (AD) using the publicly available AD Neuroimaging Initiative (ADNI) database, yielding an Alzheimer’s DPS or ADPS score for each subject and each time-point in the database. In addition, a common description of biomarker changes was produced allowing for an ordering of the biomarkers. The Rey Auditory Verbal Learning Test delayed recall was found to be the earliest biomarker to become abnormal. The group of biomarkers comprising the volume of the hippocampus and the protein concentration amyloid beta and Tau were next in the timeline, and these were followed by three cognitive biomarkers. The proposed methodology thus has potential to stage individuals according to their state of disease progression relative to a population and to deduce common behaviors of biomarkers in the disease itself.
Keywords: Neurodegenerative diseases, Alzheimer’s disease, biomarkers, disease progression score
1. Introduction
Neurodegenerative diseases such as Alzheimer’s Disease (AD), Parkinson disease (PD), Huntington Disease (HD) and Amyotrophic Lateral Sclerosis (ALS) involve the loss of structure or function of neurons, including neuronal death (see Martin (2002); Shaw (2005)). During the earliest stages of these diseases, the progression is slow, on the time scale of years, (see Sperling et al. (2011) for the case of AD). It is widely believed that these early stages are the most promising for therapeutic intervention, before irremediable neuronal loss occurs. Developing a therapeutic remedy requires a precise measure of disease progression, i.e., a quantity which would be specific to a particular disease and sensitive to subtle changes. However, obtaining accurate measures of disease progression during the earliest phases of the disease is difficult. Indeed, these phases are essentially non-symptomatic and the clinical tests which characterize the acute phase of the disease are not sensitive enough to qualify as a measure of disease progression. In response, the medical research community has contributed to developing and validating biomarkers. Biomarkers for neurodegenerative diseases include protein counts (in the cerebrospinal fluid), blood analysis, brain imaging, including molecular and MR, genetic analysis and neuropsychological tests. Structural imaging biomarkers are unique in that they allow one to characterize the size, shape, and health of various brain substructures at the organ level while being noninvasive (see e.g. Qiu et al. (2008) for AD, Rizk-Jackson et al. (2011) for HD). Functional imaging provides a spatially localized image of the physiological processes occurring in the brain. See Brooks and Pavese (2011) for a review of imaging biomarkers in PD and Turner et al. (2011) for ALS. Due to the complexity of the neurodegenerative diseases and variabilities within the human population, research efforts have been pooled in order to create datasets with a large number of subjects, time-points and biomarkers. The Alzheimer’s Disease Neuroimaging Initiative (ADNI), see http://adni.loni.ucla.edu/, was launched in 2003 by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, the Food and Drug Administration, private pharmaceutical companies and non-profit organizations, as a $60 million, 5-year public/private partnership. A related effort is taking place for PD. The Parkinson Progression Marker Initiative (PPMI), see http://www.ppmi-info.org/, is a comprehensive observational, international, multicenter study designed to identify PD progression biomarkers both to improve understanding of disease etiology and course and to provide crucial tools to enhance the likelihood of success of PD modifying therapeutic trials. Huntington disease is caused by a mutation in a single gene, HTT, with full penetrance, making it feasible to identify presymptomatic individuals who will develop the disease but do not yet show yet any clinical symptoms, see Hayden (1981). At least two large studies (Predict-HD, see https://www.predict-hd.net/ and TrackOn-HD, see http://hdresearch.ucl.ac.uk/current-studies/trackon-hd/) are underway to identify sensitive biomarkers for HD. Similar efforts are recently taking place for ALS, see Turner et al. (2009); Labbe (2012). The availability of large datasets for neurodegenerative diseases opens new opportunities for computational methods which could have a strong impact in the study, the development of therapeutics and the follow-up of patients with neurodegenerative diseases.
We present in this article a generic computational method for computing a disease progression score (DPS) by combining biomarkers. ADNI is, as of today, the largest publicly available longitudinal dataset of biomarkers related to a neurodegenerative disease. It is therefore the dataset which we have chosen to evaluate our method. Since we will work with the ADNI dataset, we recall some preliminary information on AD as well as the validated biomarkers for AD in section 2. The method for computing a DPS, which is the main contribution of this paper, is presented in section 3. Results with the ADNI dataset appear in section 4 and finally in section 5, we discuss the results in the context of ADNI, and their consequence in the study of AD and of neurodegenerative diseases.
2. Alzheimer’s disease
Although this paper describes a method applicable to any neurodegenerative disease, our current evaluation involves the ADNI dataset and therefore it is informative to use this disease as a framework for motivating the method. The classical characterization of late-onset Alzheimer’s disease progression is a time-ordered succession of three stages: normal (N), mild cognitive impairment (MCI), and AD. Physical measurements of disease progression, i.e., biomarkers, are used to classify patients into these three stages, but it has been challenging to reliably define finer stages of the disease. As a result, staging of the disease remains coarse and the evaluation of therapies are difficult at the earliest stages when intervention is most likely to be effective, see Hampel et al. (2008).
Cognitive biomarkers such as the clinical dementia rating sum-of-boxes (having scores from 0 to 18) and the mini-mental state exam (having integer scores from 0 to 30) have finer discrete levels, see Berg et al. (1988); Folstein et al. (1975). But it has been reported in Mungas and Reed (2000) and Duara et al. (2011) that these measurements have poor dynamic range in the earliest stages of AD. On the other hand, Mosconi et al. (2007) has shown that the early stages of AD can be characterized using both imaging and biochemical biomarkers. Following these observations, Jack et al. (2010) proposed that there is a single disease progression and that different biomarkers characterize the disease during different stages. They hypothesized the biomarker changes and disease progression shown in Fig. 1 (reproduced with permission from Jack et al. (2010)). In this hypothesized model, the amyloid beta (Aβ42) protein changes first, followed by changes in the protein Tau, then structural changes in the brain (gray matter loss), and lastly a deterioration of cognitive function resulting in dementia. Based on Fig. 1 we expect to find that no single biomarker has the dynamic range to cover the full spectrum of the disease. Given the limitations of any single biomarker, there is likely benefit in developing methods that can combine multiple biomarkers in a nonlinear fashion in order to represent—using a single measure—progression throughout the entire disease. This is a key motivation for the process we report in this paper. An important byproduct of this effort is a plot similar to that of Fig. 1, but derived from data using multiple biomarkers which reveal key differences in the ordering of the biomarker dynamics over the course of disease.
