

Abstract
Traditional proteomics analysis is plagued by the use of arbitrary thresholds resulting in large loss of information. We propose here a novel method in proteomics that utilizes all detected proteins. We demonstrate its efficacy in a proteomics screen of 5 and 7 liver cancer patients in the moderate and late stage, respectively. Utilizing biological complexes as a cluster vector, and augmenting it with submodules obtained from partitioning an integrated and cleaned protein–protein interaction network, we calculate a Proteomics Signature Profile (PSP) for each patient based on the hit rates of their reported proteins, in the absence of fold change thresholds, against the cluster vector. Using this, we demonstrated that moderate- and late-stage patients segregate with high confidence. We also discovered a moderate-stage patient who displayed a proteomics profile similar to other poor-stage patients. We identified significant clusters using a modified version of the SNet approach. Comparing our results against the Proteomics Expansion Pipeline (PEP) on which the same patient data was analyzed, we found good correlation. Building on this finding, we report significantly more clusters (176 clusters here compared to 70 in PEP), demonstrating the sensitivity of this approach. Gene Ontology (GO) terms analysis also reveals that the significant clusters are functionally congruent with the liver cancer phenotype. PSP is a powerful and sensitive method for analyzing proteomics profiles even when sample sizes are small. It does not rely on the ratio scores but, rather, whether a protein is detected or not. Although consistency of individual proteins between patients is low, we found the reported proteins tend to hit clusters in a meaningful and informative manner. By extracting this information in the form of a Proteomics Signature Profile, we confirm that this information is conserved and can be used for (1) clustering of patient samples, (2) identification of significant clusters based on real biological complexes, and (3) overcoming consistency and coverage issues prevalent in proteomics data sets.
Keywords: HCC (hepatocellular carcinoma), proteomics, protein networks, liver cancer, bioinformatics, systems biology
Introduction
Proteomics profiling in cancer is a very useful tool for detecting key players in oncogenic progression. Although high-throughput methods such as microarrays and RNA sequencing have been very useful in enhancing our molecular understanding, they only measure RNA level, not protein level. Thus, the evidence provided are indirect. On the other hand, there are many difficulties associated with high-throughput direct protein analysis or proteomics.
Despite improvements in mass spectrometers, protein/peptide separation approaches and MS-associated algorithms, proteomics profiling still suffers from a lack of extensive proteome coverage and consistency across samples and MS assays.1 The coverage issue—that is, the ability to profile the whole proteome—arises in part due to the limited detection range of MS instruments, as well as due to inherent sample complexity. The consistency issue—that is, whether the same results are produced in repeated runs—arises due to the high sensitivity of MS instrument, as well as stochastic ionization and sampling of ions.
The traditional post-MS analysis approach is to select and study only those proteins that are found in most of the samples, as well as having a consistently over- or under-expressed ratio. This approach is referred to as thresholding.
Since the use of thresholds increases stringency by imposing expressional and sample-support constraints, it is expected that this procedure improves data quality, and the investigator can therefore concentrate on analyzing a smaller subset of proteins. However, there are three major problems associated with the thresholding approach. First, it is an arbitrary filtering step with no fixed rules on the parameters. Second, the use of thresholds disregards most of the generated data. This is especially wasteful given already low levels of consistency between different samples. Furthermore, noticeably high or low protein expression does not necessarily imply importance or causality in the phenotype—in particular, a mutated protein that drives other proteins to change their levels may not itself report any change in expression or miss being detected. Third, for those proteins that do meet a specified threshold, there is a tendency to try to determine if the protein is over or under expressed by averaging the reported ratios. Where patient sample size is small, the averaged ratio reveals little information about the expression behavior of the proteins in the cancer population at large. The other point of contention with regards to averaging is that many proteins report “swing” ratios—that is, a mixture of both high and low ratios between samples.
Since proteins do not work singly but in groups as complexes or submodules, we previously built clusters based on the first-degree neighbors of highly differentially expressed proteins (seeds), and ranked them in an approach termed as Proteomics Expansion Pipeline (PEP).2 By building clusters around seeds, PEP allows recovery of lower confidence proteins—that is, proteins supported by only a few patients or that the proteins have ratios around 1. It also allows recovery of proteins not immediately evident from the mass spectra or filtered during the data analysis. The method is further augmented by hypergeometric test of the seeds to discover significant pathways derived from an in-house integrated database3 as well as transition tracing between early- and late-stage cancer.2 However, PEP lacks sensitivity owing to the requirement that clusters must first contain a seed. It is also dependent on the quality of the reference protein–protein interaction network (PPIN). Finally, while PEP addresses the coverage issue in proteomics partially, it does not resolve consistency issues between samples.
In this paper, we take a departure from conventional analysis approaches utilizing biological networks. We hypothesized that meaningful information is embedded in the total set of expressed proteins in every patient if appropriately contextualized. As such, it is possible to do away with the use of thresholds on the detected proteins and maximally utilize available experimental evidence.
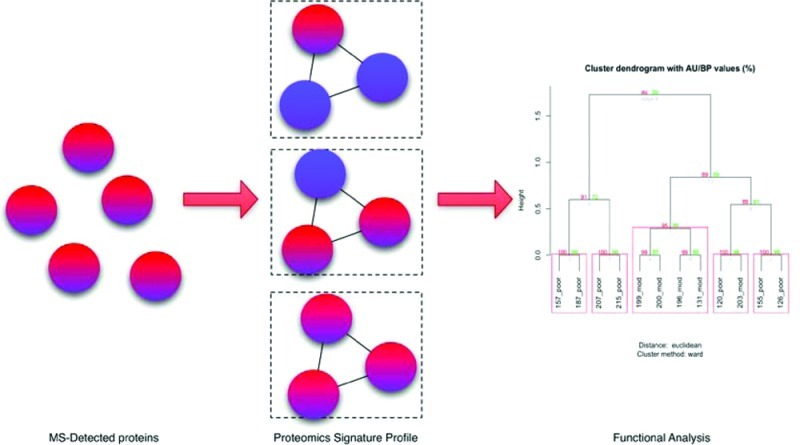
Protein complexes can be regarded as units of biological function and is suitable for contextualizing proteomics data. A given set of complexes can be represented as an unranked “cluster vector” against which we can measure the hit rate of a patient’s reported proteins. For each patient and each cluster, the hit rate = max(Np/N), where Np is the number of proteins in that specific patient found in that cluster, and N is the total number of proteins found in that cluster (Figure 1). The patient’s Proteomics Signature Profile, or PSP, is therefore simply a vector of hit rates checked against the cluster vector. Since a patient’s PSP is a vector of fixed length m, a set of n PSPs can be represented as a matrix of dimensions (n × m) on which statistical and mathematical analysis can be performed.
Figure 1.
