

Abstract
We used a high-density single-nucleotide polymorphism array to genotype 75 Plasmodium falciparum isolates recently collected from Senegal and The Gambia to search for signals of selection in this malaria endemic region. We found little geographic or temporal stratification of the genetic diversity among the sampled parasites. Through application of the iHS and REHH haplotype-based tests for positive selection, we found evidence of recent selective sweeps at a known drug resistance locus, at several known antigenic loci, and at several genomic regions not previously identified as sites of recent selection. We discuss the value of deep population-specific genomic analyses for identifying selection signals within sampled endemic populations of parasites, which may correspond to local selection pressures such as distinctive therapeutic regimes or mosquito vectors.
Keywords: Plasmodium falciparum, natural selection, SNP, genome, West Africa
Plasmodium falciparum is an obligate protozoan parasite that causes malaria worldwide, killing ∼800,000 annually (WHO 2010). It faces strong selection pressures from host immune responses and from drugs and (potentially) vaccines. Genome-wide scans for natural selection can identify genetic loci responding to these pressures. Most previous scans for selection in P. falciparum have used global collections of parasites and were thus well positioned to identify loci undergoing selection worldwide (Mu et al. 2010; Van Tyne et al. 2011). However, much natural selection occurs at smaller geographic scales, both because selective forces such as drug regimes and insect vectors vary geographically and because selected phenotypes can appear independently in different regions. Detection of localized selection is difficult in global survey but instead requires a deep survey from a single geographic region; the one study similar to this to date (Mu et al. 2007) examined primarily Asian parasites.
To search for selection in a single African parasite population, we collected 75 recent P. falciparum isolates (supplementary table S1, Supplementary Material online) from four locations in Senegal and The Gambia, a region spanning several hundred kilometers. All parasites were hybridized to a high-density Affymetrix single-nucleotide polymorphism (SNP) array with ∼17,000 assays (Van Tyne et al. 2011); after filtering out mixed infections and closely related parasites (which are uninformative for haplotype-based tests of selection), we analyzed genotypes from 70 samples for selective sweeps.
We first checked for population structure within our sampled region. Principal components analysis (fig. 1) showed little structure, whereas FST (which measures population differentiation) indicated a small, statistically significant difference between Senegal and The Gambia (FST = 0.0072, P < 0.0001). This proved to stem from subtle differences between culture-adapted parasites (present only in the Senegal set) and parasites isolated directly from patient blood (see supplementary note and supplementary fig. S1, Supplementary Material online). We found no significant structure in time across the ∼10 years of sample collection. The lack of structure suggests enough gene flow within this region (on a scale of hundreds of kilometers) that sampling for genome-wide association studies need not be finer grained spatially, at least for this sample size. We also measured linkage disequilibrium throughout the genome and found it generally consistent with previous reports (see supplementary note and supplementary figs. S2 and S3 for details and caveats, Supplementary Material online).
Fig. 1.
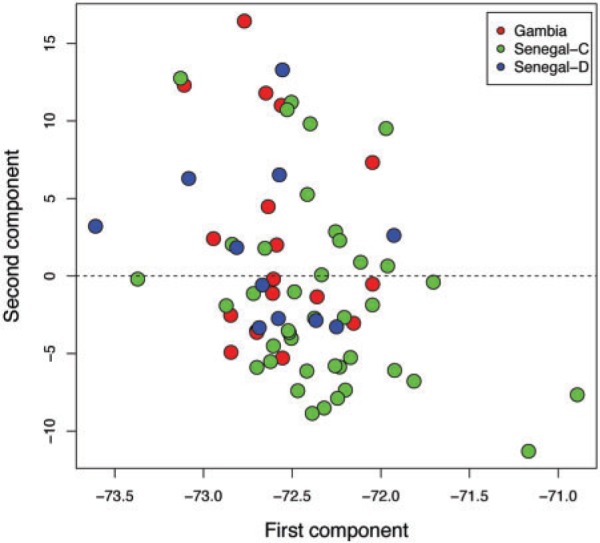
First two principal components of genotype variation. Red: Gambian samples (all directly drawn). Green: culture-adapted Senegal samples. Blue: directly drawn Senegal samples.
We identified possible selective sweeps using two haplotype-based tests for positive selection: relative extended haplotype homozygosity (REHH) (Sabeti et al. 2002) and integrated haplotype score (iHS) (Voight et al. 2006). These tests identify alleles that lie on unusually long haplotypes for that region, indicative of a recent selective sweep. We detected 11 loci with genome-wide significance, including the well-characterized pfcrt locus (Fidock et al. 2000) and a large region on chromosome 6 (fig. 2, table 1; supplementary table S2 has detailed annotation, Supplementary Material online). Five of the loci overlap with previously reported signals (Mu et al. 2010; Van Tyne et al. 2011). The signals of selection appeared consistently in both our directly drawn and our culture-adapted parasite sets (supplementary fig. S4, Supplementary Material online) and were little changed when we restricted our analysis to a 2-year time period (supplementary fig. S5, Supplementary Material online) or used a uniform recombination map. After removing the 11 loci, the remainder of the genome roughly conforms to the null expectation for test scores (fig. 2c and d).
