

Abstract
The L-phenylalanine oxidase IL4I1 inhibits T-cell proliferation in vitro through H2O2 production, and is highly expressed in tumor-associated macrophages. IL4I1 is also detected by immunohistochemistry in neoplastic cells from several B-cell lymphomas and some non-lymphoid tumors.
To evaluate IL4I1 effect on tumor growth, we developed a mouse melanoma model constitutively coexpressing IL4I1 and the GP33 epitope. After GP33 vaccination, tumors developed more frequently in mice injected with IL4I1-expressing cells in comparison to mice receiving control cells. Tumor escape was preceded by a rapid diminution of IFN-γ producing cytotoxic antitumor CD8+ T cells. Moreover, tumor incidence was already increased when only 20% of the injected cells expressed IL4I1. The minimal IL4I1 activities leading to tumor escape were close to those detected in human melanoma and mesothelioma.
Thus, we demonstrate the immunosuppressive functions of IL4I1 in vivo and suggest that IL4I1 facilitates human tumor growth by inhibiting the CD8+ antitumor T-cell response.
Keywords: Amino Acid Oxidoreductases; genetics; immunology; metabolism; Animals; Antigens, Neoplasm; genetics; immunology; metabolism; Antigens, Viral; genetics; immunology; metabolism; CD8-Positive T-Lymphocytes; immunology; metabolism; pathology; Cell Growth Processes; genetics; immunology; Glycoproteins; genetics; immunology; metabolism; Humans; Hydrogen Peroxide; metabolism; Immunization; Immunosuppression; Interferon-gamma; genetics; metabolism; Lymphocyte Activation; genetics; Melanoma, Experimental; Mice; Mice, Transgenic; Neoplasms, Experimental; immunology; Peptide Fragments; genetics; immunology; metabolism; Transgenes; genetics; Tumor Escape; Viral Proteins; genetics; immunology; metabolism
Keywords: IL4I1, tumor escape, mice, immunoediting, phenylalanine oxidase
INTRODUCTION
Neoplastic cells adopt multiple strategies to survive and grow despite tumor surveillance by the immune system. Sabotage strategies often exploit regulating cell populations, which naturally prevent autoimmunity and chronic inflammation in healthy individuals, such as regulatory T cells, myeloid-derived suppressor cells, tolerogenic dendritic cells and alternatively activated macrophages (reviewed in [1–5]). In the last decade, a family of enzymes has been described participating in the immunosuppressive capacity of the tumor-activated myeloid cell populations. The activity and functions of indoleamine-2,3-dioxygenase [6], arginase 1 [7] and inducible nitric oxide synthase [8] have been extensively explored. These enzymes share several properties [9]. First, they are produced by myeloids cells in lymphoid organs and in the tumor bed and/or by the tumor cells themselves [10–12]. Secondly, their expression involves cytokines from the Th1/Th2 family. Third, their immunosuppressive properties are based on their amino-acid catabolizing activity which leads to the depletion of essential amino-acids and to the production of metabolites which are toxic for anti-tumor effector T cells [13–15]. Several reports have established the role of these enzymes in tumor escape from immunosurveillance [10–12, 16].
Interleukin-4 Induced Gene 1 (IL4I1) was identified as an IL-4 inducible gene in B lymphocytes [17]. The human and mouse IL4I1 mRNA share a strong sequence homology and encode a secreted protein [18, 19]. We have shown that this protein is an L-amino-acid oxidase which primarily deaminates the essential amino-acid phenylalanine to produce H2O2. IL4I1 inhibits human CD4+ and CD8+ T lymphocyte proliferation in vitro via H2O2 production, with a preference towards memory T lymphocytes [18]. Monocyte-derived dendritic cells and macrophages – but not B cells, as would have been expected from the literature [17, 19] – represent the major IL4I1 producers after stimulation involving NFκB and/or STAT1 activation [20].
Tumors are often accompanied by an important myeloid infiltrate. Indeed, in a study of 315 cancers, we observed IL4I1 expression in the tumor-associated macrophage (TAM) population of most cases, independently of the tumor type. Moreover, IL4I1 was also detected in the tumor cells of several B lymphoma subtypes, comprising follicular lymphoma, Hodgkin lymphoma and primary mediastinal B cell lymphoma, and in some cases of non-lymphoid tumors, such as mesothelioma [21].
Thus, IL4I1 meets the criteria of an immunosuppressive enzyme [3, 9], suggesting that it may participate in tumor immune escape. To evaluate this hypothesis, we developed a mouse tumor model constitutively expressing IL4I1. In this work, we show for the first time that IL4I1 expression facilitates tumor growth by inhibiting the CD8+ antitumor T cell response.
RESULTS
Overexpression of IL4I1 in a murine tumor model
To assess in vivo whether IL4I1 expression in tumors favors escape from the immune response, we developed a new tumor model based on the use of the melanoma B16GP33 cell line, which expresses the GP33 CD8+ T cell epitope as a tumor antigen [22], but does not display basal IL4I1 enzymatic activity (Table I). B16GP33 cells were transfected with the mouse IL4I1 cDNA and two clones stably expressing the IL4I1 protein (B8 and B11) were selected. The A1 clone, transfected with the empty vector, was selected as a control. Production of the myc-tagged IL4I1 protein by the B8 and B11 clones was demonstrated by Western blot and immunofluorescence (Figure 1A). IL4I1 was secreted and functional in both clones with an activity on average 1.4 fold higher in B11 cells and 1.7 fold higher in B11 medium in comparison to the B8 clone (Table I). Neither IL4I1 protein, nor enzymatic activity was detected in the A1 cells. The IL4I1 enzymatic activity of B8 and B11 clones was close to those measured in human cell populations. The B11 activity (322 ±49 pmoles H2O2/h/105 cells) was nearly equivalent to the activity of the L428 Hodgkin lymphoma cell line (354 ±140 pmoles H2O2/h/105 cells; mean ±SD from 13 independent tests). It also displayed a 25% and 60% lower IL4I1 activity compared to that of unstimulated and IFNγ-stimulated monocyte-derived human macrophages respectively (432 ±101 and 804 ±129 pmoles H2O2/h/105 cells) [20].
Table I.