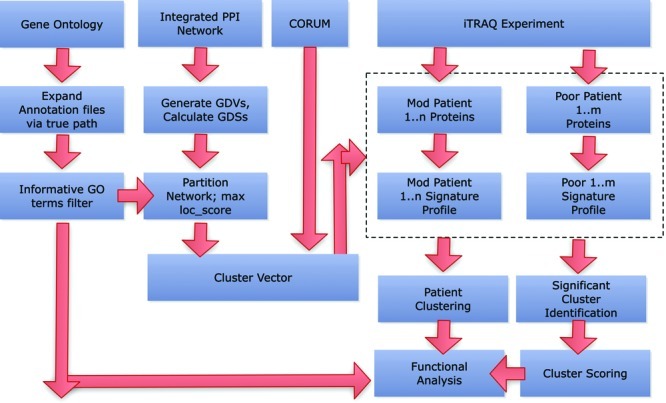
Proteomics signature profiling (PSP) pipeline. The pipeline consists of incorporating data from complex, PPI and GO. Protein lists from individual patients are converted into a proteomics signature profile (PSP) based on a vector of complexes generated from CORUM and graphlet-derived clusters. The PSP can then be used for performing sample clustering for assessing the patient samples and determining significant clusters. GO terms are used to evaluate functional significance and coherence. (Abbreviations: GDV, Graphlet degree vector; GDS, Graphlet Degree Similarity Scores). For detailed explanations, refer to Results.
The PSP can be used in two ways as illustrated in this paper. First, it can be used to understand the relationship between samples or patients. This is important because, normally, the samples are first staged according to clinical and pathological criteria rather than by their molecular profiles. Histopathological classifications may be subjective and may give rise to misclassifications. With PSP, proteomic data can be clustered and analyzed in a manner analogous to microarray data. This allows a confidence check to ensure the molecular signatures also concur with the histopathology. The second way PSP can be used is to determine the significance of each cluster within the cluster vector. This allows for selection of critical gene targets for functional studies and biomarker development. To this end, we adopted and modified the SNet algorithm (previously developed for gene expression studies), which has been shown to be extremely robust in detecting significant subnets.4 The significant clusters can then be further scored and ranked using “clusters scores” derived from the reported expression ratios.
To illustrate the efficacy of our approach, we applied PSP to a group of 12 hepatocellular carcinoma (HCC) patients, 5 of which were clinically diagnosed to be in the moderate (mod) and 7 in the poor stage. If the information embedded within the matrix is indeed meaningful, it should properly segregate patients according to the cancer stage. The returned significant clusters based on feature selection should also make biological sense and support what is currently known about liver cancer. Finally, because thresholding was not performed to filter off any reported proteins, PSP is expected to be more sensitive, therefore capturing a wider array of biological information. To illustrate how PSP compares with a more conventional network-based approach, we also compare it to the results from PEP.2
As a supplement to PSP, we also propose and show in this paper how cluster mining from a PPIN can meaningfully expand the cluster vector. This is important because the current set of known complexes is incomplete. Furthermore, more biological information can be extracted by taking advantage of network-based information.
Materials and Methods
Patient Sample Preparation and Proteomics Profiling
Briefly, liver tissues were obtained from 12 male patients diagnosed with HCC and suffered from cirrhosis with chronic Hepatitis B virus (HBV) infection. There was no reported metastasis at the point of surgery. Tissues collected were grouped according to histology report; 5 had moderately differentiated HCC (mod) and 7 had poorly differentiated HCC (poor). Paired tissues were obtained from each patient, one from the adjacent nontumor region (normal) and the other from the tumor region of the resected liver.
iTRAQ Labeling
Protein lysates from samples were first precipitated using the 2-D Clean-Up kit. The protein pellets were subsequently resuspended in either dissolution buffer (500 mM triethylammonium bicarbonate and 0.1% (w/v) SDS) for iTRAQ labeling. iTRAQ labeling and processing of the samples were carried out as described by the protocol with minor modifications and using the reagents provided from Applied Biosystems. One-hundred micrograms of protein from each sample was reduced with 50 mM of TCEP at 6 °C for 1 h, and subsequently alkylated with 200 mM of methyl methanethiosulfonate (MMTS) for 10 min at room temperature. Each sample was diluted to achieve a final concentration of 0.05% (w/v) SDS prior to trypsinization at 37 °C for 16 h. Following this, each tryptic digest was labeled for 1 h with one of the four isobaric amine-reactive tags. The labeling was carried out at random ensuring that 2 pairs of patient tissues were labeled as follows: Channel 114 (nontumor); Channel 115 (tumor); Channel 116 (nontumor); and Channel 117 (tumor samples). These four iTRAQ-labeled samples were then pooled and passed through a strong cation exchange cartridge as recommended by the manufacturer (Applied Biosystems). This eluate was further desalted using a Sep-Pak cartridge (Millipore), lypholised and reconstituted in appropriate buffers for 2-D LC
Two-Dimensional Liquid Chromatography Separation of Labeled Peptides
iTRAQ-labeled peptide mixtures was further separated using an Ultimate dual-gradient LC system (Dionex-LC Packings) with a Probot MALDI spotting device. A two-dimensional LC separation was performed as follows: the labeled peptide mixture was first dissolved in 2% (v/v) acetonitrile containing 0.05% (v/v) TFA and injected into a 0.3 × 150 mm strong cation-exchange (SCX) column (FUS-15-CP, Poros 10S; Dionex-LC Packings) for the first dimensional separation. The mobile phase A was 5 mM KH2PO4 buffer, pH 3, 5% acetonitrile and mobile phase B 5 mM KH2PO4 buffer, pH 3, 5% ACN + 500 mM KCl respectively. The flow rate was 6 μL/min. A total of 9 fractions were obtained using step gradients of mobile phase B: unbound, 0–5, 5–10, 10–15, 15–20, 20–30, 30–40, 40–50, 50–100% of B. The eluting fractions were captured alternatively onto two 0.3 × 1-mm trap column, washed with 0.05% TFA and followed by gradient elution in a 0.2 × 50-mm reverse-phase column (Monolithic PS-DVB; Dionex-LC Packings). The mobile phase used for this second-dimensional separation was 2% ACN with 0.05% TFA (A) and 80% acetonitrile with 0.04% TFA (B). The gradient elution step was 0–60% B in 15 min at a flow rate of 2.7 μL/min. The LC fractions were mixed directly with MALDI matrix solution (7 mg/mL CHCA and 130 μg/mL ammonium citrate in 75% acetonitrile) at a flow rate of 5.4 μL/min via a 25-nl mixing tee (Upchurch Scientific) before they were spotted onto a 192-well stainless steel MALDI target plate (Applied Biosystems) using a Probot Micro Precision Fraction collector (Dionex-LC Packings), at a speed of 5 s per well. ACTH (50 fmol, 18–39) peptide (m/z = 2465.199) was spiked into each well as internal standard.