Fig. 2.

Signals of selective sweeps. Significance −log10(P-value) for all SNPs for (a) REHH and (b) iHS; QQ plots (observed vs. expected P values) for (c) REHH and (d) iHS. Panels c and d also show the P value distribution with sweep loci removed (teal). Dashed lines: Bonferroni significance (0.05 level).
Table 1.
Genes in Regions with Signals of Selection.
| Chromosome | Genes | Gene annotation | Test |
|---|---|---|---|
| 2 | PFB0935w1 | Cytoadherence-linked asexual protein 2 | iHS |
| 4 | PFD0260c | Sequestrin | iHS |
| 4 | PFD0735ca | Conserved Plasmodium protein, unknown function | iHS |
| 4 | Intergenic | REHH | |
| 6 | PFF1335c | 4-methyl-5(B-hydroxyethyl)-thiazol monophosphate biosynthesis enzyme | Both |
| PFF1350c | Acetyl-coenzyme a synthetase | ||
| PFF1365c | HECT-domain (ubiquitin transferase), putative | ||
| PDD1460c | Conserved Plasmodium protein, unknown function | ||
| PFF1470c | DNA polymerase epsilon, catalytic subunit a, putative | ||
| 7 | PF07_0035 | Cg1 protein | Both |
| PF07_0036 | Cg6 protein | ||
| PF07_0038a | Cg7 protein | ||
| MAL7PI_28a | pfcrt; chloroquine resistance transporter | ||
| PF07_0042 | Conserved Plasmodium protein, unknown function | ||
| 7 | PF07_0053 | Conserved Plasmodium protein, unknown function | iHS |
| 11 | PF11_0344 | Apical membrane antigen 1 precursor, AMA1 | iHS |
| 13 | PF13_0074 | Surface-associated interspersed gene 13.1 (SURFIN13.1) | iHS |
| 13 | PF13_0201a | Sporozoite surface protein 2 | iHS |
| 14 | PF14_0726 | Conserved Plasmodium protein, unknown function | iHS |
aGenes that are no longer significant if double Bonferroni correction applied.
Senegal and The Gambia share a similar history of drug regimens, except for three years of amodiaquine use in Senegal but not in The Gambia (see supplementary note, Supplementary Material online). Given the role that loci such as pfcrt, with its large selective sweep, play in drug resistance (Fidock et al. 2000), it is reasonable to speculate that some of these novel sweeps also reflect adaptation to drug pressure. Although resistance alleles at loci such as pfcrt, pfmdr1, and dhfr contribute significantly to drug resistance in P. falciparum, it is known that other genes also affect resistance to drugs (Patel et al. 2010) or have undergone compensatory changes to offset the fitness cost of resistance alleles (Jiang et al. 2008).
Contrary to intuition, several of the loci with evidence of directional selection are known antigenic loci, which are thought to be subject primarily to balancing selection; these include ama1, trap/ssp2, clag2, and possibly PF13_0074 and PF14_0726. Similar findings have been noted previously (Mu et al. 2010). Genes under balancing selection typically exhibit short haplotypes, as their SNPs segregate for unusually long periods of time during which recombination breaks down haplotypes. If the selective sweeps do originate in these genes (and not at nearby variants absent from the array), they may reflect nonimmune pressures. Many highly polymorphic antigenic loci in P. falciparum have roles unrelated to immune evasion and presumably fix adaptive mutations related to these functions. For example, clag3 genes, in the same gene family as one of our sweep candidates (clag2), exhibit the high polymorphism typical of genes encoding surface-expressed cytoadherence proteins. Nevertheless, two clag3 genes were recently found to be associated with resistance to antimalarial drugs, suggesting that their products also mediate entry of drugs (Nguitragool et al. 2011).
Alternatively, we may be detecting positive selection at these loci as a variant unfamiliar to the host immune system rises from very low frequency. Previous studies of malaria antigenic genes (ama1 in particular) have shown complex patterns of selection (Cortes et al. 2003; Polley et al. 2003; Gunasekera et al. 2007), with evidence for both positive and balancing selection (Mu et al. 2010); a long-haplotype signal at ama1 has been detected in Asia (Mu et al. 2010). Sequencing-based study of these regions, preferably in multiple geographic regions, should provide better insight into the selective processes at work.