Figure 1.
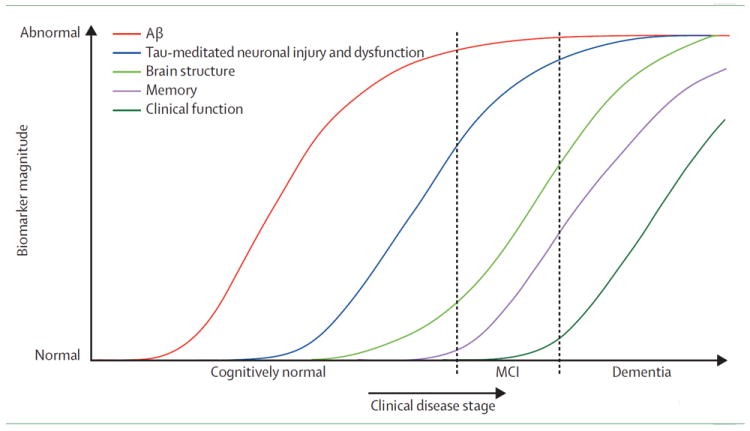
This graph represents a conceptualization of the timing of key biomarkers transitions from “Normal” to “Abnormal” as subjects go through the three stages of Alheimer’s disease: “Cognitively Normal”, “MCI”, and “Dementia.” This plot is reproduced from “Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade,” Jack CR Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ., Lancet Neurol. 2010 Jan;9(1):119-28.
3. Method
3.1. Principles for temporal standardization of multiple biomarkers
The available data are longitudinal measurements of multiple biomarkers for hundreds of subjects. Our research first describes and then evaluates a disease progression score, notated DPS, which standardizes subject time-lines onto a common temporal scale. The DPS serves as a new (derived) biomarker enabling both disease staging in single subjects and a data-driven characterization of biomarker dynamics in the entire population.
The method we use to achieve standardization is based on three assumptions:
All subjects follow a common disease progression but differ in their age of onset and rate of progression;
As the disease progresses, each biomarker changes continuously and monotonically following a sigmoid shaped curve; and
In the longitudinal period over which biomarkers are observed, the rate of progression of a given subject is constant.
The proposed computation assigns to each subject and each time-point a score denoted the DPS. Note that all subjects are expected to undergo the same biological and cognitive changes when they reach the same DPS.
3.2. Statistical model for DPS
The age t of subject i is to be transformed into the DPS si as follows
| (1) |
upon estimation of the subject dependent parameters αi and βi, which indicate rate and onset of disease, respectively. A linear transformation is justified when the interval over which longitudinal observations of subjects occur is short relative to disease duration (true at present in the ADNI database). This could be generalized to nonlinear functions in the case of cohorts with longer longitudinal base. Our objective is to standardize all I subjects by estimating α = (α1, …, αI) and β = (β1, …, βI). The subject dependent parameters α and β are deliberately modeled as fixed effects, not random effects, as the DPS may ultimately be used as a covariate.
The longitudinal dynamic of each biomarker is assumed to be the same across the population and can be represented as a sigmoidal function f of DPS s. Sigmoidal functions capture the relative quiescent states of a biomarker in the early and late parts of the disease progression while being parsimonious. Using θk = (ak, bk, ck, dk) to represent the vector of sigmoid function parameters for the k-th biomarker, we can write the form of the k-th biomarker as
| (2) |
The minimum and maximum values of the sigmoid function are dk and dk + ak, and the value of s for which the biomarker is the most dynamic, having maximum slope ak bk/4 corresponding to its inflection point, is ck. A closely related model is the trilinear model in Brooks et al. (1993). Caroli et al. (2010) and Sabuncu et al. (2011) noticed that sigmoids offer a parsimonious parametric model which is often a better fit than linear models for biomarkers. Sigmoids are also similar in form to the conceptual evolution of biomarkers envisioned in Jack et al. (2010) for AD (Fig. 1). Among parametric models, alternatives include the generalized sigmoid in Richards (1959) and polynomials of low order.
Databases for neurodegenerative diseases contain measurements yijk of biomarker k for subject i at visit j. Since there are often irregularities in data collection, we use I to denote the set of triples (i, j, k) for which measurements are available. Each biomarker observation can then be written as
| (3) |
where tij is the age of subject i at visit j. Observation noise in each biomarker is modeled for simplicity by the product of εijk, which are independent random variables with zero mean and unit variance, and σk, which is the standard deviation of biomarker k. The collection of standard deviations σ = (σ1, …, σK) comprise another unknown that must be estimated.
The unknowns in this problem are α, β, θ, and σ and the least squares problem associated with the observation model in (3) is
| (4) |
3.3. Parameter fitting
Parameter fitting is performed using alternating least squares wherein the parameters θ, α, β, and σ are optimized iteratively starting from the values computed in the previous step. The details of the fitting algorithm are shown in Alg. 1. Because of the additive form of (4), optimization over θ is done serially over each of the K biomarkers. Similarly, optimization over (α, β) is performed serially over each of the I subjects. Fitting of θ, α, and β requires optimization of continuously differentiable nonconvex functions, which is carried out using the Levenberg-Marquardt algorithm (Lines 4 and 8), see Levenberg (1944). Ik (line 4) is the number of subjects and visits available for biomarker k. The denominator in the equation of Line 5 is the number of degrees of freedom. Because unconstrained optimization can produce unfeasible parameters, parameters are projected onto the feasible space after the main loop (Lines 13–17), see (5) below. This does not change the value of the objective function in (4). Our experiments presented in section 4 confirm that successful fitting is accomplished in 15 iterations for the ADNI dataset; i.e., L = 15 on Line 2, standard optimization stopping criteria can be used otherwise. The parameters α and β are centered and rescaled in Lines 17–19 in Alg. 1 for identifiability reasons which are explained in the next section.