Mass Spectrometry Analysis and Database Search
We analyzed samples previously using a 4700 Proteomics Analyzer mass spectrometer (AB SCIEX) with MALDI source and TOF/TOF optics.5,6 Briefly, MS/MS analyses were performed using nitrogen at collision energy of 1 kV and a collision gas pressure of 1 × 10–6 Torr. The GPS Explorer software Ver. 3.6 (AB SCIEX) was used to create and search files with the MASCOT7 (version 2.1; Matrix Science) and Paragon8 (Protein Pilot version 4; AB SCIEX) search engines for peptide and protein identifications. The International Protein Index (IPI) human database (version 3.31) was used for the search and this was restricted to tryptic peptides. One thousand shots were accumulated for each MS spectrum. For MS/MS, 6000 shots were combined for each precursor ion with signal-to-noise (S/N) ratio greater or equal to 100. For precursors with S/N ratio between 50 and 100, 10000 shots were acquired. The resolution used to select the parent ion was 200. No smoothing was applied before peak detection for both MS and MS/MS, and the peaks were deisotoped. For MS/MS, only the peaks from 60 to 20 Da below each precursor mass, and with S/N greater than or equal to 10 were selected. Peak density was limited to 30 peaks per 200 Da, and the maximum number of peaks was set to 125. Cysteine methanethiolation, N-terminal iTRAQ labeling, and iTRAQ labeled-lysine were selected as fixed modifications while methionine oxidation was considered as a variable modification. One missed cleavage was allowed. Precursor error tolerance was set to 100 ppm while MS/MS fragment error tolerance was set to 0.4 Da. Maximum peptide rank was set to 2.
The average iTRAQ ratio and standard deviation (S.D.) were determined using the GPS Explorer software (version 3.6) or Protein Pilot (version 4). The ratio is taken as the tumor sample against adjacent nontumor region. For MS/MS, only the peaks from 50 to 20 Da below each precursor mass, and the minimum S/N filter was designated at 10. The mass exclusion tolerance was 3 Da around 115.5 m/z. Peak density was limited to 50 peaks per 200 Da, and the maximum number of peaks was set to 80.
Peptide Identification and iTRAQ Quantification
For each peptide, a confidence interval (CI%), corresponding to confidence of identification, was calculated using manufacturer’s recommended parameters (Mascot). Each MS/MS spectrum was searched against IPI Human. The CI% was calculated such that a CI% value of 95% is equivalent to a Mascot ion score at the significance value (Supplementary Figure 5, Supporting Information).9,10 The individual peptide identifications were grouped into protein identifications and assigned a total ion CI% by GPS Explorer.
In Goh et al., it was found that the reported proteins for both databases searches (Mascot and Paragon) corresponded well in terms of ratios and ranks.2 Most Mascot hits were also found in Paragon. In addition, Paragon consistently reported more proteins although we found that these were significantly lower ranked. Given that PSP relies on the hit rates of patient proteins against a vector of complexes, the additional proteins may (and indeed as we report in this paper) improve the performance of analysis.
CORUM
CORUM is currently among the most extensive data source for human protein complex data.11 It contains manually annotated protein complexes from mammalian organisms. Annotation includes protein complex function, localization, subunit composition, literature references and more. All information is obtained from individual experiments published in scientific articles; data from high-throughput experiments is excluded.
523 complexes (minimum size 4) were derived from CORUM. A minimum size requirement is introduced to reduce large hit-rate fluctuations.
Generation of Patient Proteomics Signature Profiles (PSP)
Each cluster C in the cluster vector, is a group of proteins derived from CORUM11 and/or the Graphlet-derived clusters. Two sets A and B of proteomic profiles from two phenotypes respectively (mod and poor) are given. For each cluster C, and each patient i in A, the hit rate H_CAi is computed, that is, the overlap between the cluster and patient. Similarly H_CBj is computed for each cluster C and patient j in B. The total set of hit-rates for each patient across the set of clusters is the patient’s PSP.
Identification of Significant Clusters
For each cluster C in the cluster vector, we can produce two lists HA = ⟨H_CA1, ..., H_CAm⟩ and HB = ⟨H_CB1, ..., H_CBn⟩, where A and B correspond to the mod and poor stages. The t-statistic score between the lists HA and HB is then computed by the standard formula:
where
If this t-statistic is significant, then the cluster C is differentially expressed between mod and poor stage. As the t-statistic may not necessarily follow an approximately normal distribution, weighted randomization via class label swapping was performed between members of moderate and poor 10000 times to produce the null distribution. If the t-statistic value is negative, the empirical p-value is determined by the percentage of null-distribution t-scores that are smaller than the actual t-statistic value. If the t-statistic value is positive, the empirical p-value is determined by the percentage of null-distribution t-scores that are larger than the actual t-statistic value. Examples of the null-distribution t-scores for 4 clusters are shown in Supplementary Figure 1 (Supporting Information).
Cluster Score
For those clusters regarded as significant (p ≤ 0.05), we calculate a score for the mod and poor stage respectively using the reported iTRAQ protein ratios. Suppose we have a complex comprised of proteins A, B, C, D. A is supported by 4 mod stage patients with ratio (1.1, 0.8, 1, 1.2), B is supported by 1 patient with ratio of 5, while C and D are not supported.
If the ratio is lower than 1, we convert it by taking its reciprocal. To find out how big is this ratio, we take difference = ratio −1. The score S, would thus be Σ(0.1, 1/0.8 – 1, 0, 0.2) + 4.
However, with this scoring approach, complexes with more proteins tend to be ranked high. For example, a complex with 10 proteins (A1, ..., A10) and patient_i has high ratio value on Ai and low ratio value on the other 9 proteins; this complex will get a higher score than a complex of size 4 with all patients have medium ratio value on all 4 proteins in this complex. To improve the scoring function for such instances, we propose dividing S by the number of unique proteins that were reported in the patient. For example, in complex (A, B, C, D), 2 patients report A - and their scores are 1.1, 1.2 while 1 patient report B and his score is 5. The cluster score is therefore (0.1 + 0.2 + 4)/2 (note that the denominator is not 4).
Gene Ontology and Cluster Functional Annotation
GO provides a controlled vocabulary for assessing cluster function and coherence.12 The annotation files and GO tree (ver. 1.2) files for Homo sapiens were downloaded from http://www.geneontology.org/ (dated 23 April 2011). UniProtKB accessions were mapped to Ensembl Gene IDs via Biomart.
Informative biological process terms were extracted from the GO OBO file.13 Significance testing for each cluster was performed using the hypergeometric test with Bonferroni correction (p ≤ 0.05).
Clustering of Patient Proteomic Signature Profiles
The patient proteomic signature profiles can be used to examine the consistency and confidence of the derived relationships between samples.
Hierarchical clustering was used to understand links between samples. Euclidean distance was used to generate distance matrix based on an m × n matrix (where m are the samples, and n is the patient proteomic signature profile). Ward’s14 was used to evaluate distance between groups derived from the distance matrix.
To gain confidence on the structure of the tree, we used the R bootstrap resampling package pvclust. For each cluster in hierarchical clustering, p-values (between 0 and 1) are calculated via multiscale bootstrap resampling. pvclust provides two types of p-values: AU (Approximately Unbiased) and BP (Bootstrap Probability). AU, which is computed by multiscale bootstrap resampling, is a better approximation to unbiased p-value than BP value computed by normal bootstrap resampling.
Reference PPI Network
As a supplement to CORUM, we used a reference protein interaction network compiled by Bossi and Lerner1 to mine for additional clusters. Briefly, human protein interactions were extracted from 21 different databases. To improve the confidence of an edge, each had to be supported by at least one direct experimental source confirming the physical interaction. The complete network consists of 80922 interactions between 10229 human proteins.
