

Abstract
Background
Protein-protein, cell signaling, metabolic, and transcriptional interaction networks are useful for identifying connections between lists of experimentally identified genes/proteins. However, besides physical or co-expression interactions there are many ways in which pairs of genes, or their protein products, can be associated. By systematically incorporating knowledge on shared properties of genes from diverse sources to build functional association networks (FANs), researchers may be able to identify additional functional interactions between groups of genes that are not readily apparent.
Results
Genes2FANs is a web based tool and a database that utilizes 14 carefully constructed FANs and a large-scale protein-protein interaction (PPI) network to build subnetworks that connect lists of human and mouse genes. The FANs are created from mammalian gene set libraries where mouse genes are converted to their human orthologs. The tool takes as input a list of human or mouse Entrez gene symbols to produce a subnetwork and a ranked list of intermediate genes that are used to connect the query input list. In addition, users can enter any PubMed search term and then the system automatically converts the returned results to gene lists using GeneRIF. This gene list is then used as input to generate a subnetwork from the user’s PubMed query. As a case study, we applied Genes2FANs to connect disease genes from 90 well-studied disorders. We find an inverse correlation between the counts of links connecting disease genes through PPI and links connecting diseases genes through FANs, separating diseases into two categories.
Conclusions
Genes2FANs is a useful tool for interpreting the relationships between gene/protein lists in the context of their various functions and networks. Combining functional association interactions with physical PPIs can be useful for revealing new biology and help form hypotheses for further experimentation. Our finding that disease genes in many cancers are mostly connected through PPIs whereas other complex diseases, such as autism and type-2 diabetes, are mostly connected through FANs without PPIs, can guide better strategies for disease gene discovery. Genes2FANs is available at: http://actin.pharm.mssm.edu/genes2FANs.
Background
Studies that utilize genome-wide profiling methods which attempt to explain the differences between two or more experimental conditions such as cells treated with a drug vs. control, diseased tissue vs. normal, gene or protein expression at different time points during cellular differentiation or reprogramming, or candidate gene lists harboring mutations associated with a particular disease, produce lists of genes/proteins without apparent functional relationship. These lists are commonly analyzed using software tools and databases that map genes to known pathways or construct subnetworks that connect input lists of genes using known protein-protein or other types of molecular interactions [1-10]. Such methods have been instrumental for organizing and reusing prior knowledge to understand new high-content experimental results. Prior knowledge networks, in particularly protein-protein interaction networks, have been useful for predicting unknown functions for genes [11,12], new interactions between proteins [13], novel disease genes [14], and guiding experimental research efforts by prioritizing the most likely regulators to test at the bench [15]. The resultant subnetwork diagrams from these analyses are useful because this prior knowledge, displayed as a network diagram, contains information about the relationships between the genes identified experimentally. This approach also abstracts the genes from the query list to higher order biological functions, allowing for the identification of novel relevant genes.
Software tools that provide users the ability to build subnetworks from lists of genes using prior knowledge networks are continually gaining popularity. For instance, a system that we developed a few years ago, Genes2Networks, utilizes twelve protein-protein interaction databases to connect lists of mammalian gene products using a shortest path algorithm [1]. Similarly, the software VisAnt version 3.5 goes a step further to automatically compute enrichment for gene ontology (GO) terms in identified PPI subnetworks [2]. Integrating PPIs, gene regulatory interactions, metabolic networks, and cell signaling networks, ConsensusPathDB provides methods to find connections between human, mouse and yeast genes [3]. Cytoscape, one of the leading academic platforms for building and visualizing networks, through its modular plug-ins, provides ways to construct networks, find paths between nodes, and compute network properties in an integrative manner [16]. Similar functionality is available in PatikaWeb [4], a web application with an underlying large protein interaction database. STRING, arguably the most comprehensive molecular interaction database, contains many different interactions including protein-protein and co-expression with assigned confidence scores [5]. Similar functionality is also available in BioPixie, initially developed for yeast but more recently extended to cover the mouse [17]. Visualization tools such as N-Browse [6], AVIS [18], FNV [19], and Cytoscape Web [20] display subnetworks from heterogeneous types of data sources with different color edges and nodes to represent different types of links and nodes on the web. GeneMANIA [7], another subnetwork generation tool, utilizes Cytoscape Web to display known and predicted protein-protein interactions, co-expression interactions, interactions based on shared pathways, and genetic interactions. So far, most subnetwork building software tools only utilize a few types of prior knowledge networks, mostly protein-protein interactions, co-expression, metabolic, and cell signaling pathway networks. Here we extend on these efforts by generating 14 functional association networks (FANs) from gene set libraries and combine them with a large-scale network of mammalian protein-protein interactions. The FANs were systematically generated by converting gene set libraries to networks, connecting pairs of genes based on their shared functional annotations. These functional association networks (FANs) together with protein-protein interaction networks are our background knowledge database for building and visualizing subnetworks from input lists of genes. Keeping functional relationships separate, we allow users to control what layers of functional associations they wish to integrate into their analysis. This system is delivered as a web based interactive tool called Genes2FANs. To demonstrate the utility of the Genes2FANs approach we applied the software to connect lists of disease genes for 90 diseases that have many known mutated genes. We find an inverse correlation between the number of protein-protein interaction links and the number of functional annotation links identified when connecting lists of disease genes. This inverse correlation separates complex diseases into two classes: those that are protein interaction centric, including many cancers, and those that are functional centric, including complex spectral disorders such as autism and type-2-diabetes.
Implementation
Methods for constructing the functional association networks
The first step in assembling the FANs was to gather data spread across a wide variety of databases and online sources. Besides collecting a comprehensive list of available protein-protein and cell signaling networks (see below), we also collected and generated gene set libraries that we later converted to FANs. Gene set libraries store sets of genes in a gene matrix transposed (GMT) file with rows containing a set of genes symbols associated with a given functional term. Using this format we were able to quantify the relationships between pairs of genes based on their co-occurrence membership in sets of the same gene set library using two different similarity measures: the Jaccard index and a Binomial Proportion test. The process of creating FANs from GMT files is outlined (Figure 1).
Figure 1.
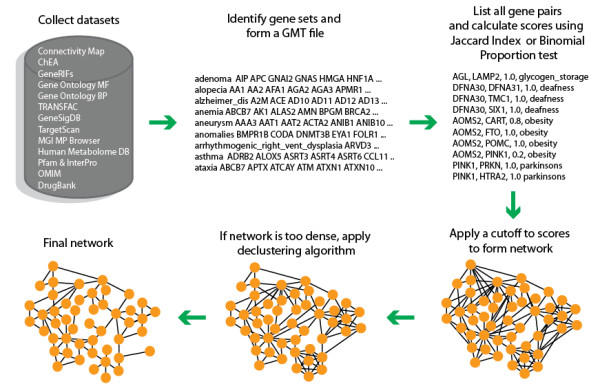
Process of creating FANs. The process of creating FANs involves gathering datasets and processing them into GMT files. Using these GMT files, networks are created using either the Jaccard index or a Binomial Proportion test. Large and dense networks are filtered using a declustering method and a cutoff is applied to produce the final FANs.
