

Abstract
Bronchopulmonary dysplasia (BPD) is a multifactorial chronic lung disease of premature infants. BPD can be attributed to the dysregulation of normal lung development due to ventilation and oxygen toxicity, resulting in pathologic complications of impaired alveolarization and vascularization. MicroRNAs (miRNA) are small noncoding RNAs that regulate gene expression posttranscriptionally and are implicated in diverse biological processes and diseases. The objectives of this study are to identify the changed miRNAs and their target genes in neonatal rat lungs in response to hyperoxia exposure. Using miRNA microarray and real-time PCR analyses, we found downregulation of five miRNAs, miR-342, miR-335, miR-150, miR-126*, and miR-151*, and upregulation of two miRNAs, miR-21 and miR-34a. Some of these miRNAs had the highest expression during embryonic and early postnatal development. DNA microarray analysis yielded several genes with conserved binding sites for these altered miRNAs. Glycoprotein nonmetastatic melanoma protein b (GPNMB) was experimentally verified as a target of miR-150. In summary, we identified seven miRNAs that were changed in hyperoxia-exposed neonatal lungs. These results provide a basis for deciphering the mechanisms involved in the spatial and temporal regulation of proteins that contribute to the pathogenesis of BPD.
Keywords: bronchopulmonary dysplasia, microRNA, lung development
bronchopulmonary dysplasia (BPD) is a chronic lung disease of infancy. It results from a complex interplay among systemic maternal infections, surfactant deficiency, ventilation (barotrauma or volutrauma), and oxygen toxicity. Despite improvements in ventilation strategies and surfactant therapy, alveolar arrest, impaired vascularization, and inflammation are inexorable complications in BPD (4, 6). An estimated affliction of 10,000–15,000 infants with BPD has been reported every year in the United States alone (31). Neonatal rodent models of hyperoxia exposure serve as a valuable tool to understand the mechanisms of BPD pathogenesis. Alveolarization occurs postnatally in rodents, which is comparable with the lung morphogenesis of premature babies. The mechanisms regulating the alveolar growth and vascularity and the contributing factors leading to their impairment are largely unknown. Spatially and temporally controlled cascades of signaling through multiple pathways and transcription factors are important in the normal development of any organ systems. The dysregulation of one or more of these factors contribute to the pathogenesis of various diseases.
The discovery of microRNAs (miRNAs) has added a new dimension into regulatory mechanisms that control development and disease (3, 33). These 21- to 24-nucleotide RNAs inhibit protein expression at the posttranscriptional level through direct binding to the target mRNA at 3′-untranslated region (3′-UTR), resulting in mRNA degradation or translation repression. By regulating different signaling cascades miRNAs control multiple biological processes in a wide array of organisms. Accumulated evidence supports that the dysregulation of multiple proteins by miRNAs contributes to the pathogenesis of various diseases (13).
The objectives of the present study are twofold: 1) to identify the miRNAs changed in neonatal lungs after hyperoxia exposure and 2) to identify target genes of the changed miRNAs. We identified seven such miRNAs (miR-342, -335, -150, -126*, -151*, -21, and -34a) using miRNA microarray and real-time PCR and a dozen of target genes using DNA microarray analysis and computational approaches. These studies provide candidate miRNAs and their targets for further studying their roles in BPD pathogenesis.
MATERIALS AND METHODS
Exposure of neonatal rat pups to hyperoxia.
All animal procedures were carried out according to the protocol (VM1025) approved by the Animal Care and Use Committee at Oklahoma State University. Timed pregnant Sprague-Dawley rats were bred in-house. Pups from two mother rats born naturally at the same day were mixed and divided into two groups. On postnatal day 3 (P3), a group of mixed pups were placed in a sealed Plexiglas chamber (90 × 45 × 45 cm). The chamber was filled with 100% oxygen and maintained at ∼95% oxygen using an oxygen flow rate of 4 l/min as described previously (22). The oxygen concentration was continuously monitored using an oxygen sensor (Vacu-Med, Ventura, CA). Soda lime was used to remove excess CO2. The dams were switched between litters every day to avoid oxygen toxicity. The control group was kept at room air for 10 days (P13). On P13, both hyperoxia- and room air-exposed animals were anesthetized, trachea was cannulated, and lungs were fixed with 4% paraformaldehyde by instilling endotracheally at 30 cmH2O pressure. For RNA and protein samples, the lungs were excised and immediately frozen in liquid nitrogen until further use.
Collection of lung tissues with different developmental stages.
To determine which miRNAs are expressed highly in alveolarization stage, a critical stage for the development of BPD, we collected lung tissues at different developmental stages for measuring miRNA levels. Pregnant rats were killed and lungs collected at embryonic stages of 16 days (E16), 19 days (E19), and 21 days (E21) or postnatal 0 day (P0), 6 days (P6), 14 days (P14), and 60 days (adult). The day of birth was considered as the postnatal day 0.
Lung morphometric analysis.
Paraformaldehyde-fixed lungs were embedded in paraffin. Four-micrometer-thick sections were made and stained with hematoxylin and eosin for morphometric analysis. Mean alveolar diameter (MAD) and mean alveolar intercept (MLI) were measured to quantify interalveolar distance. Images, devoid of major bronchi and large blood vessels, were captured with a mounted digital camera under a ×10 objective of a Nikon Eclipse E600 microscope. Alveolar diameter was measured as the longest distance between walls of a single alveolus using MetaVue software (Molecular Devices, Sunnyvale, CA). At least 20 alveoli per field were measured, and at least eight fields were counted per lung section. MAD was calculated as the average of alveolar diameters and expressed as relative units. MLI measurement was done as described by another group (2) with some modifications. Using the MetaVue software, we drew five lines across each image: two connecting opposite vertices, two bisecting the opposite sides, and one at a random position. The MLI was calculated by dividing the length of each line by the total number of alveolar intercepts for that line. Twenty-five lines were used per lung to calculate the MLI, and there were at least four lungs per treatment. For measurement of secondary septa, the slides were stained with resorcin-fuchsin solution and counterstained with tartarazine solution. Elastin was stained purple to black and tartarazine provided a yellow background. For quantification, the number of secondary crests per ×20 field was counted and at least five fields were counted per slide and measured as average of four animal lung samples per treatment.
miRNA microarray analysis.