Calculation of Graphlet Degree Similarities from the GDVs
The Graphlet degree vector (GDV) is a generalization of the degree property, that is, the number of connections of any node in a network.15 Graphlets are all possible combinations for subgraphs size 2 to 5. However, some positions in a graphlet are topologically equivalent. For example, the three points in a closed triangle.
To eliminate redundancy, the notion of graphlet orbit is introduced. That is, if the node of interest is involved in a graphlet position of topological equivalence (e.g., one point of a closed triangle), it only counts once. As there are 73 graphlet orbits from size 2 to 5, this can be represented as a vector of length 73, with each position indicating the number of counts the node of interest is found in an orbit.
Relationships between nodes can be established by measuring their GDV similarities. Although the GDVs can be translated into a distance matrix, typical distance measure such as Euclidean or Manhattan cannot be used directly due to dependencies between various orbits.
Milenkovic and Przulj introduced a distance measure for this purpose.15 A 73-dimension vector W containing the weights wi corresponding to orbits i ∈{ 0, ...72 } was defined. To compute weights wi, each orbit i is assigned an integer oi that is obtained by counting the number of orbits that affect orbit i. wi as a function of oi is computed as:
The distance for orbit i between two nodes is computed as:
Total distance between two nodes based on all 73 orbits is simply:
Since the distance is a value between 0 and 1, the similarity can be calculated as:
If two nodes have very similar connection patterns between them, it is plausible they are involved in similar processes. This is checked using functional coherence measures as shown in the next section.
Cluster Generation and Functional Evaluation Using LOC Scores
GDV similarity scores are calculated for all connected nodes in the reference PPIN. Since a high GDV similarity score between two adjoined nodes implies same cluster membership, we partitioned the network into clusters based on establishing a range of thresholds between from 0.8 to 0.9. That is, at threshold 0.8, we keep edges in the PPIN with a score of at least 0.8, and keep connected components of size 4 and above as clusters.
A cluster needs to be biologically relevant. To evaluate if the clusters have “reason” to exist, we measured their localization coherence. Cellular component terms from Gene Ontology12 were extracted and filtered for informative GO localization terms (that is, this term is annotated to at least 30 proteins and none of its descendant terms are annotated to at least 30 proteins).16 We let L = {L1, L2,..., Lk} be a set of localization groups, where each group contains a set of colocalized proteins. The colocalization score of a complex is defined as the maximal fraction of proteins in this complex that are in the same localization group among those proteins with localization annotations.17 It is a measure of functional coherence based on whether the members in a cluster tend to localize to the same functional complex or subcellular component.13 The colocalization score of a set of complexes loc_score (C) is defined as the weighted average score over all complexes c ∈ C:
By using different levels of similarity scores (from 0.6 to 0.95), the PPIN is fragmented into clusters where we monitored the effect on the localization score (loc_score) (Supplementary Figure 3, Supporting Information). Although the best loc_score is observed at similarity score of 0.95, the number of graphlet-derived clusters (minimum size 4) are far too few. We chose to use a similarity score cut off of 0.85 where the number of returned clusters did not drop too drastically and where the loc_score was acceptably high (Supplementary Figure 3, Supporting Information). Seventy-three graphlet-derived clusters were obtained.
Results
PSP clustering reveals strong associations within phenotype classes
Despite high variability and low consistency in reported proteins for each patient (Supplementary Figure 4; Supplementary Tables 1 and 2, Supporting Information), we find that meaningful information can be extracted by contextualizing the reported proteins in each patient using the PSP approach.
PSP was performed using the identified proteins from Mascot and Paragon respectively. In both cases, Paragon and Mascot generated tree structures that are similar. This indicates that PSP produces results that are stable. Also, Paragon consistently outperforms Mascot (Figure 2) due to its higher sensitivity.2 Since hierarchical clustering of patient PSPs is an unsupervised method (i.e., no class label of patients was used), there can be no overfitting with regards to class label of the patients.
Figure 2.

Comparison of bootstrapped HCL trees generated via pvclust. Values on the edges of the clustering are p-values (%). Red values are AU p-values, and green values are BP values as explained early under methods. Clusters with AU larger than 95% are highlighted by red boxes and are very strongly support by the data. With only 73 graphlet-derived clusters, this did not provide sufficient dimensions for clearly resolving the mod and poor patients (left column) although Paragon fared much better because of better hit rates. The right column shows that with the use of a much larger set of dimensions or clusters, in this case, derived from CORUM, the trees are virtually identical despite that Paragon reports a considerably larger number of proteins. It is also noteworthy in all cases; mod patient #203 is clustered with other poor patients.
In all the HCL trees in Figure 2, we noticed that mod patient #203 is consistently linked to poor patient #120. Also, mod patient #203 was always found in the cluster corresponding to the poor patients. Since the poor and mod patient groups were resolved with very high confidence from the bootstrap analysis, mod patient #203 might have been misclassified by the clinician, or has not yet presented the histological phenotype for classification as poor stage. A further check revealed that mod patient #203 reported 667 proteins whereas the other mod patients reported on average ∼300 proteins. Analysis of the histopathology data (reported in 2002) did not reveal any useful or interesting information on mod patient #203. Unfortunately, it was impossible to follow up on whether mod patient #203 developed poor-stage cancer.
Significant Clusters Are Functionally Congruent; Expressionally Silent Clusters May Also Play Key Roles
Using a modified SNet approach,4 we identified 159 CORUM complexes as significant (p-value ≤ 0.05).
To guide functional analysis, that is, whether a cluster has a propensity to show obvious expressional changes, expression information is incorporated to score and rank the significant clusters (see “cluster score” under Methods)
Since the clusters are scored in a similar manner in PEP,2 this facilitates correspondence analysis to check if the best matched clusters score and rank similarly.
Among the top ranking clusters in PSP (Table 1), which also corresponded closely to the top reported clusters in PEP, is a cluster comprising XRCC5, XRCC6, WRN, PARP1 and PRKDC.2 This correspondence further supports that this group of proteins is likely key drivers for HCC progression.
Table 1. Top Ranked Clusters.
| cluster ID | p-value | mod score | poor score | cluster name |
|---|---|---|---|---|
| 5179 | 0.000300541 | 0.513951977 | 3.159758312 | NCOA6-DNA-PK-Ku-PARP1 complex |
| 5235 | 0.000300541 | 0.513951977 | 3.159758312 | WRN-Ku70-Ku80-PARP1 complex |
| 1193 | 0.000300541 | 0.513951977 | 3.159758312 | Rap1 complex |
| 159 | 0 | 0 | 2.810927655 | Condensin I-PARP-1-XRCC1 complex |
| 2657 | 0.008815869 | 0 | 2.55616281 | ESR1-CDK7-CCNH-MNAT1-MTA1-HDAC2 complex |
| 3067 | 0.00911641 | 0 | 2.55616281 | RNA polymerase II complex, incomplete (CDK8 complex), chromatin structure modifying |
| 1226 | 0.013323983 | 0.715352108 | 2.420592827 | H2AX complex I |
| 5176 | 0 | 0.513951977 | 2.339059313 | MGC1-DNA-PKcs-Ku complex |
| 1189 | 0 | 0.513951977 | 2.339059313 | DNA double-strand break end-joining complex |
| 5251 | 0 | 0.513951977 | 2.339059313 | Ku-ORC complex |
| 2766 | 0 | 0.513951977 | 2.339059313 | TERF2-RAP1 complex |
We also ranked the most commonly occurring GO BP terms among all 159 significant clusters (Table 2). The results are congruent with expectations. The top ranking term is “viral reproduction”, which is in line with the fact that all 12 patients are Hepatitis B infected. The subsequent ranking terms corresponding to cellular reproduction—, for example, mitotic cycle, S phase, etc.—is in line with the unregulated division of cancer cells. Interestingly, “base-excision repair”, “telomere maintenance” and “apoptosis” also comes among the top-ranking terms.
