

Abstract
Direct chemical modifications of helical peptides have provided a simple and effective means to ‘translate’ the bioactive helical peptides into potential therapeutics targeting intracellular protein-protein interactions. Previously, we have shown that the distance-matching bisaryl cross-linkers can reinforce peptide helices containing two cysteines at the i.i+7 positions and confer cell permeability to the cross-linked peptides. In this work, we report the first crystal structure of a biphenyl cross-linked Noxa peptide in complex with its target Mcl-1 at a 2.0 Å resolution. Guided by this structure, we remodeled the surface of this cross-linked peptide through side chain substitution and N-methylation, and obtained a pair of cross-linked peptides with substantially increased helicity, cell permeability, proteolytic stability, and cell-killing activity in Mcl-1-overexpressing U937 cells. The success of this structure-based design of Mcl-1 inhibitors underscores the value of synergistic use of multifaceted modifications in developing peptide-based therapeutics.
BH3-only proteins are pro-apoptotic factors that induce cell death through selective binding to anti-apoptotic Bcl-2 family proteins.1 In a majority of cancers, the interactions between pro-apoptotic BH3 proteins and anti-apoptotic Bcl-2 family proteins are deregulated because of the elevated expression of some Bcl-2 proteins such as Bcl-2, Bcl-xL, and Mcl-1,2 which not only contributes to cancer progression but also renders cancer cells resistant to chemo- and radiotherapies.3 Therefore, a proven strategy in cancer therapeutic development is to design BH3 mimics as selective Bcl-2 inhibitors.4 To this end, two approaches have been successfully employed: (i) use of small molecules to mimic BH3 peptide side chains involved in binding,5 and (ii) chemically modify BH3 peptides to improve their pharmaceutical properties.6 In the former, a potent small-molecule Bcl-2 inhibitor, ABT-737, was designed;7 ABT-737 binds tightly to Bcl-2 and Bcl-xL with subnanomolar affinity but poorly to Mcl-1.8 In the latter, the chemically modified BH3 peptides containing α/β-amino acid backbones, 9 side chain cross-linking, 10 and main chain-to-side chain cross-linking11 have been reported to show improved cell permeability and/or serum stability.
Mcl-1 is a member of Bcl-2 family that undergoes frequent somatic amplification in multiple cancers and functions as a key driver of cancer cell survival.12 Although small-molecule Bcl-2-selective inhibitors, e.g. ABT-263, have entered clinical trials, these agents generally lack efficacy in tumors with elevated levels of Mcl-1.13 Since a NoxaB-(75-93)-C75A peptide derived from BH3-only Noxa protein binds to Mcl-1 with high affinity and selectivity,14 an attractive approach to develop Mcl-1-selective inhibitors is to optimize the pharmaceutical properties of Noxa BH3 peptide. Recently, we have reported a new dicysteine alkylation-based side chain cross-linking chemistry using a pair of distance-matching bisaryl cross-linkers that lead to reinforced peptide helices and improved cellular uptake.15 Herein, we report the first crystal structure of a biphenyl cross-linked Noxa BH3 peptide in complex with Mcl-1, and subsequent design of a pair of proteolytically stable, cell-permeable, peptide-based Mcl-1 inhibitors by combining structure-based peptide side chain cross-linking with peptide surface remodeling.