The Jaccard index is a measure of the similarity of two sets, A and B, which is given by the ratio of their intersection to their union:
| (1) |
Scores range in values from 0 to 1, where indices of 1 indicate exact similarity and indices of 0 indicate no relation between the sets. In our case, to score similarity between gene pairs, we divided the number of sets for both genes by the number of unique sets each gene belongs to. If we identify the sets A and B with the set of all lines of the GMT file, in which each of two respective genes are present, the the Jaccard index can be taken as a measure of the degree of association between the genes. The Jaccard index scoring method was applied to gene set libraries (GMT files) that contain a small number of genes per functional term with many different functional terms. Eight FANs were created using this method: miRNAs, mouse phenotypes, metabolites, structural domains, GO biological processes, disease genes, and drug targets. For each network we chose a cutoff that maximizes the tradeoff between coverage (maximizing the number of nodes) and sparseness (minimizing the number of links) (Tables 1 and 2).
Table 1.
FAN properties
| Network | Scoring Method | Network Cutoff | Data Source | Nodes | Edges |
|---|---|---|---|---|---|
| CMAP co-expression |
Binomial Proportion* |
130 |
Connectivity Map Database |
8,924 |
62,382 |
| Transcription Factors (ChIP-X) |
Binomial Proportion* |
27 |
ChEA database |
13,223 |
70,347 |
| GeneRIF |
Binomial Proportion* |
2000 |
NCBI GeneRIF |
3,777 |
27,487 |
| GO Molecular Function |
Binomial Proportion* |
160 |
Gene Ontology |
2,944 |
23,356 |
| TRANSFAC |
Binomial Proportion |
27 |
TRANSFAC |
15,252 |
94,642 |
| GeneSigDB |
Binomial Proportion |
350 |
GeneSigDB |
10,536 |
65,776 |
| MicroRNA |
Jaccard* |
0.3 |
TargetScan |
6,590 |
46,161 |
| Mouse Phenotype |
Jaccard* |
0.5 |
MGI MP Browser |
7,553 |
52,637 |
| Metabolites |
Jaccard* |
0.35 |
Human Metabolome Database |
3,577 |
28,617 |
| Structural Domains |
Jaccard* |
0.5 |
Pfam and InterPro |
6,746 |
46,463 |
| GO Biological Process |
Jaccard* |
0.99 |
Gene Ontology |
4,287 |
29,988 |
| OMIM Expanded |
Jaccard |
0.99 |
OMIM Morbid Map |
2,051 |
23,191 |
| OMIM Disease |
Jaccard |
0.99 |
OMIM Morbid Map |
1,618 |
22,643 |
| Drug Target |
Jaccard |
0.5 |
DrugBank |
2,121 |
16,807 |
| PPI | None | N/A | 13 Databases | 15,548 | 64,741 |
Properties of all the FANs along with their scoring method, scoring cutoff, data source, edge and node totals. * indicates that the declustering method was applied.
Table 2.
Declustering Details
| Network | Declustering Constant (Iterations) | Nodes Before | Nodes After | Edges Before | Edges After |
|---|---|---|---|---|---|
| CMAP co-expression |
2,000 |
8,924 |
8,924 |
119,420 |
61,362 |
| Transcription Factors (ChIP-X) |
1,500 |
13,223 |
13,223 |
110,901 |
70,347 |
| GeneRIFs |
2,000 |
3,777 |
3,777 |
52,512 |
27,487 |
| GO Molecular Function |
3,000 |
2,969 |
2,944 |
81,895 |
23,356 |
| MicroRNA |
3,000 |
6,590 |
6,590 |
176,766 |
46,161 |
| Mouse Phenotype |
3,300 |
7,795 |
7,553 |
290,381 |
52,637 |
| Metabolites |
3,500 |
3,692 |
3,577 |
205,468 |
28,617 |
| Structural Domains |
3,500 |
7,115 |
6,746 |
247,885 |
46,463 |
| GO Biological Process | 2,300 | 4,305 | 4,287 | 65,669 | 29,988 |
Declustering constants and node and edge counts before and after the declustering algorithm was applied on nine FANs.
The Jaccard index is biased with respect to our desired measure of similarity when comparing two lists with a large difference in size. For example, if one gene appears in 50 sets, A, and the other in 5 sets, B, but all of these 5 sets are contained within the 50 containing the first gene (B ⊂ A), the Jaccard index is 0.1, a low similarity index even though there is 100% overlap between the two genes. To correct for this we also applied the Binomial Proportions test to measure similarity between gene pairs based on their membership in gene sets. This method was applied to GMT files with a large number of genes per set. We used the z-score from a Normal approximation to the Binomial Proportion test to quantify the similarity between pairs of genes. Z-scores were calculated using the following equation:
| (2) |
where a is the number of gene sets the two genes are members of, b is the number of gene sets gene1 is a part of, c is the number of gene sets gene2 is a part of, and d is the total number of gene sets in the GMT file. A threshold for z-scores was chosen individually for each FAN to balance gene coverage and network sparseness (Table 2). Six functional association networks were created using this method: GeneRIF, CMAP co-expression [21], transcription factor co-regulation using ChEA [22] or TRANSFAC [23], GO molecular function [24], and GeneSigDB [25]. More details about each FAN are described below.
Declustering algorithm
Initially, many of the networks generated using the Jaccard index or the Binomial Proportion test were very dense, containing many interactions between highly connected genes. This made it difficult to generate specific subnetworks for input gene lists. To reduce the edge clutter of the FANs while preserving the majority of nodes and the most relevant interactions, we computed a score for each gene pair as follows:
| (3) |
where w is the weight of the edge; a is the connectivity degree of gene1; and b is the degree of gene2. Scores were sorted and the highest scoring edges were iteratively removed until there was a minimal loss of nodes and maximal loss of edges (Table 2).