Table 2. Top Ranked GO BP Terms Found in Significant Clusters.
| GO ID | description | no. of clusters |
|---|---|---|
| GO:0016032 | viral reproduction | 36 |
| GO:0000398 | nuclear mRNA splicing, via spliceosome | 34 |
| GO:0000278 | mitotic cell cycle | 28 |
| GO:0000084 | S phase of mitotic cell cycle | 28 |
| GO:0006366 | Transcription from RNA polymerase II promoter | 26 |
| GO:0006283 | Transcription-coupled nucleotide-excision repair | 22 |
| GO:0006369 | Termination of RNA polymerase II transcription | 22 |
| GO:0006284 | base-excision repair | 21 |
| GO:0000086 | G2/M transition of mitotic cell cycle | 21 |
| GO:0000079 | regulation of cyclin-dependent protein kinase activity | 20 |
| GO:0010833 | Telomere maintenance via telomere lengthening | 20 |
| GO:0033044 | regulation of chromosome organization | 19 |
| GO:0006200 | ATP catabolic process | 18 |
| GO:0042475 | Odontogenesis of dentine-containing tooth | 18 |
| GO:0034138 | toll-like receptor 3 signaling pathway | 17 |
| GO:0006915 | Apoptosis | 17 |
| GO:0006271 | DNA strand elongation involved in DNA replication | 17 |
| GO:0031145 | anaphase-promoting complex-dependent proteasomal ubiquitin-dependent protein catabolic process | 17 |
| GO:0006261 | DNA-dependent DNA replication | 17 |
| GO:0048015 | phosphatidylinositol-mediated signaling | 16 |
| GO:0006986 | Response to unfolded protein | 16 |
| GO:0000077 | DNA damage checkpoint | 16 |
| GO:0008063 | Toll signaling pathway | 16 |
| GO:0043488 | regulation of mRNA stability | 16 |
| GO:0006338 | chromatin remodeling | 16 |
| GO:0002756 | MyD88-independent toll-like receptor signaling pathway | 16 |
| GO:0000216 | M/G1 transition of mitotic cell cycle | 16 |
| GO:0071103 | DNA conformation change | 16 |
| GO:0000724 | double-strand break repair via homologous recombination | 16 |
| GO:0034142 | toll-like receptor 4 signaling pathway | 16 |
| GO:0010212 | Response to ionizing radiation | 16 |
| GO:0051301 | cell division | 15 |
| GO:0006333 | chromatin assembly or disassembly | 15 |
| GO:0071445 | cellular response to protein stimulus | 15 |
| GO:0002755 | MyD88-dependent toll-like receptor signaling pathway | 14 |
| GO:0043487 | regulation of RNA stability | 14 |
The loss of repair mechanisms gives rise to mutations that fuel cancer progression while telomeres and control over cell death or apoptosis are essential for cellular immortality and continued division. These findings indicate that, even without artificially filtering the proteins by the expression ratios or by consensus from at least half of the patients, the threshold-free PSP approach produced significant clusters that were functionally congruent with GO BP terms expected in cancer.
There were 24 clusters for which the mod and poor cluster scores are both zero, that is, the reported protein ratios for patients in both groups had no change from the normal state. This means the cluster score is zero in both mod and poor samples for these clusters. However, each of these clusters is still significant because proteins in them tend to occur mostly in mod group and not in poor group (or vice versa).
We took the view that significantly differing patterns of expression is important even if the proteins are not necessarily driven to excessively higher or lower levels. Not all mutations function in such a manner. On the other hand, it can also be argued that coverage issues due to small sample size may also introduce many false positives, in which case, it may be more feasible to focus on complexes with either high poor scores, low moderate scores, or a high poor/moderate score ratio.
Given that this group may be potentially interesting, we describe two significant complexes therein with the lowest p-values, the Wave2 complex and the Bloc1-Bloc2 complex. The Wave2 complex plays a role in controlling cellular movement and is implicated in metastasis of liver cells to the colon.18 Bloc1-Bloc2 functions in secretion and lysosomal functions. While this is an associated function of the liver, whether it plays a key role in liver cancer is not clear.
Comparisons with Proteomics Expansion Pipeline (PEP) Approach
In PEP, we obtained a smaller number of clusters at 70. This is hardly surprising because in PEP, clusters were built around seed proteins. That is, proteins which are supported by more than half of patients, and which exhibit fold change ratios above 1.3 and below 0.75.2 With such thresholds in place, (Supplementary Figure 4, Supporting Information), fewer proteins can meet such a stringent requirement.
To understand how well results match between PEP and PSP, we presorted the files in a similar order (that is, in descending order of poor cluster scores, followed by mod cluster scores).
Since the complexes will not match completely, we find the best similarity (max Jaccard score or J score) of at least 10% similarity for each PSP complex to PEP complex. The reason is because PEP clusters are built using the clique percolation method19 where similar or overlapping groups are merged and hence are more likely to be unique.
PEP ranks and PSP ranks obeyed a linear correlation (p-val = 0.00058) and the adjusted fit is acceptable (adjusted R-squared = 0.5) (Figure 3). This imperfect correlation is not unexpected given how methodologically different the two methods are.
Figure 3.
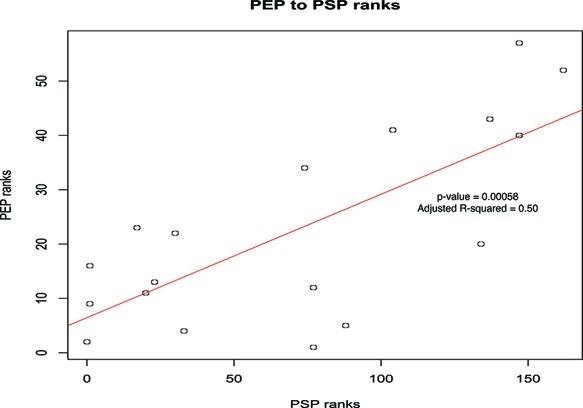
Ranks correlation between PEP and PSP. Although PEP and PSP clusters were derived from very different methods, it can be seen that their results correlate well. To reduce the level of noise, we required a Jaccard score of at least 0.1 (10% similarity).
