

Abstract
Cilia are necessary for sonic hedgehog (Shh) signaling, which is required to pattern the neural tube. We know that ventral neural cell fates are defined by a specific cohort of transcription factors that are induced by distinct thresholds of Shh activity mediated by opposing gradients of Gli activator (GliA) and Gli repressor (GliR). Despite this understanding, the role of Shh as an instructive morphogen is viewed as increasingly complex, with current models integrating positive inputs in terms of ligand concentration and time, along with negative feedback via the downstream gene regulatory network. To investigate the relative contributions of the positive and negative inputs from Shh signaling in neural patterning, we took advantage of a protein that uncouples the regulation of GliA and GliR: the cilia protein ADP-ribosylation factor-like 13b (Arl13b). By deleting Arl13b in mouse, we induced low-level constitutive GliA function at specific developmental stages and defined a crucial period prior to E10.5 when shifts in the level of GliA cause cells to change their fate. Strikingly, we found that improperly patterned cells in these mice converted to the wild-type pattern by E12.5. We further showed that the recovery of patterning did not occur when we also deleted Gli3, the primary GliR in the neural tube, revealing a crucial role of Gli3 in the maintenance of neural patterning.
Keywords: Arl13b, Cilia, Neural tube patterning, Sonic hedgehog, Mouse
INTRODUCTION
Cells in the ventral neural tube interpret Shh signaling levels over time to specify five different ventral cell fates (Briscoe et al., 2000; Briscoe et al., 1999; Chiang et al., 1996; Ericson et al., 1997a; Ericson et al., 1997b). Continuing study of the mechanism by which Shh patterns the neural tube has revealed increasing levels of complexity in this system. The initial conception was that Shh acts as a diffusible morphogen, whereby its activity is defined by its concentration at a given cell, which is a function of the distance of the cell from the morphogen source (Briscoe et al., 2000; Briscoe et al., 1999; Ericson et al., 1997a; Ericson et al., 1997b). However, as the tissue being patterned was simultaneously growing, the reliance of this model on distance was difficult to reconcile with the stable pattern observed. Hence, the current model incorporates the duration of Shh signaling in addition to concentration (Dessaud et al., 2010; Dessaud et al., 2007; Ribes et al., 2010). In this temporal adaptation model, cells become progressively less sensitive to Shh ligand as it induces expression of the negative regulator of the pathway Patched1 (Ptch1) (Chen and Struhl, 1996; Goodrich et al., 1997; Jeong and McMahon, 2005; Marigo and Tabin, 1996). This negative-feedback loop demands that cells be stimulated with higher concentrations of Shh over a longer period of time to achieve the highest Shh response. This implies that Shh signaling must be continuous and, indeed, when Shh signaling is not maintained, ventral neural cell fates are lost (Dessaud et al., 2010). Recent data show that the downstream gene regulatory network (GRN), induced by Shh activity, is also crucial for the ultimate cell fate decision in the neural tube, as it helps create a transcriptional circuit that can reinforce the ultimate fate of a cell (Balaskas et al., 2012). This transcriptional circuit insulates each cell from the normal fluctuations in Shh activity so that reliable patterning occurs.
Vertebrate hedgehog signaling requires the primary cilium, and components of the pathway are localized to cilia (Corbit et al., 2005; Haycraft et al., 2005; Huangfu et al., 2003; Rohatgi et al., 2007). Ptch1 is a Shh receptor and is localized to the cilium in the absence of Shh, whereas Smoothened (Smo) enters the cilium upon Shh stimulation (Corbit et al., 2005; Marigo and Tabin, 1996; Rohatgi et al., 2007). Gli2 and Gli3 proteins mediate the transcriptional response to Shh signaling and are processed to either an activator form (GliA) in the presence of Shh, or to a repressor form (GliR) without Shh ligand (Aza-Blanc et al., 2000; Ruiz i Altaba, 1998). Normally, the relative localization of Ptch1 and Smo shifts upon Shh stimulation, permitting Gli proteins to be enriched in cilia (Chen et al., 2009; Haycraft et al., 2005; Rohatgi et al., 2007; Tukachinsky et al., 2010).