IL4I1 enzymatic activity in B16GP33-derived cell clones and tumor biopsies
| Sample | Sample name | ADNc | IL4I1 activity (pmoles H2O2/h) | |||
|---|---|---|---|---|---|---|
| Cells | Medium | |||||
|
| ||||||
| per 105 cells | per 100μg proteins | per 40μl beads | ||||
| Cell clones | B16GP33 | - | 0 (0)a | 0 (0) | NDb | |
| A1 | mock | 0 (2.5) | 0 (8) | 3 (0.3) | ||
| B8 | IL4I1 | 234 (60) | 317 (75) | 295 (11) | ||
| B11 | IL4I1 | 322 (49) | 330 (86) | 489 (18) | ||
|
| ||||||
| Tumor biopsies | A1 | mock | NAc | 5 (1) | NA | |
| B8 | IL4I1 | NA | 722 (533) | NA | ||
| B11 | IL4I1 | NA | 913 (515) | NA | ||
mean from 4 to 5 measurements (SD)
Not Done
Not Available
Figure 1. Characterization of IL4I1-expressing B16GP33 clones.
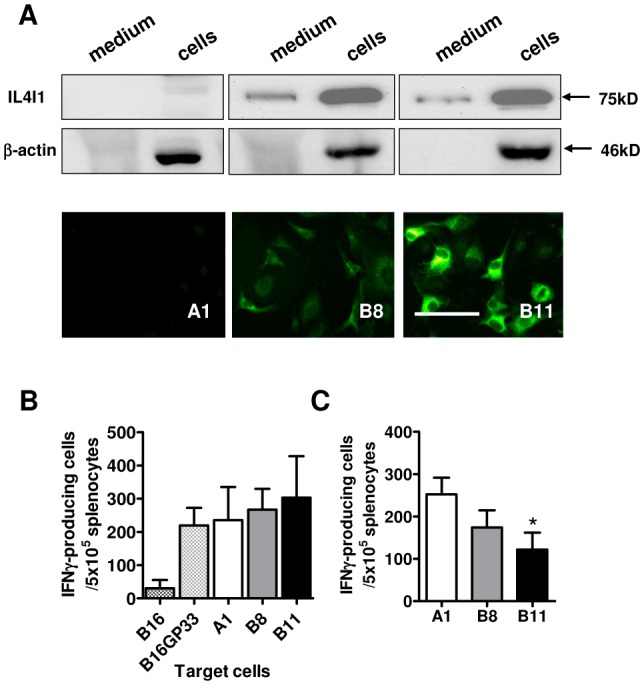
(A) Culture medium and whole cell lysate proteins from B16GP33 cells either transfected with an empty vector (A1) or with a vector coding for the myc-tagged murine IL4I1 protein (B8 and B11) were analyzed by Western blot (upper panel). IL4I1 was revealed by immunofluorescence (lower panel). Magnification ×400, scale bar = 100 μm. (B) ELISpot-IFNγ of anti-GP33 effectors against B16-derived cells. Splenocytes from GP33-vaccinated mice were cultured 24h with tumor cells, then the number of IFN-γ-producing cells was measured (mean from three experiments ±SD). (C) Ex-vivo ELISpot-IFNγ in tumor cell conditioned medium. Three-day conditioned media from 106 cells/ml tumor clones were used as culture medium for freshly isolated splenocytes from GP33-vaccinated mice. The number of IFN-γ-producing cells was measured after a 24h- incubation with GP33 (mean from six experiments ±SD; A1 vs B11, *p=0.020 p value of Mann-Withney test).
No significant difference in proliferation kinetics between A1, B8 and B11 was observed in vitro (Supplementary figure 1). Finally, the three clones displayed similar capacities to present GP33 to specific CD8+ T cells (Figure 1B).
T cell inhibiting IL4I1 properties in vitro
We previously showed that the human IL4I1 inhibits T cell proliferation in vitro. The murine form shares 80% homology with the human form in the putative enzymatic and FAD-binding domain. In agreement with this, murine IL4I1-expressing clones inhibited significantly the in vitro GP33-specific proliferation of TCR transgenic splenocytes from P14 mice (Supplementary figure 2). Moreover, the number of IFNγ− producing anti-GP33 T cells was markedly decreased when splenocytes were cultured in the 3-day conditioned medium of B11 cells (Figure 1C). In these conditions, in contrast to the experiment in Figure 1B, where a few irradiated tumor cells were used as targets, the splenocytes were immediately exposed to a high IL4I1 activity. These results, thus, indicate that IFNγ production was directly affected by IL4I1.
Resistance to immune rejection of IL4I1-expressing tumors
B16 melanoma behaves as an aggressive poorly immunogenic tumor cell line when injected in naïve C57BL/6 mice. However, expression of GP33 by B16GP33 cells leads to tumor rejection in mice adoptively transferred with anti-LCMV cytotoxic T cells [22, 23]. We first tested whether IL4I1 expression by B16GP33 cells would modify their in vivo tumor growth after s.c. injection into naïve mice. The first tumors appeared between day 11 and 18 and developed with a similar kinetic, independently of their IL4I1 expression level (Figure 2A and 2B). Thus, B16GP33 tumors are not naturally controlled by the immune system, in accordance with the literature [22].
Figure 2. Resistance to immune rejection of IL4I1-expressing tumors.