To apply our cysteine-mediated cross-linking chemistry to NoxaB-(75-93)-C75A peptide (referred to as Noxa peptide hereafter), we replaced two solvent-exposed i,i+7 residues (Gln-77 and Lys-84) in Noxa with either D- or L-cysteine, and subjected the 19-mer peptide to the 4,4′-bisbromomethyl-biphenyl (Bph)-mediated cross-linking (see Table S1 in Supporting Information for peptide characterizations). The inhibitory activities of the cross-linked peptides were then evaluated by a competitive fluorescence polarization (FP) assay. Compared to parent Noxa peptide, the Bph-cross-linked peptides 1 and 2 showed 65-fold and 12-fold increase in inhibitory activity, respectively (Table 1). To verify that the Mcl-1 targeting selectivity remains intact after cysteine substitution and subsequent side chain cross-linking, the N-terminal fluorescein-conjugated, Bph-cross-linked Noxa peptides, Fl-1 and Fl-2, were prepared and their binding affinities toward Mcl-1 and Bcl-xL were measured by FP assay. Gratifyingly, the modified Noxa peptides showed comparable nanomolar binding affinity toward Mcl-1 (Kd = 4.9 ± 1.5 nM for Fl-1 and 3.4 ± 0.2 nM for Fl-2 vs. 6.7 ± 1.0 nM for Noxa) but no measurable affinity toward Bcl-xL (Kd > 1,000 nM) similar to Noxa, indicating >200-fold selectivity towards Mcl-1. As a control, the BH3 domain of the BH3-only protein Bim showed essentially equal potency toward Mcl-1 and Bcl-xL in the same assay (Table S3). To our surprise, similar to the parent Noxa peptide, the cross-linked Noxa peptides 1 and 2 showed no activity in a cell viability assay in which the Mcl-1-overexpressing U937 cells were treated with 20 μM of the peptides for 48 hours, suggesting that Bph-mediated side chain cross-linking is inefficient to allow sufficient cytosolic transport.
Table 1.
Sequence and biological activities of the native and chemically modified Noxa BH3 peptides.
| Name | Sequencea | Charge | FP assayb Ki (nM) | Cell viabilityc (%) |
|---|---|---|---|---|
| Noxa | AAQLRRIGDKVNLRQKLLN | +4 | 648±128 | 97.6±0.9 |
| 1 | AAC′LRRIGDC′VNLRQKLLNd | +3 | 10±1 | 98.6±4.0 |
| 2 | AAc′LRRIGDC′VNLRQKLLNe | +3 | 54±14 | 100.3±0.2 |
| 3 | AAc′LRAIGDC′VNLRQKLLN | +2 | 23±8 | 85.9±2.2 |
| 4 | AAc′LRAIGDC′VNLAQKLLN | +1 | 28±11 | 72.9±3.2 |
| 5 | AAc′LRAIGDC′VNLAQALLN | 0 | 29±4 | 44.3±0.2 |
| 6 | AmAc′LRRIGDC′VNLRQKLLNf | +3 | 32±3 | 87.3±2.8 |
| 7 | AmAmc′LRRIGDC′VNLRQKLLN | +3 | 22±4 | 80.5±4.7 |
| 8 | AmAmc′LRAIGDC′VNLAQALLNg | 0 | 22±8 | 34.8±0.5 |
Peptides with N-terminal Ala were acetylated while those with N-terminal N-methyl-Ala (Am) were capped with methoxycarbonyl (Moc); all peptides were amidated at the C-termini.
Competitive fluorescence polarization (FP) assay was performed three times to derive average IC50 values along with standard deviations.
Cell viability was measured with ATP assay by treating Mcl-1-overexpressing U937 cells (cultured in RPMI1640 supplemented with 5% FBS) with 20 μM peptides for 48 hours.
C′ denotes Bph-linked L-cysteine.
c′ denotes Bph-linked D-cysteine.