To evaluate the differentially expressed miRNAs during hyperoxia exposure of neonatal rats, we performed miRNA microarray analysis using in-house developed miRNA microarray platform as previously described (34). In brief, total RNA and enriched small RNA were isolated using mirVana miRNA isolation kit (Ambion, Austin, TX). The enriched small RNA (600 ng) was labeled with the NCode miRNA Labeling System (Invitrogen, Carlsbad, CA) and purified with the MinElute PCR Purification Kit (Qiagen, Valencia, CA). Labeled RNA recovered from 120 ng small RNA was used for hybridization. After hybridization, the slides were scanned with ScanArray Express (PerkinElmer Life and Analytical Sciences, Boston, MA), and the images were analyzed with GenePix 5.0 pro (Axon Instruments, Union City, CA). The signals were further analyzed with the software Realspot (11). MiRNAs with an average quality index < 1 were excluded from further analysis. The miRNAs that passed the quality test were then analyzed with the software SAM (Significant Analysis of Microarrays, Stanford University, http://www-stat.stanford.edu/∼tibs/SAM/).
Analysis of miRNAs by quantitative real-time PCR.
The differentially expressed miRNAs were confirmed by quantitative real-time PCR (qPCR) as described (27). The Poly(A) tail was added to total RNA (1 μg) by E. coli Poly(A) Polymerase (Ambion) at 37°C for 1 h. The first-strand cDNA was generated with M-MLV Reverse Transcriptase (Invitrogen) and Poly T adaptor (5′-GCGAGCACAGAAT TAATACGACTCACTATAGGTTTTTTTTTTTTVN-3′, where V = A, G or C, N = A, T, G or C). The first-strand cDNA was diluted 50 times. For quantitative PCR, cDNA was mixed with qPCR Mastermix Plus for SYBR Green I - Low Rox (Eurogentec, San Digeo, CA), the universal reverse primer, and the forward primer specific for each miRNA. The list of primers is given in Table 1. The reactions were incubated at 95°C for 10 min, followed by 40 cycles of 95°C for 15 s and 60°C for 60 s. All PCR reactions were carried out in duplicate. U2 small nucleotide RNA was used as an internal reference. The relative expression of each miRNA was calculated with the equation, 2−(CTmiRNA−CTU2).
Table 1.
Primers used for miRNA quantification by real-time PCR
| Primers | |
|---|---|
| rno-miR-21 | 5′-TAGCTTATCAGACTGATGTTGA-3′ |
| rno-miR-34a | 5′-TGGCAGTGTCTTAGCTGGTTGTT-3′ |
| rno-miR-126* | 5′-CATTATTACTTTTGGTACGCGAA-3′ |
| rno-miR-141 | 5′-TAACACTGTCTGGTAAAGATGG-3′ |
| rno-miR-150 | 5′-TCTCCCAACCCTTGTACCAGTG-3′ |
| rno-miR-151* | 5′-TCGAGGAGCTCACAGTCTAGTA-3′ |
| rno-miR-290 | 5′-CTCAAACTATGGGGGCACTTTTT-3′ |
| rno-miR-335 | 5′-TCAAGAGCAATAACGAAAAATGT-3′ |
| rno-miR-342 | 5′-TCTCACACAGAAATCGCACCCGTCA-3′ |
| U2 | 5′-GTTGGAATAGGAGCTTGCTCCGTCC-3′ |
| Universal reverse primer | 5′-GCGAGCACAGAATTAATACGAC-3′ |
DNA microarray analysis.
We performed DNA microarray analysis on the same samples used for miRNA microarray analysis by an in-house printed 10 K rat oligonucleotide array as previously described (10). Total RNAs from control and hyperoxia-exposed lungs were split into two aliquots; each (10 μg) was labeled with Cy3/Alexa Fluor 546 or Cy5/Alexa 647-specific RT primers through reverse transcription. The primer sequences are 5′-TTC TCG TGT TCC GTT TGT ACT CTA AGG TGG A -T(17)- 3′ for Cy3/Alexa Fluor 546 and 5′-ATT GCC TTG TAA GCG ATG TGA TTC TAT TGG A-T(17)-3′ for Cy5/Alexa Fluor 647. The cDNAs were purified with Microcom YM-30 columns (Millipore, Billerica, MA). Two-step microarray hybridization was performed with the 3DNA 50 Expression kit (Genisphere, Hatfield, PA). The DNA microarray slides were hybridized with paired cDNAs at 42°C for 24 h. After being washed, the slides were rehybridized with Cy3 and Cy5 capture reagents at 42°C for 3 h. Hybridized slides were scanned with ScanArray Express. Data were analyzed using GenePix Pro 5 and SAM.
qPCR analysis of mRNA expression.
Total RNA (1 μg) was reverse-transcribed into cDNA using 200 U of M-MLV reverse transcriptase and a mixture of random primers. All primer sequences (Table 2) were designed using the primer express 3.0 software (Applied Biosystems, Foster City, CA) and confirmed for specificity by using the nonredundant basic local alignment search. The reverse transcribed cDNA was further diluted 100 times and qPCR was done using an ABI prism 7500 system. The reactions were carried out on 96-well plates. 18S rRNA was amplified on the same plates and used to normalize the data. Each sample was prepared in duplicate. The thermal cycling conditions used were: 95°C for 10 min followed by 40 cycles at 95°C for 15 s, 60°C for 60 s. A dissociation curve was generated for each gene to check the specificity of PCR products.
Table 2.
Primers for mRNA quantification by real-time PCR
| Gene Name | Primer Sequences |
|---|---|
| Connective tissue growth factor (CTGF) | forward: ATCAAGACCTGTGCCTGCCATTAC |
| reverse: TGTCCCTTACTCCCTGGCTTTACG | |
| ATPase, Na+/K+ transporting, beta (ATP1B1) | forward: TGTCCTCCAGTGTGCTCCGAAG |
| reverse: CCAGCACATGATGCCTCCAGAG | |
| Brain acid soluble protein (BASP1) | forward: GGCCGGACGACACCAGACAC |
| reverse: CCTTGGCCTTCTCGTCGTTCAC | |
| Phosphoglycerate kinase 1 (PGK1) | forward: ATGGGCTTGGACTGTGGTACTGAG |
| reverse: GCAAAGGCTTCCCATTCAAATACC | |
| Hydroxyprostaglandin dehydrogenase (HPGD) | forward: GAATTCCAGTGTAGCCATGTCCCAG |
| reverse: TTCCTTGATTTGAGGCTGAACTGTG | |
| Glycoprotein nonmetastatic melanoma protein B (GPNMB) | forward: GATGCCAGAAGGAAGATGCCAAC |
| reverse: GTGTTGTCTTCCCAGTCCTCATC |
3′-UTR reporter assay.