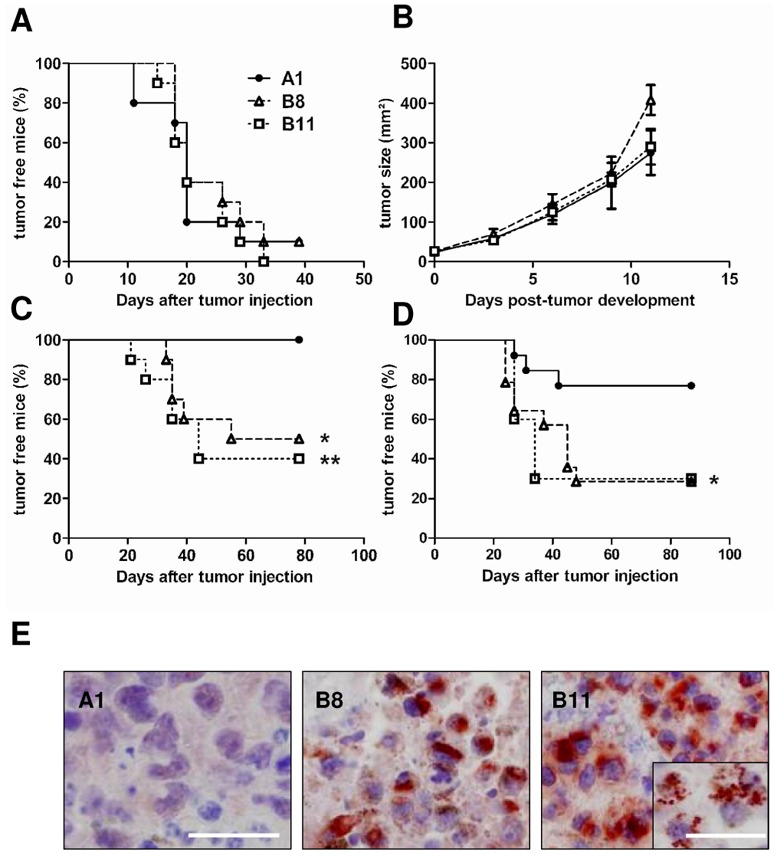
(A and B) 103 tumor cells were injected s.c. into the left flank of naïve C57BL/6 mice (n = 10 mice/group, representative experiment out of 2). (A) The survival without tumor and (B) tumor growth were evaluated twice a week during forty days after challenge. (C and D) Mice were vaccinated s.c. with GP33. (C) Seven or (D) 28 days later, 105 tumor cells were injected s.c. into the controlateral flank. (C) Representative experiment out of 5 with n = 10 mice/group and (D) representative experiment out of 3 with n = 14 mice/group. Curve comparison was performed using the Log-Rank test, A1 ●; B8 △; B11 □. (C); B8 vs B11, NS. *p=0.012; **p=0.004 (D) A1 vs B8 or vs B11, *p = 0.049. (E) IL4I1 immunostaining on tumor sections. Slices from formalin-fixed paraffin-embedded A1, B8 and B11 tumors were stained with an anti-IL4I1 antibody. Magnification x400, scale bar = 100 μm; inset magnification x630, scale bar = 50 μm.
In order to induce a strong GP33-specific T cell response, C57BL/6 mice were immunized with GP33 in IFA seven days before tumor challenge. As expected, mice were fully protected towards clone A1 (Figure 2C). In contrast, more than 50% of the mice transplanted with the IL4I1-expressing clones developed tumors (p= 0.012 B8 versus A1, p= 0.004 B11 versus A1).
We have previously demonstrated that the IL4I1 effect on human T cell proliferation is predominantly on CD45RO+ T cells [18]. To determine the impact of IL4I1 on tumor development during the memory phase of the immune response, we injected B8 and B11 tumor cells 28 days after immunization (Figure 2D). Under these conditions, 77% of A1 challenged mice controlled tumors at day 80, whereas more than 70% B8 and B11 challenged mice developed a melanoma (p= 0.049). After sacrifice of the tumor-bearing animals, melanoma biopsies were preserved for IL4I1 analysis. Expression and activity of the enzyme were detected in both B8 and B11 tumors (Figure 2E and Table 1) with B11 still displaying the strongest activity. Surprisingly, these activities were highly variable and on average two to three fold higher than those measured from the transfected cells in culture. The increase of IL4I1 activity in tumors could not be ascribed to the TAM infiltrate, which was very poor in both IL4I1-expressing and control tumors. As observed in human tumors, most of these rare TAM expressed IL4I1 (supplementary figure 3).
Thus, our data demonstrate that IL4I1 facilitates in vivo immune escape of the tumor cells.
Quantitative and functional defects of antitumor CD8+ T cells in mice challenged with IL4I1-expressing tumors
In order to establish whether escape of IL4I1-expressing tumors was associated with an alteration of the anti-tumor T cell response in vivo, we further analyzed GP33-specific CD8+ T cells after tumor challenge (Figure 3A). To increase the number of GP33-specific T cell precursors before vaccination, we adoptively transferred naïve splenocytes from P14 transgenic mice. We also increased the number of injected tumor cells to overcome the immune response in A1 challenged mice and allow simultaneous tumor appearance in all groups. Blood was regularly taken until tumor development 11 to 14 days after challenge to measure the number of circulating tumor-specific CD8+ T cells. Mice were then sacrificed and GP33-specific splenocytes were tested for (i) IFNγ production, (ii) acquisition of an effector/memory phenotype and (iii) cytotoxic function.
Figure 3. Mechanisms of IL4I1-induced immune escape.
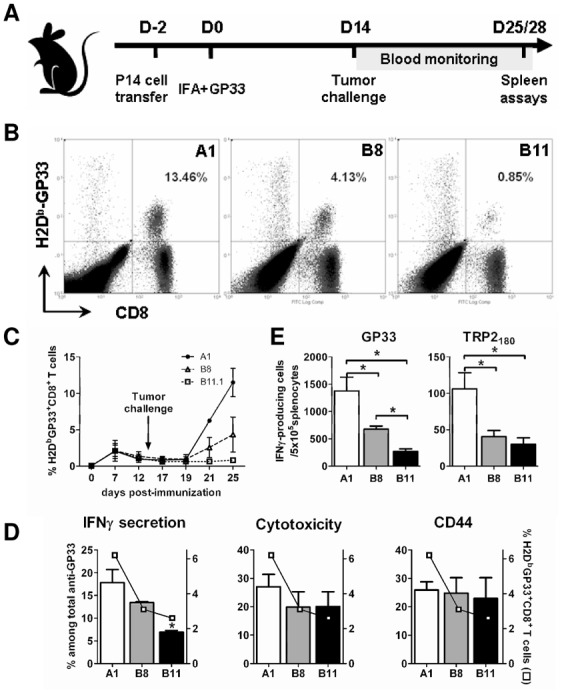
(A) Schematic representation of the experimental protocol. Naïve mice (2 to 5 mice per group) were immunized against GP33 s.c. on day 0, two days after adoptive transfer of P14 splenocytes. At day 14, mice were challenged with tumor cells and blood was taken three times a week for anti-GP33 immunomonitoring. Mice were sacrificed between day 25 and 28 for anti-tumor T-cells analyses in the spleen. (B and C) The percentage of circulating GP33-specific T-cells was measured using H2-Db-GP33 tetramers. A representative experiment out of 4 is shown with dot plots from one mouse per group (A1 ●, B8 △, B11 □; p= 0.038 A1 vs B8, p= 0.029 A1 vs B11, Mann-Withney test. (D) Percentage of IFNγ-secreting (left panel histogram), cytotoxic (CD107a+ after in vitro stimulation, central panel histogram) and effector/memory (CD44+, right panel histogram) T-cells among the GP33-specific splenic CD8+ T-cells (□), mean ± SD of two experiments, * p = 0.033, Mann-Withney test. (E) Ex-vivo ELISpot anti-IFNγ response of the same mice 11 or 14 days after challenge. Freshly isolated spleen cells of GP33-vaccinated and A1, B8 or B11 challenged mice were cultured with 1 μg/ml GP33 (left panel) or 10 μg/ml TRP2180 (right panel). Results were expressed as IFNγ-producing cells/5×105 splenocytes (mean from four experiments ± SD, * p < 0.030, Mann-Withney test).