Data extraction and FAN assembly
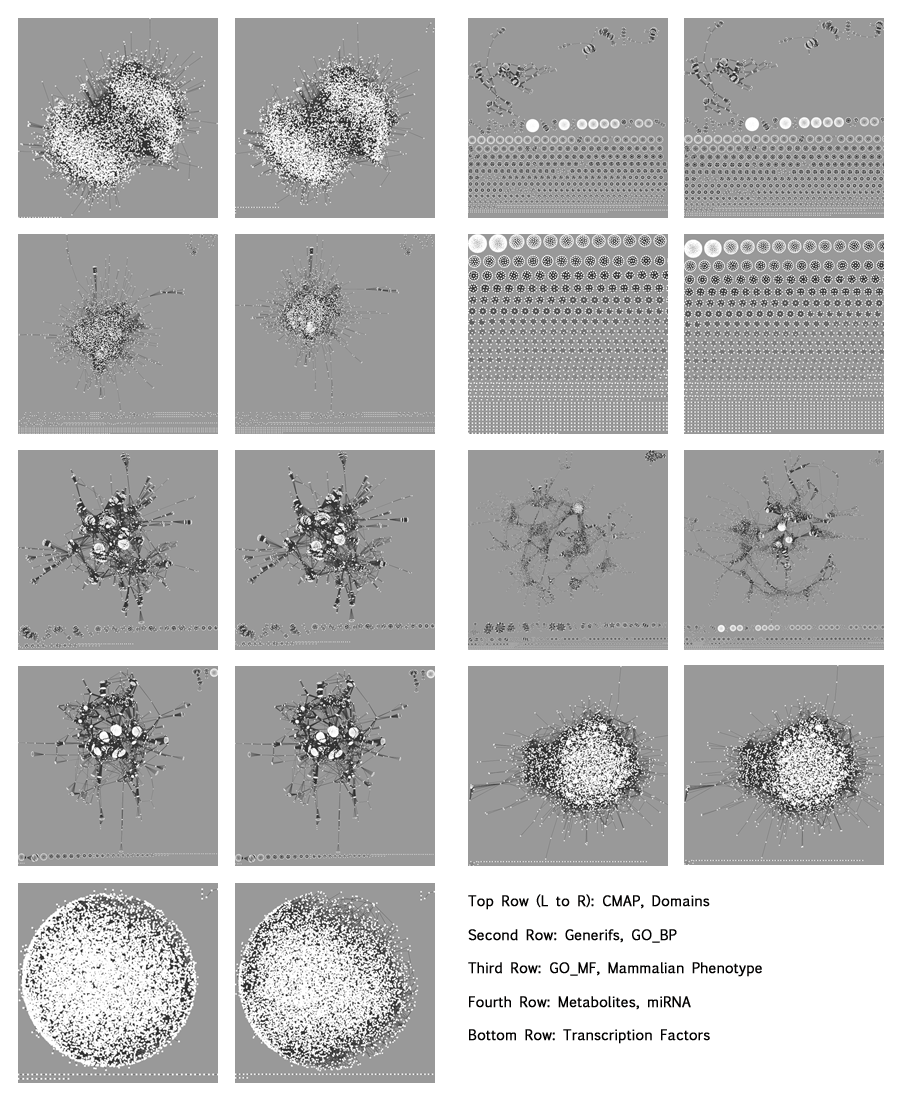
The Genes2FANs database contains 14 different FANs. Some FANs are made purely from human data whereas others are from data collected in mouse. All interactions taken from the mouse are converted to their human orthologs using NCBI’s homologene. Data for the miRNA network was taken from the TargetScan database [26]. Mouse phenotype gene sets were obtained from the Mouse Genome Informatics’ Mammalian Phenotype (MGI-MP) Browser [27]. The ontology of the MGI-MP Browser has a tree structure with the most general phenotypes represented by the root nodes and increasingly specific terms at each additional level down the tree. Starting at the lowest, most specific phenotypes, we merged descendents with their ancestor terms up to the fourth level of the tree producing a condensed set of relations between phenotypes and genes. For the metabolites FAN we derived a GMT file from the Human Metabolome database [28]. Structural domains and their associated Entrez gene symbols were extracted from Pfam [29] and InterPro [30]. The FANs made from GO Biological Process (BP) and GO Molecular Function (MF) terms [24] were assembled using GO Slim. Both OMIM FANs were created from the Online Mendelian Inheritance in Man (OMIM) [31] morbid map. These two GMT files were originally created from OMIM for the Lists2Networks project [32], where the expanded file includes neighboring genes in the PPI. The smallest FAN, drug target, is made using annotated FDA approved drug target relationships extracted from DrugBank [33]. The CMAP co-expression FAN is made from the Connectivity Map (CMAP) which reports drug induced gene expression signatures applied to human cancer cell lines [21]. We created a GMT file containing the top 1000 genes that either increased or decreased in expression after drug perturbation from all the experiments in the CMAP database. Each gene set has an equal size of 1000 genes per experiment in CMAP, 500 up-regulated genes, and 500 down-regulated. Data for the GeneRIF FAN was downloaded from NCBI’s gene reference into function dataset which links PubMed IDs to Entrez gene symbols based on manual curation. The transcription factors ChIP-X FAN is made from the ChEA database [22] which is already stored in a GMT-like file, where the functional terms are transcription factors profiled by ChIP-seq/chip experiments and the genes for each term are putative targets for the profiled factor in each experiment. To create a GMT file from TRANSFAC we identified putative target genes for all the human transcription factor binding matrices in TRANSFAC. We scanned the promoter regions of all annotated human coding genes from the −2000 to +500 nucleotides relative to the transcription start site (TSS) using the Patch program provided by TRANSFAC, and then set arbitrary cutoffs to associate transcription factors to their putative targets. GeneSigDB contains thousands of gene lists from supporting material tables manually curated from gene expression studies, mostly cancer related [25]. A summary of all FANs is provided in Table 1 along with node and edge counts, and network creation cutoffs. A more detailed summary of the effects of declustering can be seen in Table 2 with declustering coefficients and node and edge count listings, before and after declustering, for each of the nine declustered FANs. Additionally, the effects of the declustering algorithm on the global network topology can be seen in Additional file 1: Figure S1.
One of the strengths of FANs is the broad coverage of genes and their interactions. Thus, to quantify the overlap between the different types of FANs we assessed their similarity both at the gene and interaction levels, as well as comparing the FANs to the PPI network (Figures 2 and 3). Similarity was measured using the Jaccard index of the total genes and undirected edges in each of the FANs. Unsurprisingly, the largest FANs: ChEA, TRANSFAC, GeneSigDB, CMAP, PPI, and domains, contain many common genes (Figure 2). The diversity of the FANs can also be seen from the network visualization plots. Most of the networks have a large highly connected component while some networks clearly display a modular structure (Figure 4 and Additional file 1: Figure S1).
Figure 2.
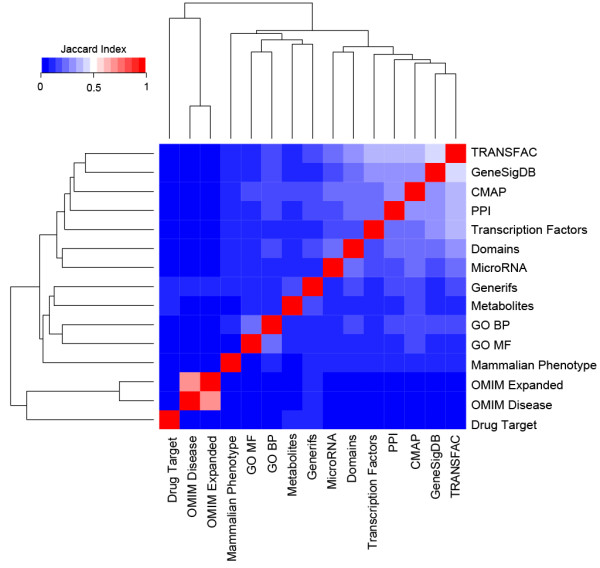
Heatmap of genes. Heatmap showing the similarity of the genes within each of the FANs and PPI network. Similarity was calculated using the Jaccard index.