3′-UTR encompassing two putative miR-150 binding sites of GPNMB was amplified from rat lung genomic DNA (BioChain Institute). The PCR fragment with XbaI and PspOMI was digested and inserted into the pRL-TK vector containing Renilla luciferase (Promega, Madison, WI). For the construction of mutated GPNMB 3′-UTR, 3 nt mutations were introduced into the putative two miR-150 binding sites by site-directed mutagenesis using overlap PCR. For a dual-luciferase assay, wild-type (WT) and mutated 3′-UTR at the binding site 1 and/or 2 (M1, M2 and M1 + M2) were transfected into HEK 293T or H441 cells. The cells (2 × 104) were seeded in each well of a 96-well plate. After 24 h, 1 ng of 3′-UTR reporter vector was cotransfected into the cells together with 30 ng of pGL-3 with Lipofectamine 2000 (Invitrogen). The pGL-3 expresses a firefly luciferase and was used as a transfection control. Forty-eight hours after transfection, Firefly and Renilla Luciferase activities were measured using Dual-Luciferase Reporter Assay System (Promega) and FLUOstar Optima (BMG Labtech, Durham, NC).
Adenoviral miR-150 vector construction.
pENTR/CMV-EGFP-miR-150 vector was constructed as previously described (5). pENTR/CMV-EGFP that only expresses EGFP was used as a control vector. The insert was switched to an adenoviral vector by the Gateway technique. The adenovirus was amplified and the virus titer was determined in HEK 293A cells.
Western blot.
We separated 20 μg of protein on 12% SDS polyacrylamide gels, transferred it to a nitrocellulose membrane, and blocked it with 5% dry nonfat milk in Tris-buffered saline (TBS) for 1 h. The membranes were incubated with primary antibodies [rabbit anti-glycoprotein nonmetastatic melanoma protein b (GPNMB): 1:200, rabbit anti-β-actin: 1:1,000 and mouse anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH): 1:4,000] overnight at 4°C. After being washed with TBS containing 0.05% Tween 20, the membrane was incubated with corresponding horseradish peroxidase-conjugated secondary antibodies (1:2,000) for 1 h. Finally, the membrane was developed with enhanced chemiluminescence reagents (Amersham Biosciences, Piscataway, NJ) and exposed to X-ray film.
Immunostaining.
This was carried out as previously described (22). We used rabbit anti-Clara cell secretory protein (CCSP; United States Biologicals, Swampscott, MA) and anti-SP-C antibodies at 1:100 dilutions (Santa Cruz Biotechnology, Santa Cruz, CA) to detect Clara cells and alveolar type II cells, respectively. Fluorescent-labeled secondary antibodies, anti-mouse Alexa 488 or anti-rabbit Alexa 555 (Molecular Probes, Eugene, OR) were used at 1: 300 dilutions. The slides were examined under a Nikon Eclipse E600 fluorescence microscope.
RESULTS
Hyperoxia exposure increases alveolar damage and decrease the secondary septa formation in neonatal rat pups.
When rat pups were exposed to 95% hyperoxia for 10 days, the survival rate was 44%. This is consistent with the previously reported survival rates in hyperoxia exposure studies in neonatal rats (32). The survival pups had a greater weight loss and were more sluggish than the controls. The 10-day hyperoxia exposure has been used in literature for gene profiling. Compared with 3-day and 6-day exposure, the neonatal lungs after 10-day exposure have a bigger airway enlargement and greater changes in gene expression (32).
Histopathologically enlarged alveoli with damaged epithelium were observed in hyperoxia-exposed animals (Fig. 1A). There was a decrease in alveolar septa formation in hyperoxia-exposed rat lungs. An unequivocal and consistent difference in the cellularity or thickness of alveolar septa was not discernible. The alveolar size was quantified by measuring MAD and MLI. There were 50 and 40% increases in MAD and MLI in the hyperoxia-exposed lungs compared with the control lungs, respectively (Fig. 1B). Hart's stain for elastin was used to detect secondary crests (Fig. 2A). The formation of secondary crests was significantly reduced in hyperoxia-exposed lungs compared with that in control lungs (Fig. 2B). There was no significant inflammation or signs of prominent fibrosis. All these results demonstrated that the lesions in this model are consistent with these described for “new” BPD pathology.
Fig. 1.
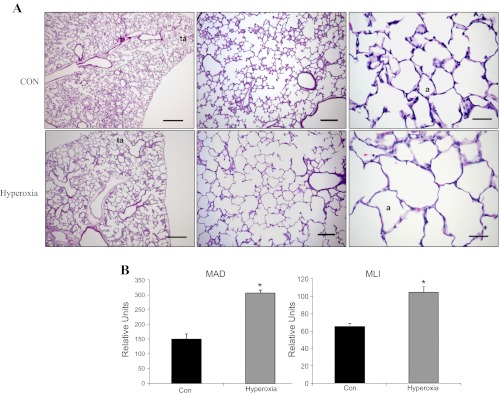
Histological and morphometric analyses of hyperoxia-exposed lungs. A: hematoxylin and eosin staining of postnatal day 3 rat pups exposed to ∼95% oxygen (hyperoxia) or room air (CON) for 10 days. a, alveolus; ta, terminal airway. Scale bars: left panels, 500 μm; middle panels, 150 μm; right panels, 35 μm. B: mean alveolar diameter (MAD) and mean linear intercept (MLI). Alveolar diameter was measured as the longest distance between walls of a single alveolus. MAD was the average of alveolar diameters from at least 20 alveoli of 8 different fields per lung section. MLI was determined by dividing the length of lines across the image field by the total number of alveolar intercepts from 25 lines per lung section. The lengths were measured with MetaVue software, which generates an arbitrary unit rather than an absolute value. Data shown are means ± SE from 4 different rats. *P < 0.005 vs. CON.
Fig. 2.
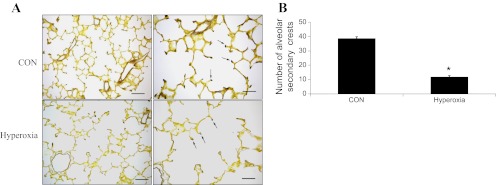
Evaluation of the formation of alveolar secondary crests: alveolar secondary crest formation was evaluated using the Hart's elastin staining. Elastin, which is abundant in the secondary crests, can be visualized as purple stained areas in the sections (arrows). A: elastin staining in CON and hyperoxia-exposed lungs. Scale bars: left panels 75 μm, right panels 35 μm. B: quantification of secondary crests. The number of secondary crests per ×20 field was counted. At least 5 different fields per section were counted. The data are from lung sections of 4 different rats per treatment. Values are expressed as means ± SE from 4 different rats. *P < 0.005 vs. CON.