The kinetics of GP33 specific responses were monitored from blood. At the peak of the immune response (day 7 post-immunization), the circulating GP33-specific population reached 1 to 5 % of the circulating CD8+ T lymphocytes and diminished to 0.4 to 3 % two days before tumor challenge with A1, B8 or B11 cell. A few days post-tumor challenge with A1 cells, a considerable increase of the circulating GP33-specific T cells was observed that reached 12 fold over the pre-challenge value, indicating a boost effect of the tumor. In contrast, challenges with B8 and B11 were associated respectively with a small (increase of 3 fold) or absent recall response to the tumor epitope (Figure 3B and 3C). Similar results were obtained on splenic CD8+ T cells with a mean of 6.2% versus 3.1% and 2.6% tetramer-positive cells, for A1, B8, and B11 challenged mice respectively (Figure 3D). These results suggest that IL4I1 expression in vivo inhibits T lymphocyte proliferation as observed in vitro.
Splenocytes were further analyzed for functionality. Mice challenged with IL4I1-expressing tumor cells presented a significantly lower anti-GP33 IFNγ response than mice challenged with A1 cells (Figure 3E). The response inhibition was correlated to the level of IL4I1 production by the tumors (respective decrease of 53% and 84% in splenocytes from B8 and B11 challenged mice). Moreover, the IFNγ response to the melanoma TRP2180 epitope was also affected, with a 30 to 50% decrease in B8 and B11 challenged mice in comparison to A1 challenged mice (Figure 3E). Interestingly, in the groups challenged with IL4I1-expressing tumors, when calculating the percentage of IFNγ-secreting cells amongst the GP33 tetramer labeled cells, we observed that the decrease in the IFNγ-producing anti-GP33 effectors was more profound than the decrease in the total anti-GP33 T cell population (Figure 3D, left panel). Since the total GP33-specific cells diminished, in animals challenged with IL4I1-expressing tumors, both anti-GP33 cytotoxic T cells (CD107a+) and effector/memory T cells (CD44+) also decreased in their spleens. However, the percentage of these two cell populations amongst the anti-GP33 effectors remained stable under the influence of IL4I1 (Figure 3D, middle and right panels). Altogether, our results suggest that IL4I1 not only diminishes the number of anti-tumor specific T cells, but also affects their capacity to produce IFNγ.
In vivo impairment of cytotoxic capacities in mice challenged with IL4I1-expressing tumors
We next evaluated the in vivo impact of the decrease of anti-GP33 cytotoxic T cells induced by IL4I1. For this purpose, we monitored the elimination of CFSE-labeled GP33-loaded cells by specific CD8+ T cells in vivo (Figure 4A). Mice were vaccinated with GP33 and challenged seven days later with A1 or B11 cells. Ten days later, splenocytes pulsed either with an irrelevant peptide or GP33 were adoptively transferred in recipient mice. Lysis of GP33-pulsed target cells was observed from day 1 in the blood of all mice (Figure 4B). After day 2, this specific lysis was less important in mice challenged with B11 tumors in comparison to control animals. This difference was stable (15% on average) and significant from day 3 to day 8. A similar difference was observed in the spleen and lymph nodes (Figure 4C and 4D), indicating a slight defect of cytotoxic activity in mice challenged with IL4I1-expressing tumors. We also confirmed the impairment of the IFNγ response to GP33 and TRP2180 in the spleen and draining lymph nodes of the B11 challenged mice (data not shown).
Figure 4. Cytotoxicity inhibition of GP33 specific T cells in IL4I1-challenged mice.
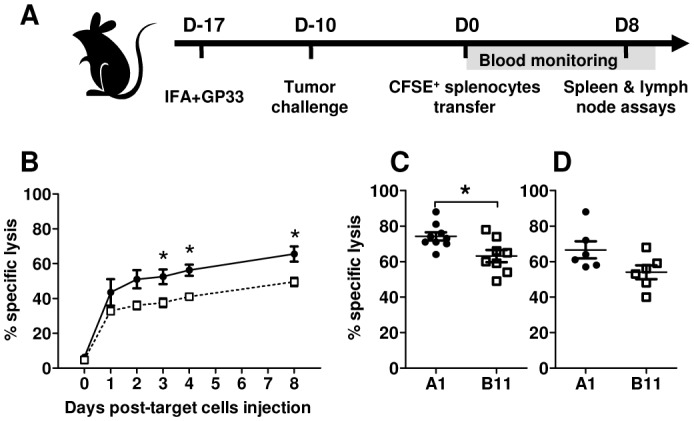
(A) Schematic representation of the experimental protocol. Naïve mice were immunized against GP33 s.c. and challenged 7 days later with A1 (●) or B11 (□) tumor cells. Ten days later (day 0), splenocytes from syngeneic naive mice were loaded with GP33 or irrelevant peptide NP50 and labeled with a high and low dose of CFSE, respectively. A mix of 1:1 GP33- and NP50-loaded splenocytes was then adoptively transferred. Blood was taken from day 0 to day 4 and at day 8 after transfer. Mice were sacrificed at day 8 for analysis in the spleen and draining lymph nodes. (B, C and D) The percentage of residual GP33- and NP50-loaded cells was measured by FACS. The rejection level is expressed as the percentage of specific lysis of GP33-loaded target cells in (B) the blood, (C) spleen and (D) lymph nodes. *p < 0.03, Mann-Withney test.