Mouse mutants that lack specific intraflagellar transport (IFT) proteins do not have cilia, which results in an absence of ventral neural cell fates owing to a lack of both GliR and GliA function (Houde et al., 2006; Huangfu et al., 2003; Liu et al., 2005; May et al., 2005; Tran et al., 2008). By contrast, we have found that mouse mutants lacking the ciliary protein Arl13b [called Arl13bhennin(hnn)] exhibit a constitutive low-level of Shh activity owing to loss of modulation of Gli2 activator. This defect in GliA function corresponds to the specification of progenitors of motoneurons (pMN cells) through most of the neural tube. However, in contrast to other mouse mutants that disrupt cilia, we found via both biochemical and genetic analyses that Gli3 repressor activity was unaffected in Arl13bhnn mutants (Caspary et al., 2007). Arl13bhnn mutants possess abnormal cilia in which components of Shh signaling are not regulated properly: Ptch1 and Smo localize to cilia regardless of Shh stimulation, and there is no Gli enrichment in cilia upon Shh stimulation (Larkins et al., 2011). This is consistent with the constitutive activation of Shh signaling in the Arl13bhnn mutant neural tube being Shh ligand independent (Caspary et al., 2007).
Here, we use a targeted conditional null allele of Arl13b to temporally control Shh signaling activity and experimentally uncouple GliA from GliR function in the mouse neural tube. Our analysis supplies in vivo evidence that cells in the mouse neural tube are sensitive to shifts in the level of functional GliA prior to E10.5. In contrast to the complete ablation of Shh signaling after the establishment of morphogen gradient, when ventral cell fates are initially specified and then lost (Dessaud et al., 2010), we show that mispatterned cells experiencing constitutive low levels of Shh signaling are able to correct their fates if the cells are initially exposed to a normal Shh gradient. As this phenotype contrasts with the Arl13bhnn germline null phenotype, our data define the timeframe during which Shh acts via GliA as an instructive morphogen. Strikingly, we found the recovery of patterning is Gli3 dependent, indicating GliR plays a crucial role in sustaining normal neural tube patterning.
MATERIALS AND METHODS
Mouse strains
All mouse work was performed under the approved guidelines of the Emory University IACUC. All mice were maintained on a C3H/HeJ background. Analysis was performed only after lines were crossed to C3H/HeJ for at least three generations. Mouse strains used were: Brn4-Cre [Tg (Pou3f4) 32Cren; from B. Crenshaw, Philadelphia, PA, USA], CAGG-CreER® (JAX 004682), EIIa-Cre (JAX 003724), Ptch1-lacZ (D allele; from M. P. Scott, Stanford, CA, USA) and conditional Gli3 allele (JAX 008873). Genotyping was performed as previously described (Ahn et al., 2001; Goodrich et al., 1997; Hayashi and McMahon, 2002; Lakso et al., 1996).
Generation of conditional Arl13b allele
Three fragments of Arl13b containing exon 1, 2 and 3 were amplified from the bMQ55p06 (the Wellcome Trust Sanger Institute) and cloned into pFlexible (van der Weyden et al., 2005). Exon 2 of Arl13b was flanked with LoxP sites, so exon 2 could be deleted upon Cre recombinase (supplementary material Fig. S1A). We screened 288 ES cell clones and identified 18 clones by Southern blotting that had undergone homologous recombination. Primers for genotyping the Arl13bfloxed allele (forward, AGGACGGTTGAGAACCACTG; reverse, AAGGCCAGCTTGGGTTATTT) amplify products of 526 bp, 679 bp and 109 bp for the wild-type, targeted and deleted alleles, respectively.
Phenotypic analysis
Immunofluorescence, RNA in situ hybridization, X-Gal staining and BrdU injection were carried out as previously described (Belo et al., 1997; Dubois et al., 2006; Horner and Caspary, 2011; Yamada et al., 1991). Primary antibodies used were rabbit anti-Olig2 (Chemicon AB9610, 1:300); mouse anti-FoxA2, -HB9, -Nkx2.2, -Nkx6.1 and -Shh (Developmental Studies Hybridoma Bank, 1:10); mouse anti-Cre (Sigma C7988, 1:500); rabbit anti-Smoothened (from K. V. Anderson, New York, NY, USA; 1:500); mouse anti-acetylated α-tubulin (Sigma T6793, 1:2500); rabbit anti-Arl13b (serum, 1:1500) (Caspary et al., 2007); mouse anti-BrdU (Roche 11770376001, 1:100); and rabbit anti-phospho-histone H3 (Millipore 06-570, 1:1000). Ptch1 probe was used for in situ hybridization (Goodrich et al., 1996). Control embryos used were: Cre-positive Arl13bfloxed/+, Cre-negative Arl13bfloxed/floxed or Cre-negative Arl13bfloxed/hnn. Images were taken on a Leica DM6000B upright fluorescence microscope and processed using the SimplePCI program. Confocal images were taken on a Zeiss LSM510 META confocal at 63× with optical zoom and were processed by LSM Image browser.