3.4. Identifiability
The units of DPS are arbitrarily defined, which implies that we must choose two specific numerical values in order to fully specify the DPS. This situation is analogous to the selection of a scale for temperature, where the numerical values of the freezing and boiling points of water determine the scale. Note that calibration is not specific to the DPS. It is in fact needed for most if not all biomarkers (see Hughes et al. (1982)). In our experiments with ADNI, we chose to fix the DPS such that after computation of DPS for the entire population, the computed DPS for all visits of subjects with normal clinical assessment had a median (mN) and a median absolute deviation (σN) which are set respectively to zero and one. This is accomplished in Lines 17–19 in Alg. 1.
Algorithm 1
Algorithm for the fitting of the parameters
Inititialize α(0), β(0)
for l = 1 to L do
for k = 1 to K do
end for
for i = 1 to I do
end for
α(0) = α(1), β(0) = β(1)
end for
for k = 1 to K do
if bk < 0 then
end if
end for
for i = 1 to I do
end for
Note that (3) is invariant with respect to the following 2 transformations, for 2 constant γ1 ≠ 0 and γ2:
Note also that the sigmoid function verifies
| (5) |
In order to build an identifiable model, we define the restricted parameter set
for some α0 ≠ 0 and β0. Necessary conditions on the available data I for guaranteing the identifiability of the parameters are as follows:
For each biomarker, there is at least 1 subject i with αi ≠ 0 and with at least 4 distinct time-points in I.
For each subject, there is at least 1 biomarker which is available at 2 time points in I
A proof is provided in the appendix. In practice, a sufficient number of data points per parameter are needed in order to obtain tight estimators. Examining first the case with no missing data, the number of equations in (3) is IJK. The number of parameters is 2I + 5K, counting two parameters per subject, and five per biomarkers: four for the sigmoid and one for the sd. In applications where I is large compared to K, the number of data points per parameter is close to JK/2. Note that longitudinal data (J > 1) is critical for such modeling. However, a small number J of time-points together with a small number K of biomarkers is acceptable. The subset of ADNI presented in section 4 has numerous missing data. Nevertheless, the identifiability conditions are met. The tightness of the estimators of the biomarker parameters is measured using bootstrap and reported in section 4.1.
3.5. The ADNI dataset
Data used in the preparation of this article were obtained from the ADNI database (adni.loni.ucla.edu). The ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and non-profit organizations, as a $60 million, 5- year public-private partnership. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), positron emission tomography (PET), other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of mild cognitive impairment (MCI) and early Alzheimers disease (AD). Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials.
The Principal Investigator of this initiative is Michael W. Weiner, MD, VA Medical Center and University of California San Francisco. ADNI is the result of efforts of many co-investigators from a broad range of academic institutions and private corporations, and subjects have been recruited from over 50 sites across the U.S. and Canada. The initial goal of ADNI was to recruit 800 adults, ages 55 to 90, to participate in the research, approximately 200 cognitively normal older individuals to be followed for 3 years, 400 people with MCI to be followed for 3 years and 200 people with early AD to be followed for 2 years. For up-to-date information, see www.adni-info.org.
The ADNI, ADNI GO, and ADNI 2 biomarker datasets were downloaded from the ADNI server (http://adni.loni.ucla.edu/) on November 24, 2011. The following seven biomarkers were selected for use based on their relevance in assessing the progression of AD. HIPPO is the sum of the two lateral hippocampal volumes (Freesurfer version 4.4.0 for longitudinal data http://surfer.nmr.mgh.harvard.edu) normalized by dividing by the intra-cranial volume. ADAS is the Alzheimer’s Disease Assessment Scale-cognitive subscale. MMSE is the Mini-Mental State Examination score. TAU and ABETA (our abbreviation for Aβ42) are protein levels measured from the cerebrospinal fluid. CDRSB is the Clinical Dementia Rating Sum of Boxes score and RAVLT30 is the Rey Auditory Verbal Learning Test, 30 minute recall. A detailed description of the ADNI population, protocols and biomarkers is provided at http://adni.loni.ucla.edu/. Of the seven biomarkers, only ADAS and RAVLT30 were available at the time of download from the ADNI 2/GO dataset. The protocol for these biomarkers is the same in ADNI, ADNI 2, and ADNI GO. All visits without date information were removed. Subjects not having at least two measurements for at least one of the seven biomarkers were also removed. Finally, subjects not having at least two measurements of the HIPPO biomarker were removed. The total number of subjects remaining was 687, where 389 were male, 275 were female, and 23 had unknown gender. The total number of visits was 3658, and the clinical diagnoses at these visits were 1103 N, 1513 MCI, and 1010 AD. There is an average of 26.92 (sd=5.52) and a minimum of 11 data points available per subject for estimating the parameters of the model.