In conclusion, IL4I1 modulates in vivo the number of anti-tumor cytotoxic T cells and affects their IFNγ-secreting capacity.
Threshold of IL4I1 immune escape effect
In human tumors, the IL4I1-expressing cells are generally diluted in IL4I1 negative populations, even when tumor cells express the enzyme [21]. B11 cells display an IL4I1 activity close to the activity measured in Hodgkin cells and 60% lower than that of activated macrophages. To mimic a partial expression in the tumor, we diluted graded numbers of B11 cells in A1 cells before challenge, to obtain mixes of 0, 20, 50, 80 and 100 percent B11/A1.
Only 4% of the mice developed a tumor in the control group receiving 100% A1 cells, whereas tumors occurred in 68% of mice challenged with B11 tumors (Figure 5A). Most interestingly, mice challenged with tumors containing at least 20% B11 cells still developed tumors with a significantly higher frequency (25%) than the A1 control mice. The incidence of tumor development increased with the proportion of IL4I1-expressing cells. Enzymatic activities of the tumors developed in these experiments ranged from 34 ±16 (20% B11) to 660 ±178 (100% B11) pmoles H2O2/h/100μg proteins and remained proportional to the amount of IL4I1-expressing cells injected (Figure 5B). Of importance, we also detected IL4I1 activity before tumor development in the sera of animals from all groups who had received IL4I1-expressing cells. These activities seemed to increase with the number of B11 cells injected (Figure 5C). Thus, IL4I1 expression in a small proportion of tumor cells is sufficient to drive tumor escape from the T cell response.
Figure 5. Threshold of immune escape effect.
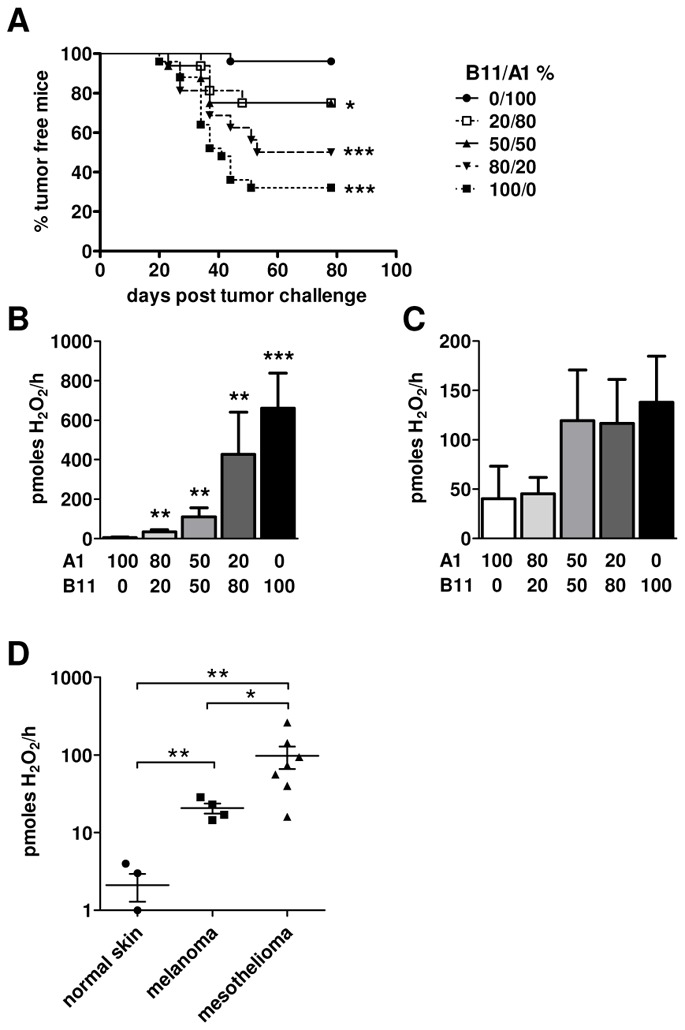
(A) Mice were vaccinated s.c. with GP33. Seven days later, graded doses of 0 (●), 20 (□), 50 (▲), 80 (▼) and 100 (■) percent of clone B11 admixed to clone A1 were injected s.c. into the controlateral flank. Cumulative results from three experiments with 5 to 8 mice per group are shown (*p < 0.05; ***p < 5×10−4, Mann-Withney test). (B) IL4I1 activity of the tumors developed in A. Results are expressed as pmoles H2O2/h/100μg proteins. Statistics compare activities of each group to A1 100% (**p < 0.006; ***p = 10−4, unpaired t-test). (C) IL4I1 activity of sera obtained before tumor development (between day 30 and 55). Results are expressed as pmoles H2O2/h/100μl serum. (D) IL4I1 activity on frozen tumor samples from 4 melanomas with corresponding normal skin and 7 mesotheliomas. Results are expressed as pmoles H2O2/h/100μg proteins (*p < 0.05; **p < 0.007, Mann-Withney test).
IL4I1 activity in human tumors
IL4I1 expression in the TAM infiltrate seems to be a constant of human tumors. In some cancers, IL4I1 is also often expressed in tumor cells, suggesting that depending on the tumor type, variable levels of IL4I1 activity may be present in the tumor bed. We decided to analyze IL4I1 activity of human tumor types that either express IL4I1 exclusively in the TAM compartment – melanomas – or both in TAM and tumor cells – mesotheliomas – [21]. Four melanoma biopsies, with corresponding normal skin and seven mesotheliomas were evaluated (Figure 5D). In all melanoma cases, we measured an IL4I1 activity ranging from 15 to 29 pmoles of H2O2/h/100μg of proteins, which significantly differed from the negligible activity detected in the normal skin. As expected, high IL4I1 activities were measured in all but one cases of mesothelioma (mean, 98; range, 16 to 261 pmoles H2O2/h/100μg proteins). Activities of both melanomas and mesotheliomas were in the range of, or even higher than, those measured in murine tumors developed after injection of 20% B11 cells mixed in A1 cells.
DISCUSSION
We have previously shown that the IL4I1 enzyme, which presents immunosuppressive properties in vitro, is expressed in most tumors, by the TAM and/or the neoplastic cells [18, 21]. Here, we establish the role of IL4I1 expression in tumor escape from the immune system and demonstrate that IL4I1 induces a decrease of IFNγ-producing and cytotoxic anti-tumor CD8+ T cells in vivo. Moreover, partial IL4I1 expression in the tumor, at equivalent levels of those detected in human tumors, is sufficient to drive escape from the T cell response.