Identification of differentially expressed miRNAs.
To determine the miRNA expression profile in the neonatal lungs during hyperoxia exposure, we used the miRNA microarray platform developed in our laboratory (34). The miRNA microarray contained probes for 227 miRNAs including 177 rat miRNAs, 5 human miRNAs, 31 mouse miRNAs, and 14 other kinds of RNAs and controls. As shown in Fig. 3, miR-342, miR-126*, miR-335, miR-150, and miR-151* were significantly downregulated while miR-141, miR-21, and miR-34a were upregulated in the hyperoxia-exposed lungs. qPCR verified these microarray data except miR-141, which was not statistically different.
Fig. 3.
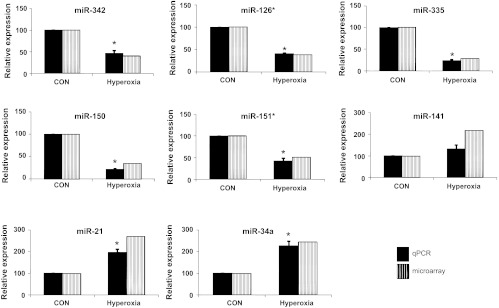
miRNA expression during hyperoxia-exposure: microarray and qPCR were done to determine the expression of various miRNAs that significantly changed in hyperoxia-exposed lungs. Total RNA from CON and hyperoxia-exposed animal lungs was isolated and used for microarray and qPCR. The expression levels of downregulated and upregulated miRNAs in both microarray and qPCR are represented as percentages in reference to control lungs (CON). Values are expressed as means ± SE from 4 different rats. *P < 0.05 vs. CON.
Since alveolarization is the critical stage for the development of BPD, we next examined whether expression levels of the identified miRNAs were high during this stage. For the upregulated miRNAs, all of the three miRNAs (miR-21, miR-141, and miR-34a) were increased during embryonic and early postnatal development and then decreased in adult lungs (Fig. 4A). miR-150 had the biggest change during lung development among the down-regulated miRNAs and exhibited the highest expression P0 and P14, the time frame for alveolar development (Fig. 4B). miR-335 expression showed a gradual increase from E16 to P6. miR-342 and miR-151 had a relatively constant expression during lung development.
Fig. 4.

Developmental regulation of the identified miRNAs during hyperoxia-mediated lung injury. The expression levels of the identified miRNAs during gestational days 16, 19, and 21 (E16, E19, and E21), postnatal days 0, 6, and 14 (P0, P6, and P14) and adult lungs (Ad) were measured by real-time PCR. The results are expressed as the relative expression to E16. Values are means ± SE from 3 rat pups. A: upregulated miRNAs. *P < 0.05 vs. E16 for miR-34a, #P < 0.05 vs. E16 for miR-21. B: downregulated miRNAs. *P < 0.05 vs. E16 for miR-150, #P < 0.05 vs. E16 for miR-335, ^P < 0.05 vs. E16 for miR-151.
Identification of target genes by DNA microarray.
To identify the possible miRNA target genes, we performed DNA microarray analysis to detect mRNA changes using the same samples for miRNA microarray analysis. A total of 5,439 genes passed the quality test in the 10K array. As shown in Table 3, the expression of 42 genes were found to be significantly changed based on the SAM test (q < 0.05) and the fold change (≥2). Among the changed genes, 35 genes were upregulated and six genes were downregulated.
Table 3.
Genes altered in the lungs during hyperoxia exposure according to DNA microarray analysis
| Gene Name | Gene ID | Description | Fold Change |
|---|---|---|---|
| Mt3 | NM_053968 | metallothionein 3 | 4.50 |
| Lcn2 | X13295 | lipocalin 2 | 4.21 |
| C4a | U42719 | complement component 4a | 3.58 |
| Lgals3 | NM_031832 | lectin, galactose binding, soluble 3 | 3.60 |
| C1qb | NM_019262 | complement component 1, q subcomponent, beta polypeptide | 2.71 |
| Gpnmb | AF184983 | glycoprotein (transmembrane) nmb | 3.29 |
| Fabp5 | U13253 | fatty acid binding protein 5, epidermal | 2.38 |
| Ctgf | NM_022266 | connective tissue growth factor | 2.81 |
| Mapk13 | NM_019231 | mitogen activated protein kinase 13 | 2.43 |
| Clca2 | AF077303 | chloride channel calcium activated 2 | 2.43 |
| Atp1b1 | NM_013113 | ATPase, Na+/K+ transporting, beta 1 polypeptide | 2.37 |
| Sod2 | NM_017051 | superoxide dismutase 2, mitochondrial | 2.61 |
| Basp1 | NM_022300 | brain abundant, membrane attached signal protein 1 | 2.25 |
| Retnla | NM_053333 | resistin like alpha | 4.12 |
| Actg2 | NM_012893 | actin, gamma 2 | 2.28 |
| Fetub | NM_053348 | fetuin beta | 2.85 |
| Prkaa2 | NM_023991 | protein kinase, AMP-activated, alpha 2 catalytic subunit | 2.00 |
| RS27 | X59375 | ribosomal protein s27 | 2.44 |
| Lr8 | AF370882 | LR8 protein | 2.00 |
| Des | NM_022531 | desmin | 2.04 |
| Sp4 | NM_012761 | Sp4 transcription factor | 2.01 |
| MT2 | M11794 | metallothionein 2 | 5.02 |
| S100 g | NM_012521 | S100 calcium binding protein G | 2.00 |
| Prdx1 | NM_057114 | peroxiredoxin 1 | 2.00 |
| Sftpd | NM_012878 | surfactant associated protein D | 2.15 |
| Ebf1 | NM_053820 | early B-cell factor 1 | 2.18 |
| Sept3 | AF111180 | septin 3 | 2.04 |
| S100a10 | NM_031114 | S100 calcium binding protein A10 (calpactin) | 2.07 |
| Homer1 | NM_031707_1 | Homer homolog 1 (Drosophila) | 1.96 |
| Gapdh | NM_017008_11 | glyceraldehyde-3-phosphate dehydrogenase | 2.12 |
| Vim | NM_031140 | vimentin | 2.16 |
| TIMP1 | NM_053819_1 | tissue inhibitor of metallopeptidase 1 | 2.16 |
| Pgk1 | NM_053291 | phosphoglycerate kinase 1 | 2.05 |
| IGF1 | M15650 | insulin-like growth factor 1 | 2.00 |
| Ldha | NM_017025 | lactate dehydrogenase A | 2.34 |
| Gstp2 | X02904 | glutathione S-transferase, pi 2 | 2.13 |
| Hpgd | NM_024390 | hydroxyprostaglandin dehydrogenase 15 (NAD) | 0.24 |
| DIDH | S57790 | 3 alpha-hydroxysteroid dehydrogenase | 0.26 |
| E41L1 | NM_021681_1 | hypothetical protein loc59317; rat brain 4.1 s; | 0.41 |
| Tas2r1 | NM_023993 | taste receptor, type 2, member 1 | 0.42 |
| Admr | NM_053302 | adrenomedullin receptor | 0.46 |
| Aoc3 | U72632 | amine oxidase, copper containing 3 | 0.45 |
We then predicted target genes by two steps: 1) we used two software systems, TargetScan (http://www.targetscan.org) and miRanda (http://microrna.org), to see whether the genes identified by DNA microarray (Table 3) have any binding sites for the identified miRNAs (Fig. 3); and 2) we inversely correlated the miRNAs with the genes identified in step 1, i.e., down- or upmiRNAs with up- or downregulated genes. These predictions are listed in Table 4. It is noted that some genes were the same as predicted by two software systems, but some were different. For example, connective tissue growth factor (CTGF) and GPNMB were identified as targets of miR-150 by both software systems. Brain acid-soluble protein 1(BASP1), early B-cell factor 1 (EBF1), and insulin-like growth factor 1 (IGF1) were only predicted as targets of miR-150 by TargetScan, and Fetuin B (Fetub) and desmin (DES) only by miRanda. Also, some genes were targeted by multiple miRNAs. For example, GPNMB were targeted by miR-150 and miR-342-3p.