Table 3 shows the best-matched PEP clusters ranked by the J score. As mentioned earlier, the third best cluster reported in PSP corresponds closely to PEP’s DNA damage cluster comprising of XRCC6, PCNA, PRKDC, WRN, XRCC5 and PARP1. As previously reported, XRCC5/6, and PCNA and PARP1 are repair factors, while WRN is a nuclear protein that could be involved in maintaining genomic stability. PRKDC is a protein kinase that is capable of targeting p53 (and we did find p53 in a larger but lower scoring variant of this cluster), and this was found to be differentially expressed in a majority of poor patients (5 out of 7). The repair factors were all low count, between 1 to 2 patients each. In PEP, we proved that even if patients report different proteins, they are likely to be linked directly to a seed, or situated in the same community as a seed and thus can be recovered by clustering the expanded neighborhood around the seed. In PSP, this was taken to a higher level where we did not consider seeds, but we found that patients expressed proteins that tend to be localized to certain complexes or submodules anyway (similar contexts).
Table 3. - Best matching PEP clusters.
| PEP rank | PSP rank | J score | members (as in PEP) |
|---|---|---|---|
| 41 | 104 | 0.4 | DHX9 SMN1 DDX20 GEMIN4 SMN2 SNRPB SIP1 |
| 23 | 17 | 0.333333333 | COL1A2 CD36 ITGB3 ITGA2B |
| 9 | 1 | 0.25 | XRCC6 PCNA PRKDC WRN XRCC5 PARP1 |
| 20 | 134 | 0.25 | PRKDC XPA RPA1 RPA2 |
| 11 | 20 | 0.222222222 | ACTR2 ACTR3 ARPC4 ARPC5 |
| 40 | 147 | 0.222222222 | PRKCD RAF1MAPK1 PRKCZ PEBP1MAP2K1 |
| 16 | 1 | 0.2 | XRCC6 PCNA PRKDC TP53 WRN NCOA6 XRCC5 PARP1 |
| 34 | 74 | 0.1875 | MAP3K14 CHUK MAP3K7 PEBP1 IKBKB |
| 4 | 33 | 0.142857143 | FUS PTBP1 SFPQ ZMYM2 |
| 5 | 88 | 0.142857143 | YWHAB HSP90AB1 IKBKB MAP3K3 |
| 22 | 30 | 0.142857143 | CANX ITGA6 ITGB1 CD82 |
| 43 | 137 | 0.133333333 | GSN AR CASP3 PXN BCAR1 FYN |
| 1 | 77 | 0.125 | YWHAQ HSPA1A HSPA8 YWHAG |
| 2 | 0 | 0.125 | TP53 NPM1 NCL PARP1 |
| 12 | 77 | 0.125 | SET APEX1 GZMA HMGB2 |
| 13 | 23 | 0.125 | RAN RCC1 XPO1 RANBP3 |
| 52 | 162 | 0.125 | PRKCD EP300 CREBBP KLF5 |
| 57 | 147 | 0.125 | PRKACA RAF1 BAD BCL2 |
| 61 | 74 | 0.117647059 | AKT1 IRS1 PRKCQ CHUK IKBKB |
| 27 | 147 | 0.111111111 | YWHAZ RAF1 CDC25A MAP3K5 YWHAE |
| 31 | 77 | 0.111111111 | HSPA1A BAG1 STUB1 HSPA8 HSF1 |
| 65 | 147 | 0.111111111 | RAF1 AR HSP90AA1MAPK1 NR3C1 |
Other top ranking clusters in PEP were also found highly ranked in PSP. For example, cluster comprising ACTR2, ACTR3, ARPC4 and ARPC5; RAN, RCC1, XPO1 and RANBP3; FUS, PTBP1, SFPQ and ZMYM2 were all found in the top 30 ranks of their corresponding PSP best match.
Using PSP with Predicted Clusters from PPIN
It was unsurprising that the CORUM-derived cluster vector performs better than the use of graphlet-derived clusters (73 clusters) alone as the former contains many more clusters (523 complexes). As the number of graphlet-derived clusters is only 73, it lacks sufficient information on its own to properly group our cancer patients. Indeed, it was found that it was less powerful (lower AU scores) in resolving poor and moderate patients although the tree structure was on the whole still stable (Figure 2, left column).
To investigate if there are significant overlaps between the information captured in CORUM and the graphlet-derived clusters, we checked the Jaccard distance between all clusters in both groups. The Jaccard distance is a measure of similarity between two groups by comparing their intersection divided over the union. At Jaccard threshold 0.9, that is, at least 90% similarity, only one graphlet-derived cluster from was dropped. When we relaxed the Jaccard threshold to 0.6, we still kept about 69 out of 75 graphlet-derived clusters. This suggests that the graphlet-derived clusters are quite distinct from the CORUM complexes and encapsulates a different set of information that is potentially interesting.
Seventeen out of 72 graphlet-derived clusters were identified as significant (24%). This is comparable to 30% from CORUM complexes. This suggests that there might be a slight bias to hit only real complexes. This also brings the total number of significant PSP complexes to 176.
Hence we merged the 72 graphlet-derived clusters (based on 0.9 Jaccard distance threshold) and the 523 CORUM complexes to be compared against the Paragon data set in the final and functional analysis. We find that the HCL tree generated from the merger (Supplementary Figure 2, Supporting Information) is identical to that of Paragon with CORUM clusters (Figure 2 bottom right). However, the approximately unbiased (AU) p-value dropped slightly. This is not surprising since the larger number of dimensions may give rise to more noise but, in turn, increase the number of potential clusters that could be implicated in cancer. CORUM complexes, while extensive, are likely to contain gaps in information that could be supplemented by the graphlet-derived clusters. Hence, for functional downstream analysis, we used Paragon with the cluster vector comprising both CORUM complexes and graphlet-derived clusters.
In any case, PSP can confidently resolve the patient groups even in the event where very few dimensions are available (using only graphlet-derived clusters) although it would be preferable in such a situation to use a database search algorithm that returns more protein hits such as Paragon. This suggests that PSP can be used in instances where only a reference PPI network and very little complex information are available.
Discussion
Here in this study, we describe a novel in silico method to functionally characterize proteome-wide data related to HCC progression by doing without predefined data thresholds on the reported protein list. Early thresholding of MS-generated proteomic data may result in a decreased volume of crucial information for statistical and biological interpretation. To yield potentially useful interpretations, we avoided the use of analysis thresholds at this level, instead opting to generate a PSP using our cluster vector.
Importantly, since PSP does not rely on fold change ratios and patient selection a priori and utilizes several lines of evidence including protein complexes and biological networks, we implicated dysregulated DNA repair and immune-evasion as two important mechanisms in the transition of moderate-stage HCC to poor-stage HCC characterized by poor survival prognosis in our patient set.
Although we are not able to follow up on mod patient #203, the results from PSP revealed fundamental weaknesses in relying solely on histopathological classifications. Compared to clinical staging, for example, TNM Classification and Barcelona-Clinic Liver Cancer (BCLC) staging which is often hampered by surgeon-to-surgeon subjectivity, heterogeneity of HCC (including borderline cases) and poor qualification, molecular signatures are objective, specific to cancer types and our results here demonstrate that PSP can deliver unprecedented characterization of HCC tumors at a medically relevant level.