Mouse embryonic fibroblasts (MEFs)
MEFs were isolated from E12.5 control Arl13bfloxed/+ and Arl13bΔCAGG-Cre embryos, and grown on gelatin-coated plates. At confluence, MEFs were split into a six-well plate containing gelatin-coated cover slips at a density of 800,000 cells/well. MEFs were then serum-starved for 24 hours and tamoxifen (2 μm, Sigma H7904) was added in serum-free media, 10% FBS-containing media or Shh-conditioned media (Larkins et al., 2011). Cover slips were collected at 24, 36, 42 and 48 hours, and MEFs were fixed and processed for antibody staining.
Quantitative real-time PCR
MEFs were isolated from E12.5 wild-type and Arl13bhnn embryos, and grown on gelatin-coated 10 cm plates. At confluence, MEFs were treated with either control 0.5% FBS-containing media or Shh-conditioned media (Larkins et al., 2011) for 24 hours. Whole RNA was extracted from MEFs using an RNeasy Mini Kit (Qiagen 74104) and QiaShredder columns (Qiagen 79654) according to manufacturer's instructions. Purified RNA was quantified using a NanoDrop ND-1000 spectrophotometer and reverse transcribed using the SuperScript III First-Strand Synthesis System (Invitrogen 18080) according to manufacturer's instructions. Quantitative real-time PCR was performed using LightCycler 480 SYBR Green I Master (Roche 04707516001) in a BioRad CFX96 Real-Time PCR detection system. The following primers were used (5′-3′): Ptch1 (CCCTAACAAAAATTCAACCAAACCT and GCATATACTTCCTGGATAAACCTTGAC); Gli1 (GCCACACAAGTGCACGTTTG and AAGGTGCGTCTTGAGGTTTTCA); Gapdh (CGTCCCGTAGACAAAATGGT and GAATTTGCCGTGAGTGGAGT) (Han et al., 2009). Each reaction was performed in triplicate. Ptch1 and Gli1 values were normalized to Gapdh within each sample. Statistical significance was evaluated by applying a two-factor ANOVA.
Tamoxifen injection
Tamoxifen (Sigma T5648) was dissolved to 10 mg/ml in 100% ethanol. For Arl13bΔE9.25 and Arl13bΔE9.5 analysis, 0.1 mg tamoxifen per 1 g body weight was dissolved into 300 μl of corn oil (Sigma C8267) by using a vacuum centrifuge at medium speed for 15 minutes. For Arl13bΔE10.5, 0.15 mg tamoxifen per 1 g body weight was administrated. Pregnant females were injected intraperitoneally once either at noon or 6 pm of the day indicated.
Western blotting
E12.5 embryos were homogenized and protein concentration was determined by Bradford assay (BioRad 500-0006). Protein (50 μg) was separated on a 10% SDS-PAGE gel and transferred onto a nitrocellulose membrane (GE Healthcare RPN203D). The membrane was incubated with affinity-purified anti-Arl13b (1:1000) (Caspary et al., 2007) and anti-actin (Sigma A5060, 1:1000), then processed for enhanced chemiluminescence.
Quantitative analysis
Cells were counted by using ImageJ software, and the three color channels were separated, so Olig2-, HB9- or Hoechst-staining cells could be counted in their own channel. Two sections were counted for one E12.5 embryo. Counts of Olig2- and HB9-positive cells were normalized to the total number of cells in the neural tube. Three E12.5 embryos were counted for each genotype to obtain an average, and the error bars represent the s.d. Significant difference from normalized Olig2 or HB9 average of mutants compared with control was calculated by Student's t-test. Arl13b-staining cilia in MEFs were counted using point selections in ImageJ software, and ten random pictures were taken from each condition to count the percentage of Arl13b-expressing cilia in total cell numbers as determined by Hoechst staining.
RESULTS
Modulation of Shh activity level and regulation of Smo localization
To alter Shh activity temporally in the neural tube during development without perturbing GliR, we generated a conditional null Arl13b allele (supplementary material Fig. S1). When we induced deletion in the germline, the conditional allele recapitulated the Arl13bhnn phenotype, indicating the same constitutive low-level Shh activity as in Arl13bhnn (supplementary material Fig. S1C-F). By combining a ubiquitous, tamoxifen-inducible Cre line, CAGG-CreER®, with the Arl13bfloxed allele and injecting the pregnant dams with tamoxifen, we controlled the timing of Arl13b deletion in the embryos (Hayashi and McMahon, 2002) (supplementary material Fig. S1I,J). Via immunofluorescence, we saw a reduction of Arl13b starting 24 hours post-injection and a complete loss of Arl13b expression at 42 hours post-injection (supplementary material Fig. S2).