Table 4.
Inverse correlation of miRNAs and their predicted mRNA targets
| MicroRNAs | TargetScan | miRnada | Genes | |
|---|---|---|---|---|
| downregulated | miR-150 | brain acid soluble protein 1(BASP1) | upregulated | |
| connective tissue growth factor (CTGF) | CTGF | |||
| early B-cell factor 1 (EBF1) | Fetuin B (Fetub) | |||
| insulin-like growth factor 1 (IGF1) | DES | |||
| glycoprotein nonmetastatic melanoma protein B (GPNMB) | GPNMB | |||
| miR-335 | phosphoglycerate kinase 1 (PGK1) | SOD2 | ||
| protein kinase catalytic subunit alpha-2 (PRKAA2) | PRKAA2 | |||
| ATPase, Na+/K+ transporting, beta 1 (ATP1B1) | ATP1B1 | |||
| miR-126* | ATP1B1 | |||
| calcium-activated chloride channel regulator 2 (Clca2) | ||||
| vimentin (Vim) | ||||
| insulin-like growth factor 1 (IGF1) | IGF1 | |||
| miR-151* | PRKAA2 | GPNMB | ||
| resistin-like molecule alpha (Retnla) | ||||
| miR-342-3p | GPNMB | |||
| PRKAA2 | lactate dehydrogenase A (LDHA) | |||
| protein S100 g (S100 g) | S100 calcium binding protein A10 (S100a10) | |||
| superoxide dismutase 2 (SOD2) | SOD2 | |||
| miR-342-5p | CTGF | CTGF | ||
| desmin (DES) | Des | |||
| EBF1 | Vim | |||
| trans-acting transcription factor 4 (SP4) | ||||
| upregulated | miR-21 | hydroxyprostaglandin dehydrogenase (HPGD) | HPGD | downregulated |
| miR-34a | none | none | ||
| miR-141 | HPGD | HPGD |
Among four miRNAs (miR-34a, miR-21, miR-150, and miR-335) that had a high expression in alveolarization stage, none of target genes was identified for miR-34a, and there was only one target for miR-21 (Table 4). However, there were seven targets and six targets for miR-150 and miR-335, respectively. We randomly selected two or three genes in these two groups for real-time PCR verification. As shown in Fig. 5, the mRNA levels of all of the selected genes [GPNMB, CTGF, ATPase, Na+/K+ transporting, beta 1 (ATP1B1), BASP1, phosphoglycerate kinase 1 (PGK1), and hydroxyprostaglandin dehydrogenase (HPGD)], as determined by real-time PCR were consistent with the DNA microarray data.
Fig. 5.
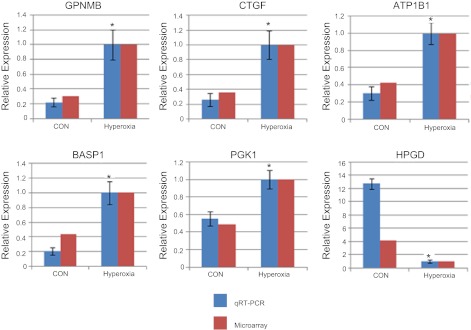
qPCR analysis of the mRNA level of target genes of the identified miRNAs. mRNA levels were determined by qPCR to verify DNA microarray data and normalized to 18S rRNA. The results are expressed as ratios to hyperoxia-exposed lungs. Data shown are means ± SE. *P < 0.05 vs. CON, n = 4.
Experimental verification of GPNMB as a target for miR-150.
The changes in miR-150 and miR-34a during alveolarization stage are both high and significant. However, our computational approach combined with DNA microarray analysis did not identify any targets for miR-34a. Therefore, we selected miR-150 for further analysis. GPNMB was chosen as a target of miR-150 for experimental verification because of our research interests in vascularization since GPNMB plays important roles in angiogenesis (23). Furthermore, TargetScan and miRanda both predict GPNMB as a target. Corresponding to mRNA level, GPNMB protein levels in the lungs were significantly increased in hyperoxia-exposed lungs (Fig. 6A). Furthermore, immunohistochemistry analyses revealed that GPNMB was expressed in bronchiolar epithelial cells, specifically in Clara cells and alveolar epithelial type II cells (Fig. 6B). GPNMB expression was also observed in alveolar macrophages.
Fig. 6.