Although mod patient #203 may be an anomaly, PSP results are stable even when performed using different protein prediction algorithms or when using predicted clusters and real protein complexes. Furthermore, with the exception of mod patient #203 and given that clustering approaches do make errors due to incomplete information, its performance is acceptable for classifying patients by their molecular signatures.
The Use of Both CORUM and Graphlet-derived Clusters in the Cluster Vector
The drop in p-value for the cluster vector comprising CORUM and graphlet-derived clusters is not unexpected. Given the larger number of dimensions included, the more likely that some dimensions might be noise/irrelevant despite controlling for biological coherence of the graphlet-derived clusters via the loc_score. Hence, it may be by sheer chance that some patients (of different phenotypes) scored identically on these irrelevant dimensions, contributing to the slight fall in p-value. It does however justify the use of our SNet-based approach for feature selection, such that analysis is focused only on significant clusters.
A second point is that graphlet-derived clusters are very different from CORUM complexes. As reported, at the minimum Jaccard score of 0.9, only one graphlet-derived cluster was dropped. This did not change significantly as the Jaccard score threshold was lowered to 0.6. Hence, it is more likely that graphlet-derived clusters capture different biological information from CORUM clusters and are therefore potentially interesting.
Fundamental Differences between PSP and PEP and How They Complement Each Other
We showed that the ranked results from PSP correlated well with PEP even though they are extremely different techniques.
PEP is dependent on the quality of the reference PPIN but produces clusters that are generally larger and more likely to contain novel interactions. It requires the initial definition of seeds, which means that it begins with a large loss of data, which is attenuated by the expansion and clustering phase.
PSP only considers whether a protein is detected or not, and uses this information to first establish a vector of hit rates to define the patient’s signature profile. The vector is limited on current knowledge of complexes, in which the exact binding configuration needs not be well-defined. Although there is substantially more knowledge on human complexes, it is by no means complete. To boost the information contained in the cluster vector, CORUM can be supplemented via the partitioning of the reference network into biologically coherent submodules.
We opted to use the graphlets approach in this paper because it has been shown to be effective in identifying clusters based on topological similarity.15 Although it gives rise to a small number of topologically coherent subnets, which may or may not be real, we found that there was only a slight propensity to report real complexes as significant. In the current literature, there exists a multitude of clustering algorithms that could also be utilized to generate a more exhaustive list of predicted clusters. However, in this instance, using one clustering approach is sufficient to demonstrate the potential of this approach in enriching PSP’s cluster vector.
The top-ranked clusters in both PEP and PSP are well matched given the surprisingly good correlations between the ranks of similar clusters. However, we also note that the complexes in PEP and PSP have a generally modest overlap. The best match is only about Jaccard score 0.4. Since PSP reports known and generated complexes whereas PEP identifies novel clusters based around seeds, matched clusters based on similarity should therefore be analyzed closely in a complementary manner. This should be a good way to discover previously unreported or novel cluster members that make for interesting biological interpretations.
Why the PSP Approach Is More Powerful and Sensitive
PSP is a very powerful technique because first, it is not constrained by the use of thresholds on the reported protein list, which is arbitrarily defined by the analyst. Instead, it uses all the information provided from the proteomics screen. It also does not use the average expression ratios of any given protein because that is likely misleading in a small sample set, especially for proteins supported by only two to three patients, or if the protein expression levels swing from low to high in different patients.
Second, it is less reliant on the reference network in which noise levels and false negative levels are not known. Instead, it uses biologically rich data sources such as complexes. It is also expandable to incorporate information from network partitions as we had done with the derivation of graphlet-derived clusters. Although we found that the sole use of graphlet-derived clusters gave poorer result, it was due to the small number of clusters. The results are improved by using a more sensitive protein search algorithm such as Paragon. The third important advantage of PSP is that by generating the signature profiles for each patient, it allows the generation of a matrix on which systematic analysis can be applied. As seen here, we showed that the poor and mod patients segregate well. We also discover a single mod-stage patient who is anomalous, and would skew the analytical results by virtue of the small sample size. PSP’s signature-based methodology will be able detect this. We have also developed a feature selection method on which to identify clusters that are significantly different in the moderate and poor phase.
Possible limitations of PSP
PSP is dependent on the quality and quantity of the cluster vector. As seen under Results, PSP performs relatively poorer in its ability to resolve moderate and poor patients using only the 73 graphlet-derived clusters although performance is greatly improved by using Paragon instead of Mascot. The obvious difference being that Paragon is more sensitive. Our previous analysis showed that Paragon and Mascot correlated well in both ranks and reported ratios for the same proteins.2 In addition, most Mascot proteins are also found in Paragon. The extra proteins reported in Paragon, however, are of lower confidence. That is, their ranks are significantly lower than expected by chance.2
On the other hand, although we used 523 CORUM complexes, there might be some redundancy. Some CORUM complexes are quite similar, with high overlaps with each other (results not shown). However, we elected not to merge these given that the merged clusters would be arbitrary and not reflective of a true biological unit. A second problem with CORUM complexes is that they do not encompass the entire protein network. Although we demonstrate how to improve this shortcoming by supplementing the cluster vector with graphlet-derived clusters, the “representative” cluster vector, encompassing maximal biological information, is probably not attainable.
We do know that the cluster vector derived using CORUM complexes gives very significant resolution in segregating mod and poor patients. Despite veering far from conventional methods, it produced results that are congruent with what is known about liver cancer, and correlates very well with PEP, which is closer to current conventional approaches.
Since we did not use any kind of thresholds to filter proteins in PSP, the effects of false-positive proteins are a legitimate concern. This is especially so on big protein complexes, because a big complex encompasses more proteins and so has a higher likelihood to be hit by false-positive proteins. However, this is mitigated as elaborated below:
The score of a complex with regards to a patient is based on hit rate, which is the percentage of proteins in the complex that got hit and not by an absolute count of the proteins that got hit. Suppose the chance of an individual protein being false positive in any patient (regardless of phenotype) is r%. And suppose a complex has n proteins. Then the expected hit rate of this complex in any patient (regardless of phenotype) due to false-positives is (n × r%)/n = r%. Thus the contribution to the hit rate of a complex with regards to any patient (regardless of his phenotype) by false-positive proteins is “independent” of the size of the complex. It follows that the t-score of a complex is unaffected by false-positive proteins. Since each randomized sample (for obtaining the null distribution for p-value calculations of whether a complex is significant) is obtained by class-label swapping, the hit rate of a complex with respect to this randomized sample also contains the same r% contribution by false-positive proteins. Therefore, the t-score of a complex with respect to randomized samples is also unaffected by false-positive proteins. Thus the r% increase in hit rates of protein complexes due to false-positive proteins is not expected to lead to an increase in false-positive protein complexes. Similarly, since the clustering distance between a pair of PSPs is based on hit rates (and not on which proteins in a complex are hit), the uniform r% contribution by false-positive proteins to the hit rates of complexes does not change this clustering distance. Therefore, the false-positive proteins are not expected to affect the hierarchical clustering of PSPs.