Am = N-methyl-alanine
The structure of 8 is shown as follows:
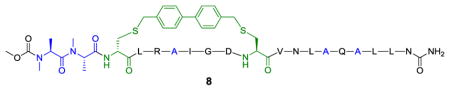
To gain a structural understanding of how the cross-linked Noxa peptide binds to Mcl-1, we solved the crystal structure of mouse Mcl-1 (mMcl-1) in complex with 2 by molecular replacement. The structure was refined to 2.0 Å resolution with Rcryst = 19.2% and Rfree = 23.9% (see Table S2 for crystal data and structural refinement). Overall, the Mcl-1 subunit in the complex is superimposable with mMcl-1 NMR structure 2JM6 with RMSD of 1.2 Å (Figure S2) and rat homolog 2NLA with RMSD of 0.6 Å.14 The bound peptide 2 adopts a helical conformation, with Bph cross-linker projecting 90º away from the deep hydrophobic binding groove (Figure 1a). Compared to 2JM6, most of the interactions between Noxa and mMcl-1 were maintained, including the hydrophobic interactions between the side chains of three canonical resides (Leu-4, Ile-7, and Val-11) of Noxa and their respective hydrophobic binding pockets on mMcl-1 formed partly by Val-216, Val-220, Phe-228, Leu-267, and Phe-270; the salt bridge between Noxa Asp-9 and mMcl-1 Arg-244; the hydrogen bonding between Noxa Asn-19 side chain and mMcl-1 backbone amide oxygen of Phe-299. A few hydrogen bonds are formed only in the mMcl-1:2 complex, i.e. between Asp-9 and mMcl-1 Asn-241 side chains, and between Arg-5 side chain and mMcl-1 His-233 and Val-234 backbone. In addition, Bph cross-linker makes an edge-to-face π-π interaction with His-205 of mMcl-1 (Figure 1a). Collectively, these new interactions may be responsible for the 12-fold increase in inhibitory activity observed in the competitive FP assay. When cross-linked peptide 2 was superimposed with linear Noxa peptide in 2JM6, significant changes in side-chain orientations in 2 were observed for the solvent-exposed, positive-charged residues (Figure 1b). For example, Arg-6 side chain disengages from the salt bridge with Asp-238 of mMcl-1 and re-orients toward the Bph cross-linker; Arg-14 becomes completely solvent exposed as the salt bridge with mMcl-1 Gly-308 carboxyl terminus was not detected; and Lys-16 side chain disengages from the salt bridge with Noxa Met-20 carboxyl terminus due to the Noxa peptide truncation. Taken together, they suggest that these three solvent-exposed, positive-charged residues can be replaced without the loss of binding affinity toward Mcl-1.
Figure 1.

Crystal structure of mouse Mcl-1 in complex with Bph-cross-linked Noxa-BH3 peptide 2. (a) Overall complex structure. The peptide and the side chains of three canonical hydrophobic residues of peptide 2, Leu-4 (h2), Ile-7 (h3) and Val-11 (h4), are colored in deep teal. The two flexible loops, Gly-1733Gly-187 (in the front; shown in dashed line) and Leu-2163Val-224 (in the back; not shown), are disordered in the electron density maps. (b) A stereo view of the superimposition of Bph-cross-linked peptide 2 (yellow-colored stick model) with mNoxa-BH3 peptide (green-colored stick model) as seen in 2JM6. The BH3-binding pocket of mMcl-1 is rendered in surface model.
Since molecules with large polar surface areas generally show poor passive membrane permeation,16 we hypothesized that by substituting the solvent-exposed charged residues with neutral ones, we could substantially improve cell permeability of the cross-linked Noxa peptides, leading to increased cellular activity. Accordingly, we replaced the three non-essential residues (Arg-6, Arg-14, and Lys-16) in 2 with Ala to obtain the cross-linked peptides 3-5, and assessed their inhibitory activities against Mcl-1 using competitive FP assay and their cell-killing activities in U937 cells using ATP assay (Table 1). To our satisfaction, roughly twofold increase in inhibitory activity was observed after Ala-substitution. More importantly, we observed progressive increases in cellular activity as the net charge decreased from +3 to 0, with only 44% U937 cells remained viable after treatment with 20 μM of the charge-neutral cross-linked peptide 5. This implies that the charge-neutral peptide surface facilitates cytosolic transport of the cross-linked peptides, presumably through passive membrane diffusion. While peptide 5 still carry two charged residues, Arg-5 and Asp-9, which contribute to Mcl-1 binding (Figure 1a), it is tempting to speculate that they may form an internal salt bridge in the lipid bilayer during membrane transport because of their favorable i/i+4 geometry.