Figure 3.
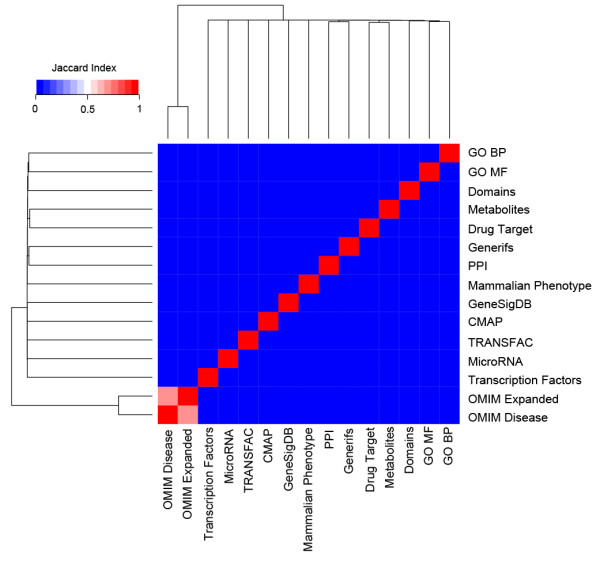
Heatmap of edges. Heatmap showing the similarity of the interactions connecting genes within each of the FANs and PPI network. Similarity was calculated using the Jaccard index.
Figure 4.
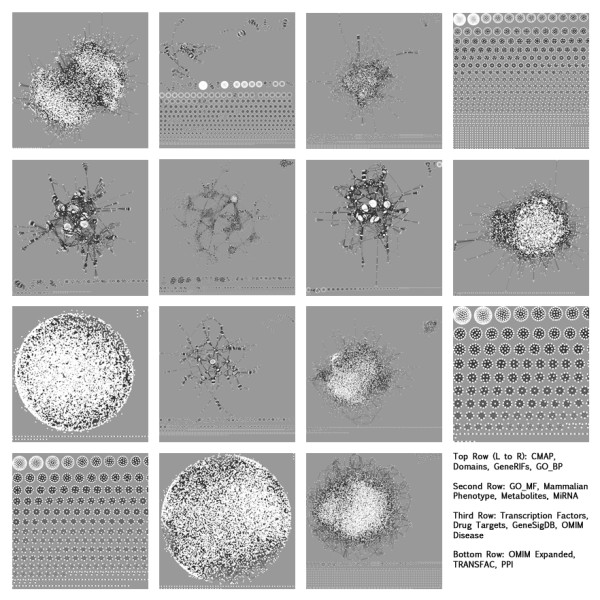
Topology of the FANs. The global structure of each of the FANs visualized with Cytoscape.
Developing the mammalian protein-protein interaction network
The protein-protein interaction network used in Genes2FANs contains physical interactions between proteins reported in the literature based on experimental evidence. For Genes2FANs we consolidated 13 databases and several published studies listing experimentally verified physical protein-protein interactions. Protein-protein interactions were combined from the following sources: MINT [34], InnateDB [35], NCBI-HPRD [36], KEGG [37], IntAct [38], BioGRID [39], PPID [40], BIND [41], DIP [42], Ma’ayan et al. [43], Stelzl et al. [44], Rual et al. [45], and Yu et al. [46]. Since high-throughput studies may contain higher degree of false positives [47] we filtered the BioGRID [39] and IntAct [38] databases to include only those interactions from studies that reported 10 or less protein-protein interactions. This removes publications that report protein interactions from mass-spectrometry proteomics and yeast-2-hybrid screens. Hence, the Genes2FANs software contains two versions of PPI datasets: filtered and unfiltered.
Web interface
The Genes2FANs web interface was developed using PHP, JavaScript, AJAX, and Perl. The core code for building subnetworks is implemented in C with a custom built hash function for fast access of network nodes and links. FNV, the subnetwork viewer, was implemented using Adobe Action Script 3.0 [19]. Currently, the application resides on a Linux server running Apache. To begin an analysis, users can enter a gene list by adding Entrez gene symbols one at a time or by pasting a list for upload. Results are presented to the user as an interactive subnetwork diagram and a table containing intermediate genes with z-scores indicating how significant the intermediates are for the input gene list. The interactive resultant subnetwork allows users to reposition nodes, hover over edges to reveal the gene sets that contributed to the edge, as well as pan and zoom. Users are presented with a choice of FANs to include and several options to control the size and aesthetics of the resulting subnetworks. Intermediate genes are displayed in a table ordered by their z-score computed using a Binomial Proportion test. There are also various export options allowing users to save the network for offline analysis. Figure 5 shows a screenshot of the web interface.
Figure 5.
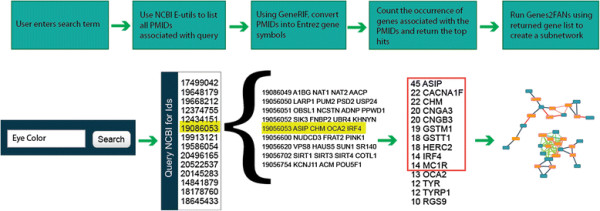
Converting PubMed queries to lists of Entrez gene symbols. PubMed queries are first converted into a list of PubMed IDs using NCBI’s e-utilities. For each PubMed ID a list of genes is obtained using GeneRIF. Genes are tallied and sorted by their occurrence and the top N genes are uploaded automatically into Genes2FANs.
PubMed search feature
If users do not have a specific gene list to enter they can query PubMed with any search term to generate a list of genes. Genes2FANs provides users with the option to choose the number of genes to return from a PubMed search, because shorter lists are more appropriate for specific queries whereas longer lists are better for ambiguous search terms. To facilitate this function we use NCBI’s e-utilities to turn search terms into their corresponding PubMed IDs and then use the GeneRIF file to convert the PubMed IDs into human genes with occurrence counts. Genes are ranked by their number of occurrences in all returned PubMed IDs. This process is summarized in Figure 6.
Figure 6.
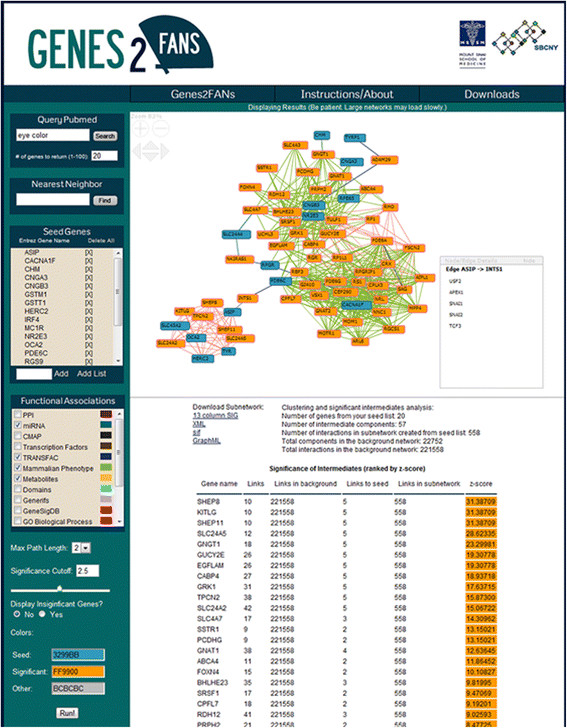
The Genes2FANs web interface. A screenshot showing the results of running Genes2FANs with the query “eye color”. On the left side of the page users can enter a PubMed query or a gene list and customize the output settings. The resulting subnetwork and a table listing ranked intermediates are shown on the right. Users can also obtain all the functional and binding interactions for a specific gene.