Another unexpected candidate locus codes for thrombospondin-related anonymous protein (TRAP), a conserved protein expressed in P. falciparum sporozoites. It contains extracellular domains associated with hepatocyte invasion (Muller et al. 1993; Wang et al. 2005) and has previously shown evidence of balancing selection (Weedall et al. 2007). TRAP has also been found to mediate the invasion of salivary glands in mosquitoes (Ghosh et al. 2009). It is possible that maintenance of long haplotypes reflects adaptation to divergent ligands in different mosquito species.
Materials and Methods
Plasmodium falciparum isolates were obtained in 2008 from three sites near Banjul in The Gambia and during the period 2001–2009 from three clinics in Senegal—in Thies (100 km from Dakar), in Pikine (10 km from Dakar), and in Vellingara (500 km from Dakar and on the other side of The Gambia). Gambian parasites were taken from clinical samples, whereas Senegal samples included both directly drawn and culture-adapted parasites; see supplementary note for details on DNA extraction, Supplementary Material online. Culture-adapted samples with evidence of multiple infection, based on polymorphism typing at the msp loci (Viriyakosol et al. 1995), were subcloned to isolate a single strain.
SNP calling was as described in Van Tyne et al. (2011): parasite DNA was hybridized to a P. falciparum Affymetrix array containing 74,656 SNP markers and genotypes called using the BRLMM-P algorithm. SNPs were validated by comparing array genotypes with Sanger sequencing genotypes for 17 reference strains (Van Tyne et al. 2011); perfect concordance was required, as was a minimum 80% call rate. Genomic positions and translations are based on the PlasmoDB v5.0 assembly and annotation (PlasmoDB.org). SNPs within var, rifin, or stevor genes were dropped to reduce artifacts from duplicate sequence. After these filters, missing SNP genotypes were imputed for use in the selective sweep search, with 12,885 SNPs successfully imputed.
FST was calculated using the method of Weir and Cockerham (1984); statistical significance was calculated by permuting population labels. iHS was computed according to the method of Voight et al. (2006) and REHH according to the method of Sabeti et al. (2002). Recombination maps for iHS were generated with the software package LDhat (McVean et al. 2002), using the program Interval with a block penalty of 5.0. As both of these tests do not tolerate missing data, SNPs were imputed with PHASE 2.1.1 (Stephens et al. 2001; Stephens and Scheet 2005). A total of 7,486 fully imputed SNPs were polymorphic among the 70 individuals.
REHH and iHS scores were normalized in 20 equal-sized bins of derived allele frequency, with ancestral alleles inferred from a P. reichenowi hybridization (Van Tyne et al. 2011). Standardized iHS and REHH scores were converted to two-tailed P values using a normal distribution. Genome-wide significance was calculated 1) separately without correction for the two tests and 2) jointly across both tests. As the two tests are not independent, a Bonferroni correction for multiple testing is quite conservative. To test whether long haplotype signals could come from differences in sample preparation, iHS scores were calculated separately for direct and culture-adapted samples.
Supplementary Material
Supplementary methods, notes, figures S1–S5, and tables S1 and S2 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
The authors thank colleagues at the MRC Unit in The Gambia including Michael Walther and Omar Janha for support in sample collection and processing. They thank colleagues at the Universite Cheikh Anta Diop and L’hopital Aristide le Dantec for support in sample collection and processing. This study was supported by the Bill and Melinda Gates Foundation, the Ellison Medical Foundation, the ExxonMobil Foundation, NIH Fogarty, the Broad Institute, and NIAID. A.A.-N. and D.J.C. were supported by funding from the UK Medical Research Council. D.J.P. was supported by an NSF Graduate Research Fellowship.