4. Results
4.1. DPS computed for ADNI subjects
The Alzheimer’s DPS (ADPS) was computed for all subject visits in the combined ADNI, ADNI 2, and ADNI GO datasets (with minimal exclusions as was described in section 3.5). Seven biomarkers—HIPPO, MMSE, TAU, ABETA, CDRSB, RAVLT30, and ADAS—were used together in the computation in order to compute an ADPS score for each visit of each subject (Fig. 2). The initial values (Line 1 of Alg. 1) are obtained as follows: firstly, we set α(0) ≡ 1 and β(0) ≡ 0; secondly, the sigmoids are replaced by linear functions. The main loop (line 2), is then executed 15 times. In this case, the optimization problems in lines 4 and 8 are least squares problems which are solved exactly. At the end of this initialization step, α(0) and β(0) are set to the corre-sponding values obtained and the sigmoids are initialized using the linear fits. The running time of the Algorithm 1, which was coded in Matlab, was 125 seconds using an Intel Core i7Q820 running at 1.73 GHz (quadcore). Overall, N subjects (black) have the smallest ADPS, MCI subjects (red) have moderate ADPS, and AD subjects (green) have the largest ADPS. Lower ADPS scores are therefore consistent with the normal population and higher ADPS scores are indicative of increased presence of dementia. Those subjects whose clinical status changes from MCI to AD (blue) are found mostly between the red and green colors.
Figure 2.
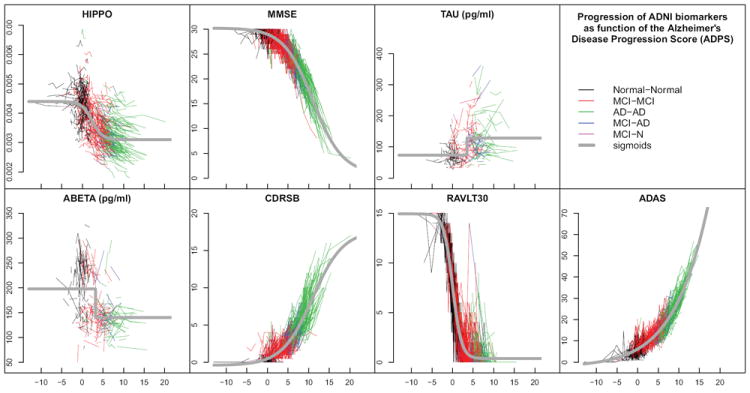
The values of seven biomarkers, measured at all visits of all ADNI subjects, are plotted on the normalized ADPS. Each connected polyline represents the consecutive visits of a single subject, and each line segment is colored according to the subject’s clinical diagnoses between visits (see legend). The gray curves are the sigmoid functions representing the fitted behavior of each biomarker in the normalized space.
The estimated sigmoidal behaviors of each biomarker were also computed as part of the normalization process (gray curves on each plot in Fig. 2). It is observed that individual subject trajectories fall near these curves and have similar slopes in most cases. This is expected due to the nature of the optimization criterion used to define ADPS. However, since ADPS is computed as a joint optimization considering all seven biomarkers, some data falls fairly far from the estimated characteristic biomarker curves.
We used bootstrapping via Monte Carlo resampling to quantify the variance of the estimated parameters. We drew 100 resamples of the observed dataset by random sampling (with replacement) from the original collection of subjects, and then recomputed the ADPS for the entire population. Bootstrap replicates of the estimated biomarker sigmoids are shown in Fig. 3 and 90% confidence intervals for the parameter ck, i.e the inflection point of each sigmoid, are presented in Fig 5(b).
Figure 3.
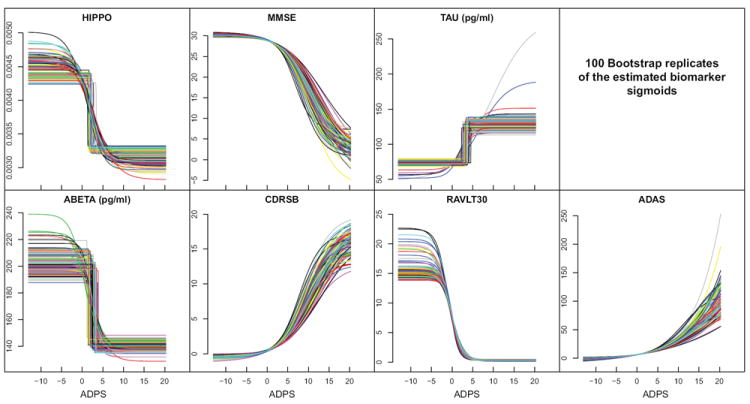
Bootstrapping yields different biomarker sigmoids with each random substitution. These plots give all the computed sigmoids over the entire bootstrapping exercise. Tight agreement overall is observed.
Figure 5.
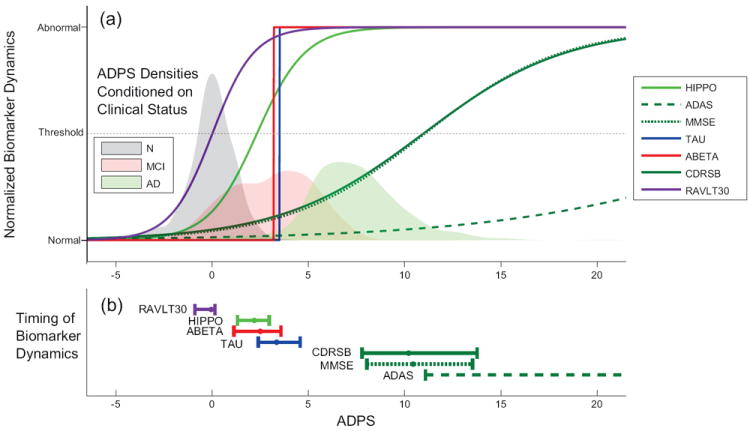
(a) Estimated biomarker dynamics as a function of the normalized ADPS. Estimation of the normalized ADPS for all ADNI subjects was carried out, and common biomarker dynamics represented by sigmoidal functions were simultaneously fitted as part of the ADPS normalization algorithm. Each sigmoidal function was scaled and flipped in order to fit on a scale going from -1 representing “Normal” to 1 representing “Abnormal”. The positions of vertical lines representing progression from Normal to MCI and MCI to AD were fitted as optimal separating thresholds between the clinical diagnoses provided in the ADNI database. (b) 90% confidence intervals for the inflection point of each biomarker
The empirical variance of the residuals εijk in (3) is the component of the variance which is unexplained by the model. It accounts for about 38% of the total variance. Hence the model explains 62% (±1.37%) of the total variance (i.e., 62% = 100% - 38%.), the standard deviation (sd) of 1.37% being computed using the bootstrap samples. If instead of the ADPS, ADAS or MMSE was used as a disease progression score, fitting sigmoid curves as previously described, the percentage of explained variance would be respectively 49.4% (± 1.4%) and 46% (± 1.4%). The percentage of explained variance is larger with the ADPS than with the ADAS (p-value < 0.01) or the MMSE (p-value <0.01); p-values being obtained using the bootstrap replicates in both cases.