Results and discussions
Analysis of disease gene FAN
To demonstrate the capabilities of Genes2FANs we applied it to find relationships between disease genes. Disease gene discovery using network approaches by pathway reconstruction has been recently proven to be very useful. Typically applications first construct a large background network and then use disease genes as seed nodes for building subnetworks that connect the seed nodes [1,48-52]. Here we implemented a similar approach to obtain a global view of subnetworks created from many disease gene lists. Using the OMIM database we compiled a list of 90 common genetic disorders. From the OMIM morbid map dataset [31] we compiled a GMT file containing all diseases with at least 10 genes (n = 90). We then used Genes2FANs to connect the genes for each disease without any intermediates using only the PPI networks or the FANs, without the OMIM FAN. We then used the disease terms from the same GMT file as input for the PubMed query tool of Genes2FANs, setting the number of returned genes to 100. The size of networks using the PPI networks only or using the FANs only (without the OMIM FAN) was then recorded. To compute the correlation between the PPI and FAN links for all the diseases, we plotted the log of the ratio of number of PPI edges against the PPI edges to functional edges. We then calculated the mean of the data points by partitioning the points into groups of 10 for the OMIM gene lists and 15 for the subnetworks made using the query PubMed function to generate a local fit. The variation was illustrated in the plot by shading the region within one standard deviation of the mean of each bin.
With both methods, directly from OMIM or through PubMed queries, diseases show an inverse correlation between protein-protein interaction (PPI) links and other types of functional annotation links, segregating diseases with many known genes into two broad categories: those with gene products that physically interact, and those that interact functionally but not physically (Figures 7 and 8). This trend is statistically significant based on a Spearman rank correlation of 0.73 which has a p-value of 2.97×10-10 for the PubMed queried lists, and 0.27 for the lists directly from OMIM (p = 0.0065). The diseases that show high level of PPI and low level of functional associations include breast, ovarian, pancreatic, colorectal, thyroid, gastric, lung, and prostate cancers, as well as ataxia and leukemia (Figure 9); whereas diseases that display high level of functional interactions and low level of PPI are: deafness, type-2 diabetes mellitus, asthma, schizophrenia, autism and epilepsy. To ensure that this is not an artifact of the declustering algorithm on the FANs we ran the same process using the nine FANs before declustering. The declustering process had little effect on these results (Additional file 2: Figure S2 and Additional file 3: Figure S3) with Spearman rank correlation of 0.38 which has a p-value of 0.00026 for the PubMed queried lists, and 0.57 for the lists directly from OMIM (p = 1.99×10-7). The finding that some diseases have disease genes that are linked mostly through PPI, while other disease genes are mostly connected through FANs, is important because many investigations attempt to use protein interactions for novel disease gene discovery, for example, prioritizing mutations in genes detected by exome sequencing. This suggests that disease gene discovery using a PPI approach would work well for diseases such as cancers where many PPIs connect the disease gene products; however, for other complex diseases such as autism and type-2 diabetes, FANs would potentially be better for disease gene discovery.
Figure 7.
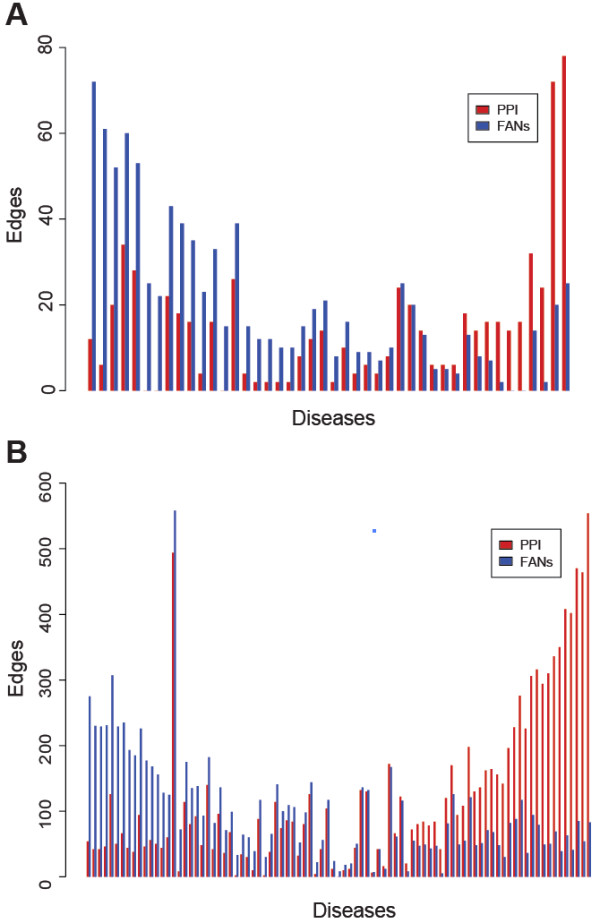
Distribution of edges for the disease gene lists. The distribution of edges for disease subnetworks created using genes directly from OMIM (A) and the disease terms with a maximum of 100 returned genes from the PubMed query tool of Genes2FANs (B). Diseases with a sum of PPI and functional edges less than 10 were omitted from both distribution plots.
Figure 8.

Correlation between subnetwork size and the edge ratio of PPIs to FANs. Scatterplots showing the correlation between the number of edges in the PPI subnetworks for each disease and the log of the ratio of PPI edges to functional edges. The red line depicts the mean of the data points (calculated by partitioning the points into groups of 10 for the OMIM disease gene lists (A) and 15 for the subnetworks made using the query PubMed function (B)). The blue dotted lines show one standard deviation away from the mean.
Figure 9.
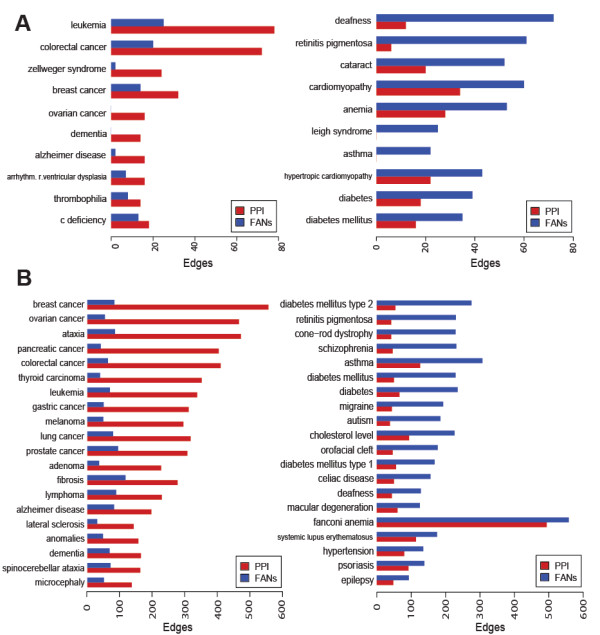
Top diseases. The top 10 diseases with the greatest difference in edge counts for the PPI vs. FANs disease subnetworks made from the OMIM disease gene lists (A) and the top 20 diseases for the subnetworks made using the query PubMed function (B).