References
- Cortes A, Mellombo M, Mueller I, Benet A, Reeder JC, Anders RF. Geographical structure of diversity and differences between symptomatic and asymptomatic infections for Plasmodium falciparum vaccine candidate AMA1. Infect Immun. 2003;71:1416–1426. doi: 10.1128/IAI.71.3.1416-1426.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock DA, Nomura T, Talley AK, et al. (14 co-authors) Mutations in the P. falciparum digestive vacuole transmembrane protein PfCRT and evidence for their role in chloroquine resistance. Mol Cell. 2000;6:861–871. doi: 10.1016/s1097-2765(05)00077-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh AK, Devenport M, Jethwaney D, Kalume DE, Pandey A, Anderson VE, Sultan AA, Kumar N, Jacobs-Lorena M. Malaria parasite invasion of the mosquito salivary gland requires interaction between the Plasmodium TRAP and the Anopheles saglin proteins. PLoS Pathog. 2009;5:e1000265. doi: 10.1371/journal.ppat.1000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunasekera AM, Wickramarachchi T, Neafsey DE, Ganguli I, Perera L, Premaratne PH, Hartl D, Handunnetti SM, Udagama-Randeniya PV, Wirth DF. Genetic diversity and selection at the Plasmodium vivax apical membrane antigen-1 (PvAMA-1) locus in a Sri Lankan population. Mol Biol Evol. 2007;24:939–947. doi: 10.1093/molbev/msm013. [DOI] [PubMed] [Google Scholar]
- Jiang H, Patel JJ, Yi M, Mu J, Ding J, Stephens R, Cooper RA, Ferdig MT, Su XZ. Genome-wide compensatory changes accompany drug- selected mutations in the Plasmodium falciparum crt gene. PLoS One. 2008;3:e2484. doi: 10.1371/journal.pone.0002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVean G, Awadalla P, Fearnhead P. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics. 2002;160:1231–1241. doi: 10.1093/genetics/160.3.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu J, Awadalla P, Duan J, McGee KM, Keebler J, Seydel K, McVean GA, Su XZ. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat Genet. 2007;39:126–130. doi: 10.1038/ng1924. [DOI] [PubMed] [Google Scholar]
- Mu J, Myers RA, Jiang H, et al. (27 co-authors) Plasmodium falciparum genome-wide scans for positive selection, recombination hot spots and resistance to antimalarial drugs. Nat Genet. 2010;42:268–271. doi: 10.1038/ng.528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HM, Reckmann I, Hollingdale MR, Bujard H, Robson KJ, Crisanti A. Thrombospondin related anonymous protein (TRAP) of Plasmodium falciparum binds specifically to sulfated glycoconjugates and to HepG2 hepatoma cells suggesting a role for this molecule in sporozoite invasion of hepatocytes. EMBO J. 1993;12:2881–2889. doi: 10.1002/j.1460-2075.1993.tb05950.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguitragool W, Bokhari AA, Pillai AD, Rayavara K, Sharma P, Turpin B, Aravind L, Desai SA. Malaria parasite clag3 genes determine channel-mediated nutrient uptake by infected red blood cells. Cell. 2011;145:665–677. doi: 10.1016/j.cell.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JJ, Thacker D, Tan JC, Pleeter P, Checkley L, Gonzales JM, Deng B, Roepe PD, Cooper RA, Ferdig MT. Chloroquine susceptibility and reversibility in a Plasmodium falciparum genetic cross. Mol Microbiol. 2010;78:770–787. doi: 10.1111/j.1365-2958.2010.07366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polley SD, Chokejindachai W, Conway DJ. Allele frequency-based analyses robustly map sequence sites under balancing selection in a malaria vaccine candidate antigen. Genetics. 2003;165:555–561. doi: 10.1093/genetics/165.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, et al. (17 co-authors) Detecting recent positive selection in the human genome from haplotype structure. Nature. 2002;419:832–837. doi: 10.1038/nature01140. [DOI] [PubMed] [Google Scholar]
- Stephens M, Scheet P. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am J Hum Genet. 2005;76:449–462. doi: 10.1086/428594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tyne D, Park DJ, Schaffner SF, et al. (31 co-authors) Identification and functional validation of the novel antimalarial resistance locus PF10_0355 in Plasmodium falciparum. PLoS Genet. 2011;7:e1001383. doi: 10.1371/journal.pgen.1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viriyakosol S, Siripoon N, Petcharapirat C, Petcharapirat P, Jarra W, Thaithong S, Brown KN, Snounou G. Genotyping of Plasmodium falciparum isolates by the polymerase chain reaction and potential uses in epidemiological studies. Bull World Health Organ. 1995;73:85–95. [PMC free article] [PubMed] [Google Scholar]
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Mu J, Li G, Chen P, Guo X, Fu L, Chen L, Su X, Wellems TE. Decreased prevalence of the Plasmodium falciparum chloroquine resistance transporter 76T marker associated with cessation of chloroquine use against P. falciparum malaria in Hainan, People’s Republic of China. Am J Trop Med Hyg. 2005;72:410–414. [PubMed] [Google Scholar]
- Weedall GD, Preston BM, Thomas AW, Sutherland CJ, Conway DJ. Differential evidence of natural selection on two leading sporozoite stage malaria vaccine candidate antigens. Int J Parasitol. 2007;37:77–85. doi: 10.1016/j.ijpara.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-statistics for the analysis of population structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
- WHO. World Health Organization World Malaria Report 2010 [cited 2012 June 15] 2010. Available from: http://www.who.int/malaria/world_malaria_report_2010/en/index.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.