Despite expression of the highly antigenic GP33 epitope, B16GP33 tumors are very aggressive in vivo. In line with this, no tumor growth difference was observed in naïve mice between the IL4I1-expressing and IL4I1-negative clones. This indicates that the IL4I1-expressing clones do not intrinsically display increased proliferative potential. It also suggests that IL4I1 does not play a major inhibitory role on naïve T cells, in accordance with our previous results [18]. On the contrary, data obtained in mice vaccinated with GP33 prior to challenge, whether at the peak or at the memory phase of the response, implicate a major effect of IL4I1 on effector/memory antitumor T cells. Indeed, in mice challenged with IL4I1-expressing cells, the incidence of tumor appearance was significantly higher than in mice challenged with control cells. This resistance to immune rejection was not due to a loss of antigen presentation.
The IL4I1-expressing clones inhibited mouse T cell proliferation and IFNγ production in vitro, confirming results obtained in a human system [18, 20]. In line with this, we observed in mice receiving IL4I1-expressing cells, that (i) GP33-specific CD8+ T cells were dramatically less frequent, suggesting that their proliferation was affected in vivo; (ii) the ratio of IFNγ-producing over GP33-specific CD8+ T cells diminished, indicating that, as observed in vitro, the IFNγ-secreting capacity of antitumor T cells was independently affected; (iii) the in vivo anti-GP33 cytotoxicity was impaired; (iv) the IFNγ response to both GP33 and the melanoma epitope TRP2180 were diminished, implying that the antitumor T cell response may be globally depressed. In accordance with a global immune suppression, the allogeneic response of splenocytes from mice challenged with IL4I1-expressing tumors in vivo was diminished in comparison to splenocytes from control mice (supplementary figure 4A). Moreover, as previously observed with human lymphocytes, mouse splenocytes exposed to IL4I1 in vivo down-regulated the expression of the TCR CD3ζ chain (supplementary figure 4B). This could explain their functional defects in proliferation and IFNγ production.
In a recent analysis of 315 biopsies from human malignancies, we detected frequent expression of IL4I1 in situ [21]. Indeed, IL4I1 was expressed in TAM from most of the tumors, particularly inflammatory tumors, and in neoplastic cells from several B cell lymphoma. IL4I1-positive tumor cells were also detected in some subtypes of non-lymphoid cancers, including mesothelioma. In follicular lymphoma, a high level of IL4I1 expression seemed associated with a better outcome, in the 23 cases studied. These results are in contradiction with data obtained in our mouse model. However, in the case of follicular lymphoma, tumor B cells depend on interactions with follicular T helper cells for survival [24], and the impact of IL4I1 expression might be directed to this T-cell population. In contrast, in non-lymphoid tumors, our data indicate that cytotoxic T cells are a major target population of IL4I1 activity. In support of this, Finak et al. identified IL4I1 mRNA expression in breast cancer as a poor outcome factor among a signature comprising 163 prognosis-predictive genes expressed in stromal cells [25]. Thus, depending on the tumor type, IL4I1 may affect different infiltrating lymphocyte populations with opposing consequences on tumor evolution.
The adoptive tumor model used here more closely reproduces IL4I1 expression in a non-lymphoid tumor. In our experimental settings, the minimal level of IL4I1 activity detected in tumors escaping immune control was in the range of activities detected in human melanomas, and on average 5 fold weaker than those measured in mesotheliomas, suggesting that the immunosuppressive effect of IL4I1 may be operative in human cancers. Melanomas express IL4I1 exclusively in the TAM compartment, while 50% of the mesotheliomas express IL4I1 both in the TAM and in the tumor cells [21], potentially due to the upregulation of the IL4I1-inducing STAT-1 and NF-κB signalling pathways [20, 26, 27]
In this work, we thus show that IL4I1 is a bona fide immunosuppressive enzyme participating in the escape of malignancies in vivo. In contrast to the enzymes described so far, IL4I1 is secreted and can be detected in the serum of the animals before tumor appearance, explaining its systemic effect. By modulating the number and IFNγ− secreting potential of cytotoxic T cells, IL4I1 severely compromises anti-tumor functions of the host immune system and may contribute to progression of human tumors. These findings provide a scientific rationale to evaluate IL4I1 inhibitors as a way to improve the efficacy of antitumor immunotherapies.
MATERIALS AND METHODS
Cell lines
B16GP33 cells expressing the immunodominant H2-Db-restricted epitope GP33 of the lymphocytic choriomeningitis virus (LCMV) glycoprotein were derived from parental B16.F10 murine melanoma cells as previously described [22]. B16GP33 were transfected with pcDNA4-TO-mIL4I1-mycHis or the same empty vector using Transfast (Promega) and were maintained in DMEM supplemented with 10% fetal calf serum, 2 mM L-glutamine, 100 U/ml penicillin, 10 μg/ml streptomycin, non essential amino-acids, 200 μg/ml neomycin and 300 μg/ml zeocine at 37°C in 5%CO2. All the reagents used were from Invitrogen.
Plasmids
Mouse IL4I1 cDNA was generated as previously described [18] and cloned into the EcoRI and NotI sites of pcDNA4-TO-mycHis plasmid (Invitrogen).
Peptides
GP33 (KAVYNFATM) and the control influenza virus nucleoprotein H2-Db-restricted peptide NP50 (SDYEGRLI) were from Polypeptide group. The tyrosinase peptide TRP2180 (SVYDFFVWL) was a gift from EMC microcollections.
Immunologic detection of IL4I1
Cells lysis and Western blot were realized as described by Boulland et al. [18]. The tagged mIL4I1 protein was revealed with an anti-myc antibody (clone 9E10, Sigma-Aldrich). The actin protein was detected using a mouse anti-actin mAb (clone C4, Millipore).
Immunohistochemistry was performed as described in [21]. For immunofluorescence, after seeding onto poly-L-lysine coated coverslips, cells were fixed with 3% paraformaldehyde and stained with a polyclonal anti-myc antibody (Cell Signaling). Revelation was performed using an Alexa fluor 488-coupled anti-rabbit IgG (Invitrogen). Photograph acquisition was performed with a Hamamatsu C8484 digital camera and Fluovision IMSTAR software.