Comparison to other similar tools
Finally, we compared Genes2FANs to other similar presently available online software tools. To compute the average number of genes returned for each of the tools we used a list of 20 randomly selected human genes and calculated the average and standard deviation of unique interactions reported by each tool. We used the nearest neighbor function of Genes2FANs and summed the number of interactions returned from each of the functional networks and the PPI. For PIPs [8], we ran the tool using the default settings and counted every interaction that had a score higher than 0. We ran HEFalMP [9] to explore a gene in relation to all genes in the context of all biological processes, only counting potentially interacting genes that had a confidence score higher than 0.5. To count the number of interactions returned by GeneMania [7] we searched for human genes with default settings and counted each edge as a separate interaction. Similarly, for STRING 9.0 [5] we ran the gene query as a human gene with default settings and counted unique edges. We also tested FunCoup [10] with its default settings. By default FunCoup applies an algorithm to reduce the number of probable links for a gene query. As a result many of our queries were capped at 60 returned genes when more significant interactions were identified.
It is difficult to quantify the accuracy of our approach compared with other similar tools since there is a lack of gold standard for functional relationships between genes. As a result, we cannot fairly compare the sensitivity and specificity of our tool against existing similar tools that integrate functional relationships. To show the differences between each tool we have chosen to focus on the number of interactions that are returned for an input gene (Table 3). The totals elucidate a clear pattern; each tool is suited for different purposes. In terms of accuracy, using a tool such as GeneMania, PIPs, or FunCoup might provide a user more reliable novel PPIs likely to interact with their query. On the other hand, for a more comprehensive analysis, STRING or HEFalMP would be the best performer. It is also worth noting that there is a great deal of overlap between Genes2FANs and these existing systems. As an example, using BRCA1 as input for each of the tools with default settings, and as input for the nearest neighbor function in Genes2FANs, we observed that most of the genes returned by STRING 9.0 (10 out of 10 genes), GeneMania (12 out of 19), and FunCoup (12 out of 25) were in our PPI dataset. All but four of our functional interactions for BRCA1 were returned by HEFalMP with varying degrees of confidence and three of these functional interactions were also returned by PIPs. Those genes identified by Genes2FANs but not in HEFalMP, are OVCAS1, FAM82A1, AIMP2, and MIR21. These genes were implicated in the literature to be associated with breast and/or ovarian cancer and may be indirectly related to BRCA1.
Table 3.
Comparison with Similar Tools
| Tool Name | Average Interactions | Background Knowledge | Unique Genes in database | Organisms |
|---|---|---|---|---|
| Genes2FANs |
72.1 ± 51 |
PPI, literature co-occurrence, miRNAs, co-regulation, domains, drug signatures & targets, gene signatures, metabolites, and phenotypes |
35,078 |
H.sapiens, M.musculus, R.norvegicus |
| PIPs |
10.1 ± 25.2 |
Co-expression, orthology, domains, co-localization, and PTMs |
5,338 |
H.sapiens |
| HEFalMp |
681.3 ± 1123.2 |
Functionally mapped data from microarray experiments and sequence comparisons |
24,433 |
H.sapiens |
| GeneMania |
78.7 ± 39.2 |
Co-expression, physical & genetic interactions, domains, co-localization, pathways, and orthology |
155,238 |
H.sapiens, A.thaliana, C.elegans, D.melanogaster, M.musculus, R.norvegicus, and S.cerevisiae |
| STRING 9.0 |
24.3 ± 14.4 |
Co-localization, fusion, co-occurrence, co-expression, literature co-occurrence, and orthology |
5,214,234 |
1,133 Organisms |
| FunCoup | 47.7 ± 21.9 | PPI, orthology, co-expression, miRNA, co-localization, phylogenetics, co-regulation, genetic interactions, and domains | 1,800,000 | H.sapiens, M.musculus, R.norvegicus, D.melanogaster, A.thaliana, C.elegans, S.cerevisiae,and C.intestinalis |
Comparison of Genes2FANs with five similar tools; examining the average number of interactions returned for single gene queries, the types of background knowledge for each tool, the number of unique genes/proteins in each knowledgebase, and the supported organisms.
Conclusions
Genes2FANs is a potentially useful tool for interpreting the relationships between gene lists in the context of their various functions and networks. Combining these functional association interactions with physical protein-protein interactions from high and low throughput datasets can be useful for revealing new biology and help form hypotheses for further experimentation. Our observation of disease gene lists commonly connected by either PPIs or FANs, but not by both, can assist with disease gene discovery strategies using network analysis and disease gene classifiers.
However, Genes2FANs is not without limitations. Currently, it does not include a confidence score for each edge. We also keep the FANs separate but all FANs can potentially be integrated into one large network. In the future we plan to constantly continue to update Genes2FANs with more FANs and to add more interactive features to the website. We also plan to develop a feature that will allow users to upload their own gene-set libraries for constructing their own functional networks. Additionally, we are working on improving our network generation process to improve the quality of the FANs.
Availability and requirements
Project name: Genes2FANs
Project home page: http://actin.pharm.mssm.edu/ genes2FANs
Operating System: Platform Independent
Programming Language: HTML, CSS, JavaScript, Perl, C, PHP, Python, Flash/Action Script
Other Requirements: Adobe Flash Player 9.0 or higher
License: GNU GPL
Competing interests
Authors declare that they have no competing interests.
Authors' contributions
RD assembled the FANs and designed and implemented the web interface and database. AM designed and supervised the project and implemented the declustering algorithm. NRC helped with computing the inverse correlation and provided helpful suggestions. AM and RD wrote the paper. All authors read and approved the final manuscript.
Supplementary Material
Figure S1.Effect of declustering on topology. The global network structure of each of the nine declustered FANs before (left) and after (right) applying the declustering algorithm; visualized with Cytoscape.