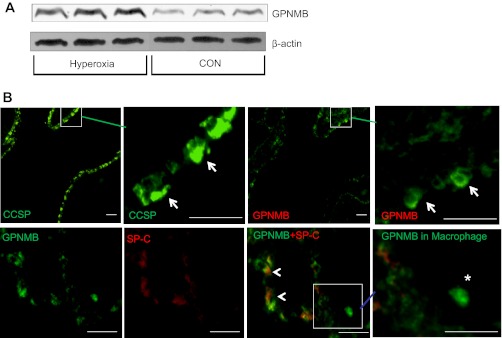
Glycoprotein nonmestastatic melanoma protein b (GPNMB) expression in the lungs. A: Western analysis of GPNMB protein levels in the CON and hyperoxia-exposed lungs. B: immunostaining in the lungs. For colocalization of GPNMB with Clara cell secretory protein (CCSP) (arrows), 2 consecutive lung sections were probed with anti-CCSP and anti-GPNMB antibodies. For colocalization of GPNMB and surfactant protein (SP)-C, double-labeling was performed with anti-SP-C (red) and anti-GPNMB (green) antibodies. GPNMB was also detected in macrophages (*). Arrowheads: overlap of GPNMB and SP-C. Square areas are enlarged in the panels immediately to the right. All scale bars: 40 μm.
We also performed a 3′-UTR reporter assay to determine whether miR-150 binds the 3′-UTR of GPNMB gene. We cloned 3′-UTR region of GPNMB (WT) and its mutations at both binding sites (M1, M2 or their combination, M1 + M2) into the pRL-TK vector containing Renilla Luciferase. When the reporters were transfected into the lung epithelial cells (H441), the mutated 3′-UTR reporter showed a much higher luciferase activity compared with the WT 3′-UTR reporter (Fig. 7). Similar results were observed in HEK 293FT cells (data not shown). The results suggest that endogenous miR-150 inhibits the GPNMB 3′-UTR activity and both binding sites are required for the binding of miR-150.
Fig. 7.

GPNMB 3′-untranslated region (UTR) reporter assay. H441 cells were transfected with wild-type (WT) and mutated at miR-150 binding sites 1 and/or 2 (M1, M2, or M1 + M2) 3′-UTR reporter vectors, and dual luciferase assays were performed. *P < 0.05 vs. WT; n = 3.
We then transduced HEK 293T and H441 cells with an adenoviruses expressing miR-150 to determine whether miR-150 depresses GPNMB expression. miR-150 reduced both mRNA and protein levels of GPNMB in HEK 293T cells and H441 cells (Fig. 8).
Fig. 8.

Effect of miR-150 on GPNMB expression. 293FT and H441 were transduced with an adenovirus expressing miR-150 (Ad-miR-150) or adenovirus control (Ad-Con) at multiplicities of infectivity (MOI) of 5, 20, and 100. A, C: mRNA levels of GPNMB as determined by qRT-PCR, n = 3 independent preparations, each assay performed in duplicate. *P < 0.05 vs. Ad-Con at the same MOI. B, D: Western blot analysis of GPNMB protein levels.
DISCUSSION
Hyperoxia-mediated lung injury is a major cause for pathologic complications in BPD. In the present study, we determined differentially expressed miRNAs in neonatal rat lungs during hyperoxia exposure using an in-house developed miRNA microarray platform. Neonatal rat pups, exposed to ∼95% oxygen for 10 days showed pathologic lesions of impaired alveoli with increased MLI and MAD values and decreased secondary alveolar crests, as described for new BPD (12). We have identified five downregulated miRNAs, including miR-150, miR-342, miR-335, miR-126*, and miR-151*, and two upregulated miRNAs, miR-21 and miR-34a, in response to hyperoxia. Some of the identified miRNAs had high expression during the early postnatal lung development, the stages that are critical for lung morphogenesis in rodents. This suggests that the identified miRNAs may have an association with molecular events in the pathogenesis of BPD.
We found that neonatal rats exposed to ∼95% oxygen from P3 to P13 showed lesions consistent with that described for new BPD. Decrease in number, large, and simplified alveoli with fewer secondary alveolar crests are the hallmark of new BPD. Other lesions such as elastin formation, fibrosis, inflammation, and airway epithelial proliferation vary from moderate to absent in new BPD (12). Although it has widely been used as an experimental model of BPD, hyperoxia exposure of neonatal rodents has its limitations. For example, it does not capture all of the BPD pathological features. Furthermore, clinical BPD is a response to both hyperoxia and mechanical ventilation.
The molecular mechanisms that contribute to both manifestation and progression of BPD are poorly understood. It has been shown that disruption of normal angiogenesis and improper development of capillaries in relation to the alveolar septa might be a major contributing factor toward manifestation of BPD (16). Abnormal signaling mediated by vascular endothelial growth factor (VEGF), fibroblast growth factors (FGF), and insulin like growth factors (IGF) are thought to contribute to BPD (17, 32, 35). All these pathways are major players in controlling proper lung development. Since impaired alveolar formation and subsequent defective development of the respiratory zone are an essential feature of BPD, understanding the molecular mechanisms that control proper septation and alveolarization can provide greater insights into the pathogenesis of BPD. The advent of miRNA as one of the major regulatory components of biological process necessitates the study of its role in BPD pathogenesis. This, in turn, can bring new insights to the finer mechanisms of BPD pathogenesis.
Involvement of miRNAs in lung development was first described using dicer knockout mice, which showed abnormal alveolarization and epithelial branching and apoptosis (15). We and other investigators have reported the involvement of microRNAs during lung development (5, 7, 19, 36). Specifically, miR-17-92 cluster and miR-127 have been shown to influence proliferation and differentiation of lung epithelial cells and branching morphogenesis during development (5, 7, 19). In the current study, we have identified several miRNAs that may have roles in hyperoxia-mediated lung injury.
Among the identified miRNAs, miR-21 was upregulated in BPD while miR-335 was downregulated. It is known that miR-335 is a potent metastasis suppressor, a proliferation inhibitor, and an apoptosis promoter (24, 29). miR-21, on the other hand, is a potent proliferation promoter, tumor promoter, and inhibitor of apoptosis (8, 28). It has been shown that knock-down of miR-21 can lead to the activation of caspases and cause increased cell death (8). More interestingly, it has also been demonstrated that miR-335 knock-down prevents cell death caused by miR-21 suppression. Thus, miR-335 might be a functional antagonist of miR-21 (24). The opposite trend of expression showed by these potentially antagonizing miRNAs in BPD might have some functional significance. Proper development of any organ involves controlled proliferation and apoptosis. Downregulation of miR-335 and concurrent upregulation of miR-21 is suggestive of a proproliferative and antiapoptotic signaling environment in BPD lungs. Sox-4 is an important transcription factor in the regulation of embryonic development and determination of cell fate and has experimentally been verified as a target for miR-335 (29). The verified targets for miR-21 are tropomyocin and BCL-2, both tumor suppressor genes (39).