4.2. Relation between ADPS and Rate of Progression
The rate of progression αi of each subject i is also computed as part of the ADPS parameter fitting algorithm. We plotted the rate of progression of each subject against their ADPS at baseline to see whether a relationship might exist (Fig. 4). A clear trend of increasing rate of ADPS as a function of ADPS is observed. The third column of Table 1 provides the mean rate of change of ADPS in unit of years for each status. AD subjects progress faster on average than MCI subjects. MCI subjects progress faster on average than N subjects. Observed during 3 years, an MCI subject would progress on average at 0.76 ADPS per year. The corresponding ADPS would then increase by 0.76 × 3 = 2.28 units. In our model, the ADPS of each subject is a linear function of age, or equivalently the rate of change of ADPS is constant over the time a subject is observed. Retrospectively, it is therefore a reasonable approximation for N and MCI subjects. It might be too simple a model for AD subjects. It is important to recall that these observations are made in light of the optimization criterion of ADPS, which uses the commonality of biomarker trends as a basis for determining rate. Thus, an increasing rate of ADPS truly means that subjects are progressing through degrading biomarkers at a faster rate.
Figure 4.
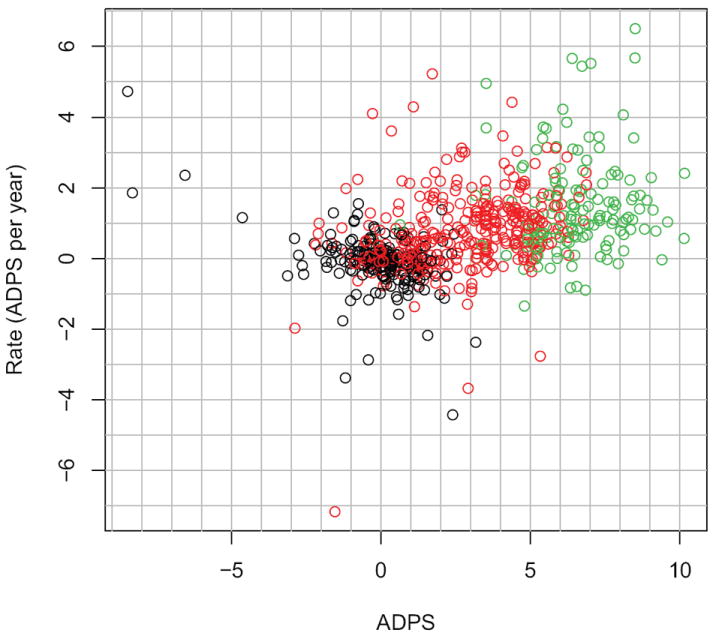
Rate of the ADPS as function of the ADPS for baseline visits. Black: Normal subjects. Red: MCI subjects Green: AD subjects.
Table 1.
Mean value (standard deviation) of ADPS and rate of change of ADPS for N, MCI and AD subjects in ADNI at baseline
| ADPS: Mean (sd) | Rate of change of ADPS: Mean (sd) | |
|---|---|---|
| N | -0.03 (1.48) | -0.08 (0.81) |
| MCI | 2.85 (1.98) | 0.76 (1.11) |
| AD | 6.49 (1.61) | 1.46 (1.38) |
4.3. Biomarker dynamics
The sigmoidal functions representing common behavior of biomarker dynamics of the entire ADNI population can be compared by scaling (and inverting if necessary) each of them independently to range from −1 (Normal) to +1 (Abnormal). Plotted as a function of the normalized ADPS (Fig. 5(a)), these scaled sigmoidal functions provide a plot similar to the conceptual plot in Jack et al. (2010) (Fig. 1). Our plot is data driven, of course, representing what the entire ADNI dataset predicts under our model assumptions. Its sigmoidal functions also provide information about the time of initial biomarker change (represented by the heels of the sigmoidal functions), the time of maximum biomarker change (represented by the inflection point of the sigmoidal functions), and the rate of biomarker change over the course of its activation (represented by the slopes of the sigmoidal functions).
In addition to their interpretation as the time of maximum biomarker change, the inflection points also could represent a threshold between normal and abnormal. Therefore, we use them as an indicator of biomarker timing in the disease process. We recomputed the inflection point of the normalized biomarker sigmoids for each bootstrap sample and plotted 90% confidence intervals (Fig 5(b)). Furthermore, counting pairwise ordering within the bootstrap samples, we find that RAVLT30 precedes all other 6 other biomarkers (p-value < 0.01) and HIPPO, ABETA and TAU precede MMSE and ADAS (p-value < 0.02).
4.4. Relation between ADPS and Clinical Status
Conditional probability densities of ADPS given the clinical status of each subject were computed using Gaussian kernel density estimation (Fig. 5(a)). Since N subjects tend to have a smaller ADPS than MCI subjects who in turn tend to have a smaller ADPS than AD subjects, this plot confirms that ADPS provides a scale that correlates strongly with clinical classification of disease. The mean and standard deviation of the baseline ADPS for N, MCI and AD subjects in ADNI is provided in table 1, column 2. The means are well separated from each other. There is overlap in the baseline ADPS value between N and MCI and also between MCI and AD, but essentially not between N and AD. It is worth restating the clinical diagnosis is not used in computing the ADPS except to determine its units.