Encouraged by the initial Arg/Lys-to-Ala substitution results, we sought to further reduce the number of polar groups on peptide surface in order to maximize the passive membrane diffusion. In this regard, a proven modification is backbone N-methylation, especially for the N-H groups that are not involved in the intramolecular hydrogen bonding.17 For the N-acyl-capped helical peptides, the first three N-terminal N-H groups are typically not engaged in the intramolecular hydrogen bonding because of the lack of preceding carbonyl groups one helical turn away. Careful examination of the Mcl-1:2 complex structure revealed that the first two Ala N-H are solvent-exposed while the 3rd residue D-Cys N-H forms a hydrogen bond with the capping acetyl group (H···O distance = 2.20 Å) (Figure S2c). Therefore, we substituted the N-terminal Ala with N-methyl-Ala to generate the cross-linked peptides 6-8, and compared their inhibitory activities to that of their parent peptide (Table 1). We found that addition of two N-methyl groups afforded higher activities both in the FP assay and in the cell viability assay (compare 7 to 2; 8 to 5). In particular, Bph-cross-linked peptide 8 containing two N-methyl groups in addition to three Ala substitutions showed the most robust activity in cell culture: only 35% cells remained viable after treatment with peptide 8 for 48 hours (Table 1). A subsequent concentration-dependent ATP assay with U937 cells for peptide 8 gave rise to an EC50 value of 13.4 μM (Figure S3).
To probe the effect of chemical modifications on the peptide secondary structure, we performed far-UV CD measurement and determined the helicity for cross-linked Noxa peptides 1, 2, 5, 7, and 8 along with the linear Noxa peptide (Figure 2). It is evident that all Bph-cross-linked peptides showed higher helicity than the linear Noxa peptide. Replacing the three positive-charged residues with Ala in the cross-linked peptides led to more than twofold increase in helicity (compare 5 to 2), presumably due to stronger helix propensity of Ala relative to Arg and Lys.18 Addition of two N-methyl groups at the N-terminus appears to destabilize the helix (compare 7 to 2);19 however, the Ala-substituted, cross-linked peptide 8 seems to tolerate N-methylation to some extent (compare 52% helicity for 8 vs. 66% for 5).
Figure 2.

CD spectra of the Bph-cross-linked and linear Noxa peptides and their calculated percent helicity values. The peptides were dissolved in ACN/H2O (1:1) for a final concentration of 50 μM. The percent helicity was calculated based on [θ]222 value.
To confirm that the increased cellular activity was a result of improved cytosolic transport, we prepared the fluorescein-labeled Bph-cross-linked peptides Fluo-2, Fluo-5, and Fluo-8, together with Fluo-Noxa (Table S1). The uptake of these cross-linked and linear peptides into HeLa cells at both 37 °C and 4 °C were analyzed by fluorescence activated cell sorting. We expect that at 4 °C, the energy-dependent active transport processes, e.g. pinocytosis previously reported to be a major membrane permeation pathway for the side chain cross-linked peptides20 will be inhibited, while the passive membrane diffusion will remain unaffected. Not surprisingly, we observed that Bph-mediated cross-linking enhances peptide cellular uptake by 26–40-fold at 37 °C and by 7–33-fold at 4°C (compare Fluo-2, -5, -8 to Fluo-Noxa in Figure 3b, 3c). However, the effect of temperature switch from 37 °C to 4 °C varies; the +3 charged, cross-linked peptide 2 showed 82% reduction whereas the Ala-substituted, charge-neutral cross-linked peptides showed much smaller reduction (48% for 5 and 45% for 8). The dramatic reduction in cellular uptake of Fluo-2 indicates that peptide 2 permeates into cells mainly through the energy-dependent endocytotic process, resulting in endosome trapping. On the other hand, the smaller reductions for 5 and 8 indicate that passive membrane diffusion represents a major pathway for the uptake of 5 and 8 because of their favorable physicochemical properties including neutral charge, reduced number of polar groups on their surfaces, and overall higher helicity. To confirm that the cross-linked peptides 5 and 8 are localized in the cytosol and not bound to the cell membrane, a confocal microscopy experiment was carried out. We found that the fluorescent peptides were predominantly localized in the cytosol (Figure S4).
Figure 3.

Flow cytometry analysis of HeLa cells after treatment with 10 μM fluorescein-labeled peptides, Fluo-Noxa, Fluo-2, Fluo-5, and Fluo-8. (a) Structure of Fluo-8; (b) Representative flow cytometry histogram at 37°C and 4°C; (c) Bar graph showing normalized relative fluorescence: filled bar = 37°C; open bar = 4°C.