IL4I1 enzymatic activity assay
Cell line lysates and mIL4I1 purification from the culture medium on nickel beads were performed as previously described by [18]. Tumors were collected in PBS containing Complete mini® protease inhibitors (Roche, France) and frozen at −80°C until use. After thawing, 100 mg of tumor resuspended in 500 μl PBS containing Complete mini® were homogenized by shaking with stainless steel beads in a Qiagen Tissuelyser (Qiagen, Courtaboeuf, France) for 2 cycles of 2 min at 20 Hz. Lysates were centrifuged 10 min at 10000g and whole cell supernatants collected. All samples were tested for phenylalanine oxidative activity according to [21]. Activities are expressed as pMoles H2O2 produced per hour by 105 cells, 100 μg proteins, 40 μl bead suspension or 100 μl mouse serum as stated in the figure legends. Permission to use human samples was given by CPP Ile de France IX.
Mice
P14 transgenic mice (line 318) were purchased from TAAM (France). Fifty to 60% of P14 CD8+ T cells express a Vα2/Vβ8 TCR specific for the LCMV-derived GP33 peptide in association with the H-2Db molecule [28]. Naïve C57BL/6 mice (6 to 10 weeks old, Charles River) were injected s.c. with 103 tumor cells or vaccinated s.c. with 50 μg of GP33 in Incomplete Freund’s Adjuvant (IFA, Sigma-Aldrich) then challenged s.c. 7 or 28 days later into the controlateral flank with 105 tumor cells. Tumor size was evaluated twice a week with a caliper and calculated as the product of bisecting tumor diameters. Mice bearing a tumor with a diameter >15 mm were sacrificed according to animal care regulations.
In the adoptive transfer protocols, 2×106 splenocytes from P14 mice were injected i.v. via the retro-orbital sinus. Two days later, C57BL/6 mice were immunized as above, then challenged s.c. 14 days later with 107 tumor cells.
For CFSE assays, 107 tumor cells were injected s.c. seven days after GP33 vaccination. Ten days after tumor challenge, mice received an intravenous injection of a half-mix of 3.107 splenocytes loaded with GP33 and with NP50 and labeled respectively with 1 μM and 0.1 μM of the fluorescent dye 5–6-carboxyfluorescein diacetate succinimidyl ester (CFSE, Molecular Probes). CFSE+ donor cells in blood, spleen and lymph nodes were determined by flow cytometry. GP33-specific donor cell rejection was calculated using the formula: 100− [(% of CFSEhigh cells/% of CFSElowcells) ×100].
All experiments were performed in compliance with the French Ministry of Agriculture regulations for animal experimentation (laboratory accreditation C 94-028-31, authorization 94-308 for VMF).
Flow cytometry
For blood T-cells analysis, GP33-specific CD8+ lymphocytes were labelled with H2-Db-GP33 tetramers coupled to phycoerythrin, an anti-CD8 antibody (clone KT15, fluorescein-coupled) and an anti-CD44 antibody (clone KM201, APC-coupled). All reagents were from Beckman-Coulter. After red blood cell lysis using iTAg MHC tetramer lysis reagent, the cells were fixed with 1% paraformaldehyde before flow cytometry analysis. For surface CD107a labelling, heparinised blood was incubated with GP33 tetramers and an anti-CD107a antibody (clone 1D4B, APC-coupled) before a 5-hour culture at 37°C. Two μM monensin was added one hour after beginning the stimulation. CD8 labelling, red blood cell lysis and cell fixation were then performed as above. At least 50,000 events were acquired on a Cyan flow cytometer (Beckman-Coulter) and data were analyzed using the Summit 4 software (DAKO-Cytomation).
ELISpot-IFNγ
The ELISpot-IFNγ assay was performed according to Mabtech’s instructions except for the revelation. Spots were revealed by adding, successively, alkaline-phosphatase-labeled ExtrAvidin and BCPI/NBT substrate (Sigma-Aldrich) and counted with a transmitted-light stereomicroscope using image-analyzing software connected to a camera (KS ELISPOT system; Carl Zeiss Vision).
In Fig. 1C, culture medium was replaced by 3-days conditioned medium obtained from 106 tumor cells/ml. Results are expressed as the number of spots per 5×105 cells after subtraction of the background obtained with the irrelevant peptide. The percentage of IFNγ-secreting T cells among total GP33-specific T cells was calculated as follows: % IFNγ+ CD8+/% H2-Db-GP33 tetramer+ CD8+ T-cells.
Acknowledgments
We are grateful to Hanspeter Pircher for the B16GP33 cell line and for relevant comments on the work. We thank William Hempel for critical reading of the manuscript. Grants support: ARC subvention fixe 4883 (FC) and the French association for therapeutic, genetic and immunologic research on lymphoma (ARTGIL) granted by Roche and Amgen (CC). We thank Chrystelle Guiter, Sophia Balustre, Mathieu Surénaud, Franck Delafond and the cytometry platform for help in this work.
Footnotes
The authors declare no financial or commercial conflict of interest.