5. Discussion
We combine multiple biomarkers to provide a neurodegenerative disease progression. In contrast, in the case of AD, Brooks et al. (1993); Stern et al. (1994); Ashford et al. (1995); Mitnitski et al. (1999) and others use MMSE or ADAS as measure of disease progression. In Yang et al. (2011a), the authors synchronize subjects onto a time-line constructed using ADAS scores. The core assumption is that the rate of change of ADAS is linear with respect to the ADAS score, resulting in an exponential model of disease progression. In Walhovd et al. (2010); Hinrichs et al. (2011), multiple biomarkers are combined to diagnose AD. In Fonteijn et al. (2011) the progression of AD is divided into discrete events based on the atrophy of different structures in the brain providing a probabilistic framework for estimating the global progression of AD as well as for estimating the position of a single subject’s measurements. Longitudinal measurements are not used. In Ververidis et al. (2010), a Bayesian classifier selects the set of biomarkers which are most informative for classifying the current state of the disease. Time-series models are used to predict the future state of the disease. Yang et al. (2011b) use independent component analysis and support vector machines to classify subjects into N versus MCI or AD. Our statistical model is related to so-called single index models (see Hardle et al. (1993); Carroll et al. (1997) and the references therein). However, our models differ from these, as we assume parsimonious parametric forms for the index function and allow for multivariate outcomes.
Our modeling technique applied to the ADNI has provided confirmation of existing results: Jack et al. (2011) binarized each biomarker into either normal or abnormal using a threshold or cut point. Cut points were determined for each biomarker at autopsy and with an independent cohort. When using these cut point to determine the ADPS at which a biomarker changes from normal to abnormal, we find that ABETA precedes both HIPPO and TAU which is consistent with the results in Jack et al. (2011). We have also obtained surprising results. The fact that the inflection of RAVLT30 precedes that of all other biomarkers, and in particular that of ABETA is surprising, compared to Fig. 1, but consistent with some predictions. Jicha and Carr (2010) refer to the study in Bennett et al. (2006) stating, “Retrospective analysis of their neuropsychological test performance demonstrated significant differences in only delayed recall tasks between subjects with pathological AD autopsy findings and those with normal autopsy findings, suggesting that memory decline may be present, albeit subtly, in persons with (preclinical) AD before sufficient cognitive decline to warrant the diagnosis of either MCI or dementia.”Also, Dubois et al. (2007) advocate that the presence of an early and significant episodic memory impairment should constitute one of the core diagnostic criteria for AD.
6. Conclusion
We report a multiple biomarker, data-driven approach to assess time-dependent changes of biomarkers in neurodegenerative disease and to localize subjects on a scale of disease progression, the DPS, over the entire range of progression. The statistical model is shown to be identifiable and bootstrap replicates show that the parameters are estimated tightly in case of the ADNI dataset. The DPS integrates information from multiple biomarkers into a single composite biomarker. Using this approach the conceptual plot of Jack et al. (2010) can be recreated using the ADNI data. The sequence of biomarkers obtained by comparing the inflection point of each biomarker is similar to that in Jack et al. (2010) with an exception: the RAVLT30 becomes dynamic before all other biomarkers. The DPS provides a continuous measure of progression over the whole course of disease, and it could therefore be used to stage individuals for prognosis and to evaluate the effects of novel drugs at all stages of the disease. The method is generic and is applicable to all neurodegenerative diseases pending availability of the data.
Highlights.
A computational neurodegenerative disease progression score (DPS) is proposed
The DPS combines measurements from multiple biomarkers
Validation with the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort
An Alzheimer’s DPS (ADPS) is computed for each subject and time-point in ADNI
Evidence for a common Alzheimer’s disease progression within ADNI subjects
Acknowledgments
Personnel costs for this research were partially supported by a grant from Pfizer Inc. Other support came from grants numbered P41EB015909 and R01EB012547 from the National Institute of Biomedical Imaging And Bioengineering as well as from an Ossoff scholar award. Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott; Alzheimers Association; Alzheimers Drug Discovery Foundation; Amorfix Life Sciences Ltd.; AstraZeneca; Bayer HealthCare; BioClinica, Inc.; Biogen Idec Inc.; Bristol-Myers Squibb Company; Eisai Inc.; Elan Pharmaceuticals Inc.; Eli Lilly and Company; F. Hoffmann-La Roche Ltd and its affiliated company Genentech, Inc.; GE Healthcare; Innogenetics, N.V.; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Medpace, Inc.; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Servier; Synarc Inc.; and Takeda Pharmaceutical Company. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. This research was also supported by NIH grants P30 AG010129 and K01 AG030514. The first author would also like to thank Patrick Slama for his insightful remarks.
Appendix A
Proof of Identifiability
Theorem 1
The model {Pρ; ρ ∈ ϱ} is identifiable as long as the following 2 conditions are verified:
For each biomarker, there is at least 1 subject i with αi ≠ 0 and with at least 4 distinct time-points at which this biomarker is available.
For each subject, there is at least 1 biomarker which is available at 2 time points.
The proof uses the invertibility of a multivalued function closely related to f. This property is deferred to lemma 1.
Proof of Theorem 1
Let us assume that the model is not identifiable. Then there exists 2 sets of parameters in ϱ, ρ = (a, b, c, d, α, β, σ) and ρ′ = (a′, b′, c′, d′, α′, β′, σ′) which differe by at least 1 component, while verifying Pρ = Pρ′. Equivalently,
| (A.1) |
for all (i, j, k) ∈ I and for all k
We proceed in steps until we verify that necessarily ρ = ρ′. Since , for all k = 1 … K, we concentrate on the other parameters. For each k, let i be a subject such that αi > 0 and for which biomarker k is observed at four different time points ti1, ti2, ti3, ti4. Notate uik = bkαi, υik = bk (βi − ck), and . Rearanging the arguments of f and using (A.1),
for j = 1 … 4. Note that since ai ≠ 0 and bk ≠ 0, uik ≠ 0 and . Now, using Lemma 1, , , , . Summing up over i and dividing by I in , we obtain , and since α0 ≠ 0, . Since bk ≠ 0, it follows that and . Replacing in and summing up over i and dividing by I, we obtain that . We have then obtained that for all biomarkers, , , , and . Now, for each subject i, there is at least one biomarker k for which two time-points ti1 and ti2 are available. Replacing in (A.1),
| (A.2) |
for j = 1, …, 2. Since ak ≠ 0 and bk ≠ 0, t → f(t; ak, bk, ck, dk) is invertible which, together with (A.2), implies that and concluding the proof.