One of the key benefits of peptide side chain cross-linking is the improved proteolytic stability. To test this, we selected the most potent Bph-cross-linked peptides 5 and 8 and compared their proteolytic stability to that of the parent Noxa peptide in the presence of chymotrypsin, trypsin, and mouse serum (Figure 4). In all three conditions, the cross-linked peptides 5 and 8 exhibited greatly improved proteolytic stabilities compared to the linear Noxa peptide, 7.2- and 8.7-fold improvement in half-life (t1/2) against chymotrypsin, and 8.9- and 14.8-fold improvement against trypsin, respectively. The higher stability of 5 relative to 8 can be attributed to its higher helicity (66% vs. 52%). The most dramatic effect was seen with mouse serum where the linear Noxa peptide showed a half-life of only 10.5 ± 2.3 min while the cross-linked peptides 5 and 8 showed half-lives of 31.6 ± 2.2 h and 21.2 ± 2.6 h, representing 180- and 121-fold increase in stability, respectively. This prolonged stability may be also partly due to the presence of the hydrophobic biphenyl cross-linker in 5 and 8, which facilitates sequestration/protection of the Bph-cross-linked peptides by serum albumin proteins.21
Figure 4.

Proteolytic stability of the linear and Bph-cross-linked Noxa peptides in the presence of (a) chymotrypsin (ChT), (b) trypsin, and (c) mouse serum. A zoom-in view of the degradation plot for the linear Noxa peptide is shown on the right in (c). The calculated half-life (t1/2) values for the various peptides are given in (d).
In conclusion, we have solved the first crystal structure of a biphenyl-cross-linked peptide in complex with its target Mcl-1. Similar to what was observed in the crystal structures involving the hydrocarbon cross-linkers,22 the biphenyl-cross-linker made an edge-to-face π-π interaction with His-205 of Mcl-1, potentially contributing to the tighter binding. With the structural insights, we then successfully remodeled the surface of the cross-linked peptide through residue substitution and backbone N-methylation, and obtained a pair of cross-linked peptides with greatly increased helicity, cell permeability, proteolytic stability, and cell-killing activity in Mcl-1-overexpressing cancer cells. While side chain cross-linking has become a major strategy for translating bioactive helical peptides into potential therapeutics targeting the intracellular protein-protein interactions,23 the work presented here illustrates the subtlety of each system and highlights the value of complementary peptide modification chemistries, e.g. N-methylation.
Supplementary Material
Acknowledgments
We gratefully acknowledge the Pardee Foundation and the Oishei Foundation (to Q.L.), and the National Institutes of Health (CA82197 to H.G.W.; GM068440 to A.M.G.) for financial support. The crystal structure of Mcl-1 in complex with 2 has been deposited into the Protein Data Bank with access code 4G35.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Crystal data and structure refinement, supplemental figures, experimental procedures, peptide characterization data, and full references 2c, 6 and 12. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Willis SN, Adams JM. Curr Opin Cell Biol. 2005;17:617. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Mol Cell. 2005;17:393. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 2.(a) Vaux DL, Cory S, Adams JM. Nature. 1988;335:440. doi: 10.1038/335440a0. [DOI] [PubMed] [Google Scholar]; (b) Minn AJ, Rudin CM, Boise LH, Thompson CB. Blood. 1995;86:1903. [PubMed] [Google Scholar]; (c) Beroukhim R, et al. Nature. 2010;463:899. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reed JC. Adv Pharmacol. 1997;41:501. doi: 10.1016/s1054-3589(08)61070-4. [DOI] [PubMed] [Google Scholar]
- 4.(a) Lessene G, Czabotar PE, Collman PM. Nat Rev Drug Disc. 2008;7:989. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]; (b) Chonghaile TN, Letai A. Oncogene. 2009;27:S149. [Google Scholar]