References
- 1.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gordon S, Martinez FO. Alternative activation of macrophages: mechanism and functions. Immunity. 2010;32:593–604. doi: 10.1016/j.immuni.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 3.Marigo I, Dolcetti L, Serafini P, Zanovello P, Bronte V. Tumor-induced tolerance and immune suppression by myeloid derived suppressor cells. Immunol Rev. 2008;222:162–179. doi: 10.1111/j.1600-065X.2008.00602.x. [DOI] [PubMed] [Google Scholar]
- 4.Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- 5.Youn JI, Gabrilovich DI. The biology of myeloid-derived suppressor cells: the blessing and the curse of morphological and functional heterogeneity. Eur J Immunol. 2010;40:2969–2975. doi: 10.1002/eji.201040895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Munn DH, Mellor AL. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest. 2007;117:1147–1154. doi: 10.1172/JCI31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munder M. Arginase: an emerging key player in the mammalian immune system. Br J Pharmacol. 2009;158:638–651. doi: 10.1111/j.1476-5381.2009.00291.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bogdan C. Regulation of lymphocytes by nitric oxide. Methods Mol Biol. 2011;677:375–393. doi: 10.1007/978-1-60761-869-0_24. [DOI] [PubMed] [Google Scholar]
- 9.Grohmann U, Bronte V. Control of immune response by amino acid metabolism. Immunol Rev. 2010;236:243–264. doi: 10.1111/j.1600-065X.2010.00915.x. [DOI] [PubMed] [Google Scholar]
- 10.Johansson CC, Egyhazi S, Masucci G, Harlin H, Mougiakakos D, Poschke I, Nilsson B, Garberg L, Tuominen R, Linden D, Stolt MF, Hansson J, Kiessling R. Prognostic significance of tumor iNOS and COX-2 in stage III malignant cutaneous melanoma. Cancer Immunol Immunother. 2009;58:1085–1094. doi: 10.1007/s00262-008-0631-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johansson CC, Mougiakakos D, Trocme E, All-Ericsson C, Economou MA, Larsson O, Seregard S, Kiessling R. Expression and prognostic significance of iNOS in uveal melanoma. Int J Cancer. 2010;126:2682–2689. doi: 10.1002/ijc.24984. [DOI] [PubMed] [Google Scholar]
- 12.Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, Boon T, Van den Eynde BJ. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9:1269–1274. doi: 10.1038/nm934. [DOI] [PubMed] [Google Scholar]
- 13.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189:1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodriguez PC, Zea AH, DeSalvo J, Culotta KS, Zabaleta J, Quiceno DG, Ochoa JB, Ochoa AC. L-arginine consumption by macrophages modulates the expression of CD3 zeta chain in T lymphocytes. J Immunol. 2003;171:1232–1239. doi: 10.4049/jimmunol.171.3.1232. [DOI] [PubMed] [Google Scholar]
- 15.Taheri F, Ochoa JB, Faghiri Z, Culotta K, Park HJ, Lan MS, Zea AH, Ochoa AC. L-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin Cancer Res. 2001;7:958s–965s. [PubMed] [Google Scholar]
- 16.Friberg M, Jennings R, Alsarraj M, Dessureault S, Cantor A, Extermann M, Mellor AL, Munn DH, Antonia SJ. Indoleamine 2,3-dioxygenase contributes to tumor cell evasion of T cell-mediated rejection. Int J Cancer. 2002;101:151–155. doi: 10.1002/ijc.10645. [DOI] [PubMed] [Google Scholar]
- 17.Chu CC, Paul WE. Fig 1, an interleukin 4-induced mouse B cell gene isolated by cDNA representational difference analysis. Proc Natl Acad Sci U S A. 1997;94:2507–2512. doi: 10.1073/pnas.94.6.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boulland ML, Marquet J, Molinier-Frenkel V, Moller P, Guiter C, Lasoudris F, Copie-Bergman C, Baia M, Gaulard P, Leroy K, Castellano F. Human IL4I1 is a secreted L-phenylalanine oxidase expressed by mature dendritic cells that inhibits T-lymphocyte proliferation. Blood. 2007;110:220–227. doi: 10.1182/blood-2006-07-036210. [DOI] [PubMed] [Google Scholar]
- 19.Chavan SS, Tian W, Hsueh K, Jawaheer D, Gregersen PK, Chu CC. Characterization of the human homolog of the IL-4 induced gene-1 (Fig 1) Biochim Biophys Acta. 2002;1576:70–80. doi: 10.1016/s0167-4781(02)00295-6. [DOI] [PubMed] [Google Scholar]
- 20.Marquet J, Lasoudris F, Cousin C, Puiffe M, Martin-Garcia N, Baud V, Chéreau F, Farcet J, Molinier-Frenkel V, Castellano F. Dichotomy between factors inducing the immunosuppressive enzyme IL4I1 in B lymphocytes and mononuclear phagocytes. European Journal of Immunology. 2010;40:2557–2568. doi: 10.1002/eji.201040428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carbonnelle-Puscian A, Copie-Bergman C, Baia M, Martin-Garcia N, Allory Y, Haioun C, Cremades A, Abd-Alsamad I, Farcet JP, Gaulard P, Castellano F, Molinier-Frenkel V. The novel immunosuppressive enzyme IL4I1 is expressed by neoplastic cells of several B-cell lymphomas and by tumor-associated macrophages. Leukemia. 2009;23:952–960. doi: 10.1038/leu.2008.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prevost-Blondel A, Zimmermann C, Stemmer C, Kulmburg P, Rosenthal FM, Pircher H. Tumor-infiltrating lymphocytes exhibiting high ex vivo cytolytic activity fail to prevent murine melanoma tumor growth in vivo. J Immunol. 1998;161:2187–2194. [PubMed] [Google Scholar]
- 23.Zimmermann C, Prevost-Blondel A, Blaser C, Pircher H. Kinetics of the response of naive and memory CD8 T cells to antigen: similarities and differences. Eur J Immunol. 1999;29:284–290. doi: 10.1002/(SICI)1521-4141(199901)29:01<284::AID-IMMU284>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 24.Roulland S, Suarez F, Hermine O, Nadel B. Pathophysiological aspects of memory B-cell development. Trends Immunol. 2008;29:25–33. doi: 10.1016/j.it.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14:518–527. doi: 10.1038/nm1764. [DOI] [PubMed] [Google Scholar]
- 26.Janssen YM, Barchowsky A, Treadwell M, Driscoll KE, Mossman BT. Asbestos induces nuclear factor kappa B (NF-kappa B) DNA-binding activity and NF-kappa B-dependent gene expression in tracheal epithelial cells. Proc Natl Acad Sci U S A. 1995;92:8458–8462. doi: 10.1073/pnas.92.18.8458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kothmaier H, Quehenberger F, Halbwedl I, Morbini P, Demirag F, Zeren H, Comin CE, Murer B, Cagle PT, Attanoos R, Gibbs AR, Galateau-Salle F, Popper HH. EGFR and PDGFR differentially promote growth in malignant epithelioid mesothelioma of short and long term survivors. Thorax. 2008;63:345–351. doi: 10.1136/thx.2007.085241. [DOI] [PubMed] [Google Scholar]
- 28.Pircher H, Burki K, Lang R, Hengartner H, Zinkernagel RM. Tolerance induction in double specific T-cell receptor transgenic mice varies with antigen. Nature. 1989;342:559–561. doi: 10.1038/342559a0. [DOI] [PubMed] [Google Scholar]