{kind=link}
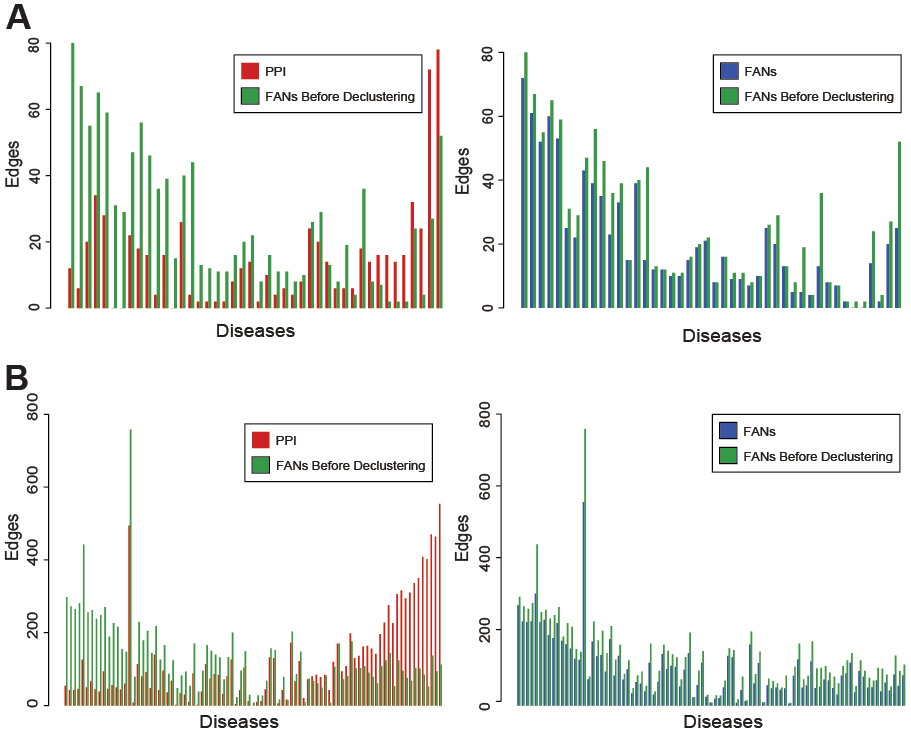
Figure S2.Edge distribution of FANs before declustering. The distribution of edges for disease subnetworks created using disease genes directly from OMIM (A) and the subnetworks made using the PubMed query tool with an maximum of 100 (B) using background FANs before declustering. Diseases with a sum of PPI and FAN edges less than 10 were omitted from both bar charts.
{kind=link}
Figure S3.Correlation between subnetwork size and the edge ratio of FANs to PPI before declustering. Scatterplots show the correlation between the number of edges in the PPI subnetworks for each disease and the log of the ratio of PPI edges to functional edges from subnetworks created from FANs before declustering. The red line depicts the mean of the data points (calculated by partitioning the points into groups of 10 for the OMIM disease gene lists (A) and 15 for the subnetworks made using the query PubMed function (B)). The blue dotted lines show one standard deviation away from the mean.
{kind=link}
Table S1.
Contributor Information
Ruth Dannenfelser, Email: ruth.dannenfelser@mssm.edu.
Neil R Clark, Email: neil.clark@mssm.edu.
Avi Ma'ayan, Email: avi.maayan@mssm.edu.
Acknowledgements
This work was supported by NIH grants R01GM098316-01A1, P50GM071558-03, R01DK088541-01A1, RC2LM010994-01, P01DK056492-10, RC4DK090860-01, KL2RR029885-0109 to AM.
References
- Berger SI, Posner JM, Ma'ayan A. Genes2Networks: connecting lists of gene symbols using mammalian protein interactions databases. BMC Bioinforma. 2007;8:372. doi: 10.1186/1471-2105-8-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu ZJ, Hung JH, Wang Y, Chang YC, Huang CL, Huyck M, DeLisi C. VisANT 3.5: multi-scale network visualization, analysis and inference based on the gene ontology. Nucleic Acids Res. 2009;37:W115–W121. doi: 10.1093/nar/gkp406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 2011;39:D712–D717. doi: 10.1093/nar/gkq1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogrusoz U, Erson EZ, Giral E, Demir E, Babur O, Cetintas A, Colak R. PATIKAweb: a Web interface for analyzing biological pathways through advanced querying and visualization. Bioinformatics. 2006;22(3):374–375. doi: 10.1093/bioinformatics/bti776. [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P. et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39(Database issue):D561–D568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HL, Gunsalus KC. Browsing multidimensional molecular networks with the generic network browser (N-Browse) Curr Protoc Bioinformatics. 2008;Chapter 9:Unit 9–Unit 11. doi: 10.1002/0471250953.bi0911s23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38(Web Server issue):W214–W220. doi: 10.1093/nar/gkq537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowall MD, Scott MS, Barton GJ. PIPs: human protein-protein interaction prediction database. Nucleic Acids Res. 2009;37(Database issue):D651–D656. doi: 10.1093/nar/gkn870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttenhower C, Haley EM, Hibbs MA, Dumeaux V, Barrett DR, Coller HA, Troyanskaya OG. Exploring the human genome with functional maps. Genome Res. 2009;19(6):1093–1106. doi: 10.1101/gr.082214.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexeyenko A, Schmitt T, Tjarnberg A, Guala D, Frings O, Sonnhammer EL. Comparative interactomics with Funcoup 2.0. Nucleic Acids Res. 2012;40(Database issue):D821–D828. doi: 10.1093/nar/gkr1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan R, Ulitsky I, Shamir R. Network-based prediction of protein function. Mol Syst Biol. 2007;3:88. doi: 10.1038/msb4100129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Keating AE. AVID: an integrative framework for discovering functional relationships among proteins. BMC Bioinforma. 2005;6:136. doi: 10.1186/1471-2105-6-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Paccanaro A, Trifonov V, Gerstein M. Predicting interactions in protein networks by completing defective cliques. Bioinformatics. 2006;22(7):823–829. doi: 10.1093/bioinformatics/btl014. [DOI] [PubMed] [Google Scholar]
- Cordeddu V, Di Schiavi E, Pennacchio LA. Ma'ayan A, Sarkozy A, Fodale V, Cecchetti S, Cardinale A, Martin J, Schackwitz W, et al: Mutation of SHOC2 promotes aberrant protein N-myristoylation and causes Noonan-like syndrome with loose anagen hair. Nat Genet. 2009;41(9):1022–1026. doi: 10.1038/ng.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg KD. Ma'ayan A, Neves SR, Iyengar R: Design logic of a cannabinoid receptor signaling network that triggers neurite outgrowth. Science. 2008;320(5878):903–909. doi: 10.1126/science.1152662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CL, Robson D, Wible A, Hibbs MA, Chiriac C, Theesfeld CL, Dolinski K, Troyanskaya OG. Discovery of biological networks from diverse functional genomic data. Genome Biol. 2005;6(13) doi: 10.1186/gb-2005-6-13-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SI, Iyengar R, Ma'ayan A. AVIS: AJAX viewer of interactive signaling networks. Bioinformatics. 2007;23(20):2803–2805. doi: 10.1093/bioinformatics/btm444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenfelser R, Lachmann A, Szenk M, Ma'ayan A. FNV: light-weight flash-based network and pathway viewer. Bioinformatics. 2011;27(8):1181–1182. doi: 10.1093/bioinformatics/btr098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CT, Franz M, Kazi F, Donaldson SL, Morris Q, Bader GD. Cytoscape Web: an interactive web-based network browser. Bioinformatics. 2010;26(18):2347–2348. doi: 10.1093/bioinformatics/btq430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN. et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Lachmann A, Xu H, Krishnan J, Berger SI, Mazloom AR, Ma'ayan A. ChEA: transcription factor regulation inferred from integrating genome-wide ChIP-X experiments. Bioinformatics. 2010;26(19):2438–2444. doi: 10.1093/bioinformatics/btq466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, Land S, Barre-Dirrie A, Reuter I, Chekmenev D, Krull M, Hornischer K. et al. TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 2006;34(Database issue):D108–110. doi: 10.1093/nar/gkj143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT. et al. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culhane AC, Schwarzl T, Sultana R, Picard KC, Picard SC, Lu TH, Franklin KR, French SJ, Papenhausen G, Correll M. et al. GeneSigDB-a curated database of gene expression signatures. Nucleic Acids Res. 2010;38:D716–D725. doi: 10.1093/nar/gkp1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, Bartel DP. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120(1):15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE. The Mouse Genome Database genotypes::phenotypes. Nucleic Acids Res. 2009;37(Database issue):D712–719. doi: 10.1093/nar/gkn886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, Knox C, Guo AC, Eisner R, Young N, Gautam B, Hau DD, Psychogios N, Dong E, Bouatra S. et al. HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 2009;37:D603–D610. doi: 10.1093/nar/gkn810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K. et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bork P, Das U, Daugherty L, Duquenne L. et al. InterPro: the integrative protein signature database. Nucleic Acids Res. 2009;37:D211–D215. doi: 10.1093/nar/gkn785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger J, Bocchini C, Valle D, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2002;30(1):52–55. doi: 10.1093/nar/30.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmann A, Ma'ayan A. Lists2Networks: integrated analysis of gene/protein lists. BMC Bioinforma. 2010;11:87. doi: 10.1186/1471-2105-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knox C, Law V, Jewison T, Liu P, Ly S, Frolkis A, Pon A, Banco K, Mak C, Neveu V. et al. DrugBank 3.0: a comprehensive resource for 'omics' research on drugs. Nucleic Acids Res. 2011;39(Database issue):D1035–1041. doi: 10.1093/nar/gkq1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceol A, Aryamontri AC, Licata L, Peluso D, Briganti L, Perfetto L, Castagnoli L, Cesareni G. MINT, the molecular interaction database: 2009 update. Nucleic Acids Res. 2010;38:D532–D539. doi: 10.1093/nar/gkp983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, Gardy JL, Roche FM, Chan THW, Shah N. et al. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol Syst Biol. 2008;4: . doi: 10.1038/msb.2008.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad TSK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A. et al. Human Protein Reference Database-2009 update. Nucleic Acids Res. 2009;37:D767–D772. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38:D355–D360. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranda B, Achuthan P, Alam-Faruque Y, Armean I, Bridge A, Derow C, Feuermann M, Ghanbarian AT, Kerrien S, Khadake J. et al. The IntAct molecular interaction database in 2010. Nucleic Acids Res. 2010;38:D525–D531. doi: 10.1093/nar/gkp878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark C, Breitkreutz BJ, Chatr-aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang XD, Shi XQ. et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermjakob H, Montecchi-Palazzi L, Bader G, Wojcik J, Salwinski L, Ceol A, Moore S, Orchard S, Sarkans U, von Mering C. et al. The HUPO PSI's molecular interaction format–a community standard for the representation of protein interaction data. Nat Biotechnol. 2004;22(2):177–183. doi: 10.1038/nbt926. [DOI] [PubMed] [Google Scholar]
- Alfarano C, Andrade CE, Anthony K, Bahroos N, Bajec M, Bantoft K, Betel D, Bobechko B, Boutilier K, Burgess E. et al. The Biomolecular Interaction Network Database and related tools 2005 update. Nucleic Acids Res. 2005;33:D418–D424. doi: 10.1093/nar/gki051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D. The Database of Interacting Proteins: 2004 update. Nucleic Acids Res. 2004;32:D449–D451. doi: 10.1093/nar/gkh086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma'ayan A, Jenkins SL, Neves S, Hasseldine A, Grace E, Dubin-Thalere B, Eungdamrong NJ, Weng GZ, Ram PT, Rice JJ. et al. Formation of regulatory patterns during signal propagation in a mammalian cellular network. Science. 2005;309(5737):1078–1083. doi: 10.1126/science.1108876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S. et al. A human protein-protein interaction network: A resource for annotating the proteome. Cell. 2005;122(6):957–968. doi: 10.1016/j.cell.2005.08.029. [DOI] [PubMed] [Google Scholar]
- Rual JF, Venkatesan K, Hao T, Hirozane-Kishikawa T, Dricot A, Li N, Berriz GF, Gibbons FD, Dreze M, Ayivi-Guedehoussou N. et al. Towards a proteome-scale map of the human protein-protein interaction network. Nature. 2005;437(7062):1173–1178. doi: 10.1038/nature04209. [DOI] [PubMed] [Google Scholar]
- Yu JY, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318(5858):1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Patil A, Nakamura H. Filtering high-throughput protein-protein interaction data using a combination of genomic features. BMC Bioinforma. 2005;6:100. doi: 10.1186/1471-2105-6-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Aronow BJ, Jegga AG. Disease candidate gene identification and prioritization using protein interaction networks. BMC Bioinforma. 2009;10:73. doi: 10.1186/1471-2105-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navlakha S, Kingsford C. The power of protein interaction networks for associating genes with diseases. Bioinformatics. 2010;26(8):1057–1063. doi: 10.1093/bioinformatics/btq076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oti M, Snel B, Huynen MA, Brunner HG. Predicting disease genes using protein-protein interactions. J Med Genet. 2006;43(8):691–698. doi: 10.1136/jmg.2006.041376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Sun F, Jiang R. Integrating multiple protein-protein interaction networks to prioritize disease genes: a Bayesian regression approach. BMC Bioinforma. 2011;12(Suppl 1):S11. doi: 10.1186/1471-2105-12-S1-S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann MG. Advances in translational bioinformatics: computational approaches for the hunting of disease genes. Brief Bioinform. pp. 96–110. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.Effect of declustering on topology. The global network structure of each of the nine declustered FANs before (left) and after (right) applying the declustering algorithm; visualized with Cytoscape.
Figure S2.Edge distribution of FANs before declustering. The distribution of edges for disease subnetworks created using disease genes directly from OMIM (A) and the subnetworks made using the PubMed query tool with an maximum of 100 (B) using background FANs before declustering. Diseases with a sum of PPI and FAN edges less than 10 were omitted from both bar charts.
Figure S3.Correlation between subnetwork size and the edge ratio of FANs to PPI before declustering. Scatterplots show the correlation between the number of edges in the PPI subnetworks for each disease and the log of the ratio of PPI edges to functional edges from subnetworks created from FANs before declustering. The red line depicts the mean of the data points (calculated by partitioning the points into groups of 10 for the OMIM disease gene lists (A) and 15 for the subnetworks made using the query PubMed function (B)). The blue dotted lines show one standard deviation away from the mean.
Table S1.