There is, however, an important caveat to the reasoning above. Let u and v respectively be the actual hit rate (i.e., not due to false-positive proteins) of a complex in positive and negative phenotypes. Suppose u + r% and v + r% both exceed 100%. Then the observed hit rates (i.e., due to both true-positive proteins and false-positive proteins) of a complex in positive and negative phenotypes would both be 100%. In such a case, the t-score of this complex is 0, regardless of whether this complex is significant, leading to a loss of sensitivity in our procedure. Similarly, this complex’s contribution to the clustering distance of any pair of PSPs would become 0, causing a loss of resolution in our hierarchical clustering procedure. In general, whenever |u – v| > | min(u + r%, 100%) – min(v + r%, 100%)|, there would be a loss of sensitivity and resolution. Hence a large value of r can have a negative impact on our analysis procedures, especially when the complexes involve have high actual hit rates.
Although theoretically, pathways could also be used as cluster vectors for PSP, there might be many limitations. We expect PSP to work well with small pathways but for pathways that are too large, the extra proteins would confound the significance testing. For example, in gene expression analysis, GSEA often indicates a pathway as insignificant. Yet, when a subnetwork identified using SNet from the same pathways is fed to GSEA, the results become significant. One possible way to get around this is to extract likely subnets from pathways; however, there is no straightforward way to perform this.
Conclusions
We introduce a novel contextualization proteomics approach that does away with thresholding at the protein list level and apply it to a case study on liver cancer. We compared the results to our analytical pipeline PEP and found that the results correlated well. Unlike PEP and other network-based method, PSP can deal with both coverage and consistency issues in proteomics. GO term analysis also indicates that the threshold-free approach select clusters that play integral roles in cancer. The PSP approach revealed many more potential clusters than PEP and is not constrained by any prior arbitrary filtering which is a common first step in conventional analytical approaches.
As PSP only considers whether a protein is present or not in a sample, it also should mean that it could be generalized for multiphase comparisons. For example, mixing data from different cancers and establishing which clusters are differential. This is likely useful for discovering cancer-type-specific complexes from which biomarkers could be derived and developed.
Acknowledgments
W.W.B.G. is supported by a Wellcome Trust Scholarship (83701/Z/07/Z). L.W. is supported in part by a Singapore National Research Foundation grant NRF-G-CRP-2007-04-082(d).
Supporting Information Available
Supplementary figures and tables. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Bossi A.; Lehner B. Tissue specificity and the human protein interaction network. Mol. Syst. Biol. 2009, 5, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goh W. W.; Lee Y. H.; Zubaidah R. M.; Jin J.; Dong D.; Lin Q.; Chung M. C.; Wong L. Network-based pipeline for analyzing MS data: an application toward liver cancer. J. Proteome Res. 2011, 10, 2261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh D.; Dong D.; Guo Y.; Wong L. Consistency, comprehensiveness, and compatibility of pathway databases. BMC Bioinform. 2010, 11, 449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soh D.; Dong D.; Guo Y.; Wong L. Finding Consistent Disease Subnetworks Across Microarray Datasets. BMC Bioinform. 2011, 12(Suppl 13), S15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan H. T.; Tan S.; Lin Q.; Lim T. K.; Hew C. L.; Chung M. C. Quantitative and temporal proteome analysis of butyrate-treated colorectal cancer cells. Mol. Cell. Proteomics 2008, 7(6), 1174–85. [DOI] [PubMed] [Google Scholar]
- Lee Y. H.; Chung M. C.; Lin Q.; Boelsterli U. A. Troglitazone-induced hepatic mitochondrial proteome expression dynamics in heterozygous Sod2(±) mice: two-stage oxidative injury. Toxicol. Appl. Pharmacol. 2008, 231(1), 43–51. [DOI] [PubMed] [Google Scholar]
- Koenig T.; Menze B. H.; Kirchner M.; Monigatti F.; Parker K. C.; Patterson T.; Steen J. J.; Hamprecht F. A.; Steen H. Robust prediction of the MASCOT score for an improved quality assessment in mass spectrometric proteomics. J. Proteome Res. 2008, 7(9), 3708–17. [DOI] [PubMed] [Google Scholar]
- Shilov I. V.; Seymour S. L.; Patel A. A.; Loboda A.; Tang W. H.; Keating S. P.; Hunter C. L.; Nuwaysir L. M.; Schaeffer D. A. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 2007, 6(9), 1638–55. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20(18), 3551–67. [DOI] [PubMed] [Google Scholar]
- Singh A.; Wirtz M.; Parker N.; Hogan M.; Strahler J.; Michailidis G.; Schmidt S.; Vidal-Puig A.; Diano S.; Andrews P.; Brand M. D.; Friedman J. Leptin-mediated changes in hepatic mitochondrial metabolism, structure, and protein levels. Proc. Natl. Acad. Sci. U.S.A. 2009, 106(31), 13100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp A.; Brauner B.; Dunger-Kaltenbach I.; Frishman G.; Montrone C.; Stransky M.; Waegele B.; Schmidt T.; Doudieu O. N.; Stumpflen V.; Mewes H. W. CORUM: the comprehensive resource of mammalian protein complexes. Nucleic Acids Res. 2008, 36(Database issue), D646–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M.; Ball C. A.; Blake J. A.; Botstein D.; Butler H.; Cherry J. M.; Davis A. P.; Dolinski K.; Dwight S. S.; Eppig J. T.; Harris M. A.; Hill D. P.; Issel-Tarver L.; Kasarskis A.; Lewis S.; Matese J. C.; Richardson J. E.; Ringwald M.; Rubin G. M.; Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25(1), 25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W. L.; Tung C. W.; Ho S. W.; Hwang S. F.; Ho S. Y. ProLoc-GO: utilizing informative Gene Ontology terms for sequence-based prediction of protein subcellular localization. BMC Bioinform. 2008, 9, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J. H. Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 1963, 58(301), 236–44. [Google Scholar]
- Milenkovic T.; Przulj N. Uncovering biological network function via graphlet degree signatures. Cancer Inform. 2008, 6, 257–73. [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Kao M. C.; Wong W. H. Transitive functional annotation by shortest-path analysis of gene expression data. Proc. Natl. Acad. Sci. U.S.A. 2002, 99(20), 12783–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G.; Wong L.; Chua H. N. Complex discovery from weighted PPI networks. Bioinformatics 2009, 25(15), 1891–7. [DOI] [PubMed] [Google Scholar]
- Iwaya K.; Oikawa K.; Semba S.; Tsuchiya B.; Mukai Y.; Otsubo T.; Nagao T.; Izumi M.; Kuroda M.; Domoto H.; Mukai K. Correlation between liver metastasis of the colocalization of actin-related protein 2 and 3 complex and WAVE2 in colorectal carcinoma. Cancer Sci. 2007, 98(7), 992–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palla G.; Derenyi I.; Farkas I.; Vicsek T. Uncovering the overlapping community structure of complex networks in nature and society. Nature 2005, 435(7043), 814–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.