- 5.(a) Kutzki O, Park HS, Ernst JT, Orner BP, Yin H, Hamilton AD. J Am Chem Soc. 2002;124:11838. doi: 10.1021/ja026861k. [DOI] [PubMed] [Google Scholar]; (b) Yin H, Lee GI, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang HG, Sebti SM, Hamilton AD. J Am Chem Soc. 2005;127:10191. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- 6.Oltersdorf T, et al. Nature. 2005;435:677. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 7.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DC. Cancer Cell. 2006;10:389. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tahir SK, Wass J, Joseph MK, Devanarayan V, Hessler P, Zhang H, Elmore SW, Kroeger PE, Tse C, Rosenberg SH, Anderson MG. Mol Cancer Ther. 2010;9:545. doi: 10.1158/1535-7163.MCT-09-0651. [DOI] [PubMed] [Google Scholar]
- 9.Horne WS, Boersma MD, Windsor MA, Gellman SH. Angew Chem Int Ed. 2008;47:2853. doi: 10.1002/anie.200705315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. Science. 2004;305:1466. doi: 10.1126/science.1099191. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, Korsmeyer SJ. Mol Cell. 2006;24:199. doi: 10.1016/j.molcel.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 11.Wang D, Liao W, Arora PS. Angew Chem Int Ed. 2005;44:6525. doi: 10.1002/anie.200501603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Inuzuka H, et al. Nature. 2011;471:104. doi: 10.1038/nature09732. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wertz IE, et al. Nature. 2011;471:110. doi: 10.1038/nature09779. [DOI] [PubMed] [Google Scholar]
- 13.(a) Lin X, Morgan-Lappe S, Huang X, Li L, Zakula DM, Vernetti LA, Fesik SW, Shen Y. Oncogene. 2007;26:3972. doi: 10.1038/sj.onc.1210166. [DOI] [PubMed] [Google Scholar]; (b) Yecies D, Carlson NE, Deng J, Letai A. Blood. 2010:3304. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DC, Fairlie WD, Hinds MG, Colman PM. Proc Natl Acad Sci USA. 2007;104:6217. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muppidi A, Wang Z, Li X, Chen J, Lin Q. Chem Commun. 2011;47:9396. doi: 10.1039/c1cc13320a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Refsgaard HH, Jensen BF, Brockhoff PB, Padkjar SB, Guldbrandt M, Christensen MS. J Med Chem. 2005;48:805. doi: 10.1021/jm049661n. [DOI] [PubMed] [Google Scholar]
- 17.(a) Biron E, Chatterjee J, Ovadia O, Langenegger D, Brueggen J, Hoyer D, Schmid HA, Jelinek R, Gilon C, Hoffman A, Kessler H. Angew Chem Int Ed. 2008;47:2595. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]; (b) White TR, Renzelman CM, Rand AC, Rezai T, McEwen CM, Gelev VM, Turner RA, Linington RG, Leung SS, Kalgutkar AS, Bauman JN, Zhang Y, Liras S, Price DA, Mathiowetz AM, Jacobson MP, Lokey RS. Nat Chem Biol. 2011;7:810. doi: 10.1038/nchembio.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pace CN, Scholtz JM. Biophys J. 1998;75:422. doi: 10.1016/s0006-3495(98)77529-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang CF, Zehfus MH. Biopolymers. 1996;40:609. doi: 10.1002/(SICI)1097-0282(1996)40:6%3C609::AID-BIP2%3E3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 20.Bird GH, Bernal F, Pitter K, Walensky LD. Methods Enzymol. 2008;446:369. doi: 10.1016/S0076-6879(08)01622-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Madden MM, Rivera Vera CI, Song W, Lin Q. Chem Commun. 2009:5588. doi: 10.1039/b912094g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.The specific binding of Fluo-8 to homologous bovine serum albumin (BSA) at protein concentrations as low as 25 μg/mL was detected by the direct FP assay; see Figure S5 for details.
- 22.(a) Stewart ML, Fire E, Keating AE, Walensky LD. Nat Chem Biol. 2010;6:595. doi: 10.1038/nchembio.391. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baek S, Kutchukian PS, Verdine GL, Huber R, Holak TA, Lee KW, Popowicz GM. J Am Chem Soc. 2012;134:103. doi: 10.1021/ja2090367. [DOI] [PubMed] [Google Scholar]
- 23.Verdine GL, Walensky LD. Clin Cancer Res. 2007;13:7264. doi: 10.1158/1078-0432.CCR-07-2184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.