Lemma 1
The vector values function R4 → R4 for fixed x1 < x2 < x3 < x4: defined by
with a ≠ 0, b > 0 is invertible.
Proof of Lemma 1
We verify that the Jacobian determinant of this function is nonzero, which is enough to prove invertibility using the inverse function theorem of multivariate calculus. Let c′ = ebc
It is equivalent to show the Jacobian determinant of
is non zero.
The ith row of the Jacobian matrix is:
Column linear transformation will not change the singularity of the Jacobian matrix. After some linear transformations, the ith row is:
Suppose the Jacobian matrix is singular, i.e. there exists (not all zero) coefficients k, l, m, n such that
then the function
must have four real roots. Differentiating twice,
would need to have 2 real roots. Since it is not the case, the Jacobian matrix is invertible, which concludes the proof.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ashford J, Shan M, Butler S, Rajasekar A, Schmitt F. Temporal quantification of Alzheimer’s disease severity: ‘Time Index’ model. Dementia. 1995;6(5):269–280. doi: 10.1159/000106958. [DOI] [PubMed] [Google Scholar]
- Bennett DA, Schneider JA, Arvanitakis Z, Kelly JF, Aggarwal NT, Shah RC, Wilson RS. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66(12):1837–1844. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- Berg L, Miller JP, Storandt M, Duchek J, Morris JC, Rubin EH, Burke WJ, Coben LA. Mild senile dementia of the alzheimer type: 2. Longitudinal assessment. Annals of Neurology. 1988;23(5):477–484. doi: 10.1002/ana.410230509. [DOI] [PubMed] [Google Scholar]
- Brooks DJ, Pavese N. Imaging biomarkers in parkinson’s disease. Progress in Neurobiology. 2011 Aug; doi: 10.1016/j.pneurobio.2011.08.009. URL http://dx.doi.org/10.1016/j.pneurobio.2011.08.009. [DOI] [PubMed]
- Brooks J, 3rd, Kraemer H, Tanke E, Yesavage J. The methodology of studying decline in Alzheimer’s disease. Journal of the American Geriatrics Society. 1993;41(6):623–628. doi: 10.1111/j.1532-5415.1993.tb06734.x. [DOI] [PubMed] [Google Scholar]
- Caroli A, Frisoni G. Alzheimer’s Disease Neuroimaging Initiative., The dynamics of Alzheimer’s disease biomarkers in the alzheimer’s disease neuroimaging initiative cohort. Neurobiol Aging. 2010;31(8):1263–74. doi: 10.1016/j.neurobiolaging.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll R, Fan J, Gijbels I, Wand M. Generalized partially linear single-index models. Journal of the American Statistical Association. 1997;92(438):477–489. [Google Scholar]
- Duara R, Loewenstein DA, Greig MT, Potter E, Barker W, Raj A, Schinka J, Borenstein A, Schoenberg M, Wu Y, Banko J, Potter H. Pre-MCI and MCI: Neuropsychological, clinical, and imaging features and progression rates. American Journal of Geriatric Psych. 2011;19(11):951–960. doi: 10.1097/JGP.0b013e3182107c69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, DeKosky ST, Barberger-Gateau P, Cummings J, Delacourte A, Galasko D, Gauthier S, Jicha G, Meguro K, O’Brien J, Pasquier F, Robert P, Rossor M, Salloway S, Stern Y, Visser PJ, Scheltens P. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. The Lancet Neurology. 2007;6(8):734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- Folstein MF, Folstein SE, McHugh PR. “mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. Journal of Psychiatric Research. 1975;12(3):189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- Fonteijn HMJ, Clarkson MJ, Modat M, Barnes J, Lehmann M, Ourselin S, Fox NC, Alexander DC. Proc Information Processing in Medical Imaging (IPMI). Lecture Notes in Computer Science. Vol. 6801. Springer; 2011. An event-based disease progression model and its application to familial Alzheimer’s disease; pp. 748–759. [DOI] [PubMed] [Google Scholar]
- Hampel H, Burger K, Teipel SJ, Bokde AL, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimer’s and Dementia. 2008;4(1):38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Hardle W, Hall P, Ichimura H. Optimal smoothing in single-index models. The annals of Statistics. 1993;21(1):157–178. [Google Scholar]
- Hayden M. Huntington’s chorea. Springer-Verlag; 1981. URL http://books.google.com/books?id=8dprAAAAMAAJ. [Google Scholar]
- Hinrichs C, Singh V, Xu G, Johnson SC. Predictive markers for AD in a multi-modality framework: An analysis of MCI progression in the ADNI population. NeuroImage. 2011;55(2):574–589. doi: 10.1016/j.neuroimage.2010.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes C, Berg L, Danziger W, Coben L, Martin R. A new clinical scale for the staging of dementia. British Journal of Psychiatry. 1982;140:566–572. doi: 10.1192/bjp.140.6.566. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CRJ, Vemuri P, Wiste HJ, Weigand SD, Aisen PS, Trojanowski JQ, Shaw LM, Bernstein MA, Petersen RC, Weiner MW, Knopman DS. Alzheimer’s Disease Neuroimaging Initiative, Evidence for ordering of alzheimer disease biomarkers. Arch Neurol. 2011;68(12):1526–1535. doi: 10.1001/archneurol.2011.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jicha GA, Carr SA. Conceptual evolution in Alzheimer’s disease: Implications for understanding the clinical phenotype of progressive neurodegenerative disease. Journal of Alzheimer’s Disease. 2010;19(1):253–272. doi: 10.3233/JAD-2010-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe A. ALS biomarkers study seeks 250 participants. MDA ALS News Magazine; Jan, 2012. [Google Scholar]