It is interesting to note that one of the upregulated miRNAs in BPD, miR-34a, is a tumor suppresser. It is antiproliferative and proapoptotic and can induce cell cycle arrest by downregulating CCND1 and CDK6 (9, 30).
miR-342 is another miRNA that is downregulated in BPD. Like miR-335, it is a proapoptotic tumor suppressor whose downregulation induces progression of colorectal cancer (14). miR-342 is located in the introns of the host gene EVL, a member of Ena/Vasp protein family that is important in controlling cytoskeleton remodeling, cell polarity, and cell migration (14). It is a member of a large cluster of imprinted miRNAs expressed only from the maternal chromosome. The cluster is present in a locus that is traditionally known to have genes important in development regulation (25).
miR-126* is also downregulated in BPD. It is located on a region that is deleted in lung cancer (38). Another study has demonstrated the association of miR-126* with abnormal T helper-2 (TH2) lymphocyte responses to inhaled antigens in asthma (21).
miR-150, another downregulated miRNA in BPD, is well studied for its role in B cell development. It regulates c-Myb protein levels through two conserved binding sites in the 3′-UTR of the c-Myb mRNA (37). Ectopic expression of miR-150 results in the downregulation of c-Myb and a reduced B1 cell number. In a recent study, the treatment with pre-miR-150 has been shown to inhibit neovascularization in a mouse model of ischemia (26). They have also shown that miR-150 suppresses VEGF and platelet-derived growth factor-B (PDGF-B). These studies suggest possible link of miR-150 in the pathologic consequences in BPD.
Using DNA microarray analysis and computational approaches, we identified several potential novel targets of the identified miRNAs. BASP-1, CTGF, EBF1, GPNMB, and IGF-1 were identified as targets for miR-150. GPNMB is a potential target not only for miR-150, but also miR-335 and miR-151*. We experimentally verified GPNMB as a target of miR-150 by 3′-UTR reporter assay and endogenous protein expression. GPNMB is a type I transmembrane glycoprotein that is functionally known for its role in differentiation of osteoblasts and osteoclasts in the bone (1). Recent studies have shown that GPNMB promotes tumor growth by promoting angiogenesis and also ameliorates inflammation through induction of autophagy in kidney (18). Among other targets identified, Na+/K+-ATPase is a target for miR-335 and miR-126* and is involved in alveolar fluid clearance (20). SOD2 has been identified as a target for miR-342 and miR-335. Thus, our studies provide a strong basis for the identification of potential miRNA signaling in hyperoxia-mediated lung injury.
As we see the functional implications of the targets of both upregulated and downregulated miRNAs in BPD, there is no clear-cut demarcation that one set has targets that downregulate one process, while the other has targets that exclusively antagonize the same process. Both upregulated and downregulated miRNAs have potential targets that can modulate a variety of processes. The regulatory mechanisms involving miRNAs seem to be much more complex and can fill the whole spectrum, ranging from functional antagonism to a significant degree of agonism.
In summary, we have identified seven miRNAs and their targets that are changed in neonatal lungs after hyperoxia exposure. The results have given us candidate miRNAs that might be important players in modulating BPD pathogenesis.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-083188, HL-087884, and HL-095383 and Oklahoma Center for the Advancement of Science and Technology Grant HR08-064 (L. Liu). Y. Wang was supported by American Heart Association predoctoral fellowship 0810016Z.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: M. Bhaskaran, D.X., and L.L. conception and design of research; M. Bhaskaran, D.X., Y.W., C.H., T.N., W.S., C.Z., X.X., and S.M. performed experiments; M. Bhaskaran, D.X., Y.W., C.H., T.N., W.S., M. Breshears, and L.L. analyzed data; M. Bhaskaran, D.X., Y.W., C.H., T.N., M. Breshears, and L.L. interpreted results of experiments; M. Bhaskaran, D.X., C.H., and T.N. prepared figures; M. Bhaskaran and T.N. drafted manuscript; M. Bhaskaran, D.X., Y.W., C.H., T.N., W.S., C.Z., X.X., S.M., M. Breshears, and L.L. approved final version of manuscript; L.L. edited and revised manuscript.
ACKNOWLEDGMENTS
We thank Dr. Gerry T. M. Wagenaar, Leiden University Medical Center, Netherlands, for suggestions and guidance on establishing the rat model for BPD and Dr. Barry Starcher at University of Texas Health Center at Irving for providing us with the materials and protocol for Hart's elastin staining. The help from Dr. Pradyumna Baviskar, Tingting Weng, and Dr. Reddy Chintagari in experimental procedures is also acknowledged. We also thank Tazia Cook for editorial assistance.
REFERENCES
- 1. Abdelmagid SM, Barbe MF, Rico MC, Salihoglu S, Arango-Hisijara I, Selim AH, Anderson MG, Owen TA, Popoff SN, Safadi FF. Osteoactivin, an anabolic factor that regulates osteoblast differentiation and function. Exp Cell Res 314: 2334–2351, 2008 [DOI] [PubMed] [Google Scholar]
- 2. Ashour K, Shan L, Lee JH, Schlicher W, Wada K, Wada E, Sunday ME. Bombesin inhibits alveolarization and promotes pulmonary fibrosis in newborn mice. Am J Respir Crit Care Med 173: 1377–1385, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, function. Cell 116: 281–297, 2004 [DOI] [PubMed] [Google Scholar]
- 4. Bhandari A, Bhandari V. Pitfalls, problems, and progress in bronchopulmonary dysplasia. Pediatrics 123: 1562–1573, 2009 [DOI] [PubMed] [Google Scholar]
- 5. Bhaskaran M, Wang Y, Zhang H, Weng T, Baviskar P, Guo Y, Gou D, Liu L. MicroRNA-127 modulates fetal lung development. Physiol Genomics 37: 268–278, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bourbon J, Boucherat O, Chailley-Heu B, Delacourt C. Control mechanisms of lung alveolar development and their disorders in bronchopulmonary dysplasia. Pediatr Res 57: 38R–46R, 2005 [DOI] [PubMed] [Google Scholar]
- 7. Carraro G, El-Hashash A, Guidolin D, Tiozzo C, Turcatel G, Young BM, De Langhe SP, Bellusci S, Shi W, Parnigotto PP, Warburton D. miR-17 family of microRNAs controls FGF10-mediated embryonic lung epithelial branching morphogenesis through MAPK14 and STAT3 regulation of E-Cadherin distribution. Dev Biol 333: 238–250, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chan JA, Krichevsky AM, Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Res 65: 6029–6033, 2005 [DOI] [PubMed] [Google Scholar]