- Levenberg K. A method for the solution of certain non-linear problems in least squares. The Quarterly of Applied Mathematic. 1944;2:164–168. [Google Scholar]
- Martin LJ. Encyclopedia of the Human Brain. Vol. 3. Elsevier Science Academic press; 2002. pp. 441–463. Ch. Neurodegeneratives disorders of the human brain and spinal cord. [Google Scholar]
- Mitnitski A, Graham J, Rockwood K. Modeling decline in Alzheimer’s disease. International Psychogeriatrics. 1999;11(02):211–213. doi: 10.1017/s1041610299005761. [DOI] [PubMed] [Google Scholar]
- Mosconi L, Brys M, Glodzik-Sobanska L, Santi SD, Rusinek H, de Leon MJ. Early detection of Alzheimer’s disease using neuroimaging. Experimental Gerontology. 2007;42(1-2):129–138. doi: 10.1016/j.exger.2006.05.016. [DOI] [PubMed] [Google Scholar]
- Mungas D, Reed BR. Application of item response theory for development of a global functioning measure of dementia with linear measurement properties. Statistics in Medicine. 2000;19(11-12):1631–1644. doi: 10.1002/(sici)1097-0258(20000615/30)19:11/12<1631::aid-sim451>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Qiu A, Younes L, Miller MI, Csernansky JG. Parallel transport in diffeomorphisms distinguishes time-dependent hippocampal surface atrophy in healthy aging and alzheimers disease. NeuroImage. 2008;40:68–76. doi: 10.1016/j.neuroimage.2007.11.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards F. A flexible growth function for emperical use. J Exp Botany. 1959;10:290–300. [Google Scholar]
- Rizk-Jackson A, Stoffers D, Sheldon S, Kuperman JM, Dale AM, Goldstein J, Corey-Bloom J, Poldrack RA, Aron AR. Evaluating imaging biomarkers for neurodegeneration in pre-symptomatic huntington’s disease using machine learning techniques. NeuroImage. 2011;56(2):788–796. doi: 10.1016/j.neuroimage.2010.04.273. [DOI] [PubMed] [Google Scholar]
- Sabuncu MR, Desikan RS, Sepulcre J, Yeo BTT, Liu H, Schmansky NJ, Reuter M, Weiner MW, Buckner RL, Sperling RA, Fischl B. ADNI, The dynamics of cortical and hippocampal atrophy in Alzheimer disease. Arch Neurol. 2011;68(8):1040–1048. doi: 10.1001/archneurol.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw PJ. Molecular and cellular pathways of neurodegeneration in motor neurone disease. Journal of Neurology, Neurosurgery & Psychiatry. 2005;76(8):1046–1057. doi: 10.1136/jnnp.2004.048652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & dementia : the journal of the Alzheimer’s Association. 2011;7(3):280–292. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern R, Mohs R, Davidson M, Schmeidler J, Silverman J, Kramer-Ginsberg E, Searcey T, Bierer L, Davis K. A longitudinal study of Alzheimer’s disease: measurement, rate, and predictors of cognitive deterioration. Am J Psychiatry. 1994;151(3):390–396. doi: 10.1176/ajp.151.3.390. [DOI] [PubMed] [Google Scholar]
- Turner MR, Grosskreutz J, Kassubek J, Abrahams S, Agosta F, Benatar M, Filippi M, Goldstein LH, van den Heuvel M, Kalra S, Lul D, Mohammadi B. Towards a neuroimaging biomarker for amyotrophic lateral sclerosis. The Lancet Neurology. 2011;10(5):400–403. doi: 10.1016/S1474-4422(11)70049-7. URL http://www.sciencedirect.com/science/article/pii/S1474442211700497. [DOI] [PubMed] [Google Scholar]
- Turner MR, Kiernan MC, Leigh PN, Talbot K. Biomarkers in amyotrophic lateral sclerosis. The Lancet Neurology. 2009 Jan;8(1):94–109. doi: 10.1016/S1474-4422(08)70293-X. [DOI] [PubMed] [Google Scholar]
- Ververidis D, Van Gils M, Koikkalainen J, Lotjonen J. Feature selection and time regression software: Application on predicting Alzheimer’s disease progress. Proc European Signal Processing Conference (EU-SIPCO).2010. [Google Scholar]
- Walhovd K, Fjell A, Brewer J, McEvoy L, Fennema-Notestine C, Hagler DJ, J, Jennings R, Karow D, Dale A. Alzheimer’s Disease Neuroimaging Initiative, Combining MR imaging, positron-emission tomography, and CSF biomarkers in the diagnosis and prognosis of Alzheimer disease. AJNR Am J Neuroradiol. 2010;31(2):347–354. doi: 10.3174/ajnr.A1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Farnum M, Lobanov V, Schultz T, Raghavan N, Samtani MN, Novak G, Narayan V, DiBernardo A. Quantifying the pathophysiological timeline of Alzheimer’s disease. Journal of Alzheimer’s Disease. 2011a;26(4):745–753. doi: 10.3233/JAD-2011-110551. [DOI] [PubMed] [Google Scholar]
- Yang W, Lui R, Gao J, Chan T, Yau S, Sperling R, Huang X. Independent component analysis-based classification of alzheimer’s disease mri data. Journal of Alzheimer’s Disease. 2011b;24(4):775–783. doi: 10.3233/JAD-2011-101371. [DOI] [PMC free article] [PubMed] [Google Scholar]