- 9. Chang TC, Wentzel EA, Kent OA, Ramachandran K, Mullendore M, Lee KH, Feldmann G, Yamakuchi M, Ferlito M, Lowenstein CJ, Arking DE, Beer MA, Maitra A, Mendell JT. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell 26: 745–752, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen Z, Chen JW, Weng T, Jin N, Liu L. Identification of rat lung-prominent genes by a parallel DNA microarray hybridization. BMC Genomics 7: 47, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Z, Liu L. RealSpot: software validating results from DNA microarray data analysis with spot images. Physiol Genomics 21: 284–291, 2005 [DOI] [PubMed] [Google Scholar]
- 12. Coalson JJ. Pathology of new bronchopulmonary dysplasia. Semin Neonatol 8: 73–81, 2003 [DOI] [PubMed] [Google Scholar]
- 13. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat Rev Genet 10: 704–714, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grady WM, Parkin RK, Mitchell PS, Lee JH, Kim YH, Tsuchiya KD, Washington MK, Paraskeva C, Willson JK, Kaz AM, Kroh EM, Allen A, Fritz BR, Markowitz SD, Tewari M. Epigenetic silencing of the intronic microRNA hsa-miR-342 and its host gene EVL in colorectal cancer. Oncogene 27: 3880–3888, 2008 [DOI] [PubMed] [Google Scholar]
- 15. Harris KS, Zhang Z, McManus MT, Harfe BD, Sun X. Dicer function is essential for lung epithelium morphogenesis. Proc Natl Acad Sci USA 103: 2208–2213, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 279: L600–L607, 2000 [DOI] [PubMed] [Google Scholar]
- 17. Kunig AM, Balasubramaniam V, Markham NE, Morgan D, Montgomery G, Grover TR, Abman SH. Recombinant human VEGF treatment enhances alveolarization after hyperoxic lung injury in neonatal rats. Am J Physiol Lung Cell Mol Physiol 289: L529–L535, 2005 [DOI] [PubMed] [Google Scholar]
- 18. Li B, Castano AP, Hudson TE, Nowlin BT, Lin SL, Bonventre JV, Swanson KD, Duffield JS. The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J 24: 4767–4781, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lu Y, Thomson JM, Wong HY, Hammond SM, Hogan BL. Transgenic over-expression of the microRNA miR-17–92 cluster promotes proliferation and inhibits differentiation of lung epithelial progenitor cells. Dev Biol 310: 442–453, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Machado-Aranda D, Adir Y, Young JL, Briva A, Budinger GR, Yeldandi AV, Sznajder J, Dean DA. Gene transfer of the Na+,K+-ATPase beta1 subunit using electroporation increases lung liquid clearance. Am J Respir Crit Care Med 171: 204–211, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mattes J, Collison A, Plank M, Phipps S, Foster PS. Antagonism of microRNA-126 suppresses the effector function of TH2 cells and the development of allergic airways disease. Proc Natl Acad Sci USA 106: 18704–18709, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Narasaraju T, Chen H, Weng T, Bhaskaran M, Jin N, Chen JW, Chen Z, Chinoy MR, Liu L. Expression profile of IGF system during lung injury and recovery in rats exposed to hyperoxia - a possible role of IGF-1 in alveolar epithelial cell proliferation and differentiation. J Cell Biochem 97: 984–998, 2006 [DOI] [PubMed] [Google Scholar]
- 23. Rose AA, Annis MG, Dong Z, Pepin F, Hallett M, Park M, Siegel PM. ADAM10 releases a soluble form of the GPNMB/Osteoactivin extracellular domain with angiogenic properties. PLoS One 5: e12093, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Sathyan P, Golden HB, Miranda RC. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J Neurosci 27: 8546–8557, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Seitz H, Royo H, Bortolin ML, Lin SP, Ferguson-Smith AC, Cavaille J. A large imprinted microRNA gene cluster at the mouse Dlk1-Gtl2 domain. Genome Res 14: 1741–1748, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shen J, Yang X, Xie B, Chen Y, Swaim M, Hackett SF, Campochiaro PA. MicroRNAs regulate ocular neovascularization. Mol Ther 16: 1208–1216, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shi R, Chiang VL. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 39: 519–525, 2005 [DOI] [PubMed] [Google Scholar]
- 28. Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY. miR-21-mediated tumor growth. Oncogene 26: 2799–2803, 2007 [DOI] [PubMed] [Google Scholar]
- 29. Tavazoie SF, Alarcon C, Oskarsson T, Padua D, Wang Q, Bos PD, Gerald WL, Massague J. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451: 147–152, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tazawa H, Tsuchiya N, Izumiya M, Nakagama H. Tumor-suppressive miR-34a induces senescence-like growth arrest through modulation of the E2F pathway in human colon cancer cells. Proc Natl Acad Sci USA 104: 15472–15477, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thebaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 175: 978–985, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wagenaar GT, ter Horst SA, van Gastelen MA, Leijser LM, Mauad T, van der Velden PA, de Heer E, Hiemstra PS, Poorthuis BJ, Walther FJ. Gene expression profile and histopathology of experimental bronchopulmonary dysplasia induced by prolonged oxidative stress. Free Radic Biol Med 36: 782–801, 2004 [DOI] [PubMed] [Google Scholar]
- 33. Wang Y, Stricker HM, Gou D, Liu L. MicroRNA: past and present. Front Biosci 12: 2316–2329, 2007 [DOI] [PubMed] [Google Scholar]
- 34. Wang Y, Weng T, Gou D, Chen Z, Chintagari NR, Liu L. Identification of rat lung-specific microRNAs by microRNA microarray: valuable discoveries for the facilitation of lung research. BMC Genomics 8: 29, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development 125: 3615–3623, 1998 [DOI] [PubMed] [Google Scholar]
- 36. Williams AE, Moschos SA, Perry MM, Barnes PJ, Lindsay MA. Maternally imprinted microRNAs are differentially expressed during mouse and human lung development. Dev Dyn 236: 572–580, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, Rajewsky N, Bender TP, Rajewsky K. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 131: 146–159, 2007 [DOI] [PubMed] [Google Scholar]
- 38. Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, Yi M, Stephens RM, Okamoto A, Yokota J, Tanaka T, Calin GA, Liu CG, Croce CM, Harris CC. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 9: 189–198, 2006 [DOI] [PubMed] [Google Scholar]
- 39. Zhu S, Si ML, Wu H, Mo YY. MicroRNA-21 targets the tumor suppressor gene tropomyosin 1 (TPM1). J Biol Chem 282: 14328–14336, 2007 [DOI] [PubMed] [Google Scholar]