We previously showed that the expansion of pMN cells in the neural tube of Arl13bhnn embryos is independent of Shh ligand, as Shh Arl13bhnn double mutants resemble Arl13bhnn single mutants (Caspary et al., 2007). However, it is unclear whether cells lacking Arl13b can increase their level of signaling if exposed to Shh ligand. To address this, we performed quantitative real-time PCR in wild-type and null Arl13bhnn mouse embryonic fibroblasts (MEFs) to detect transcriptional levels of two Shh target genes: Gli1 and Ptch1. In wild-type MEFs, we found Gli1 and Ptch1 transcription were induced after 24 hours of Shh stimulation (Fig. 1A). In Arl13bhnn MEFs, however, Gli1 and Ptch1 transcription remained at baseline levels regardless of Shh stimulation, indicating that, consistent with the in vivo phenotype, Arl13bhnn MEFs were not responsive to Shh in vitro (Fig. 1A).
Fig. 1.
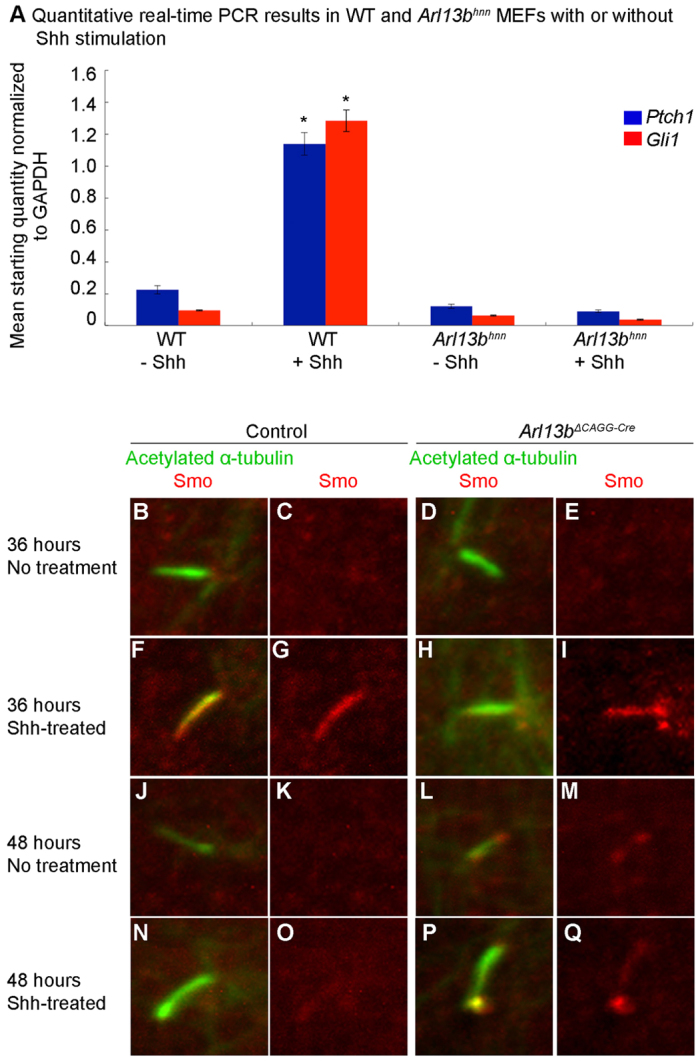
Response to Shh ligand and the localization of Smo are abnormal in the absence of Arl13b in MEFs. (A) Quantitative real-time PCR analysis demonstrates the expression of Ptch1 (blue) and Gli1 (red) in wild-type or Arl13bhnn MEFs. The expression of Ptch1 and Gli1 in wild-type MEFs after Shh stimulation is significantly higher than without Shh stimulation, but Arl13bhnn MEFs do not show any response after Shh stimulation. *P<0.05. (B-I) Confocal images of Smo (red) expression in the cilium, marked by acetylated α-tubulin (green) in control (B,C,F,G) or Arl13bΔCAGG-Cre (D,E,H,I) MEFs after cells are treated with tamoxifen for 36 hours in the absence of Shh-conditioned media (B-E) or in the presence of Shh stimulation (F-I). (J-Q) After treating control MEFs with Shh-conditioned media for 48 hours, Smo is no longer able to respond, so there is no cilium localization of Smo (J,K,N,O). By contrast, Smo is localized to Arl13bΔCAGG-Cre cilia and enriched at the tip when Arl13b is deleted, even without Shh stimulation (L,M). Smo is also enriched in the cilia upon Shh stimulation of Arl13bΔCAGG-Cre at the 48-hour time point (P,Q).
To confirm that the temporal loss of Arl13b affected Shh signaling in the same mechanistic manner as constitutive loss of Arl13b, we turned to Arl13b deletion in cell culture with control Arl13bfloxed/+ and Arl13bhnn/floxed; CAGG-Cre/+ (Arl13bΔCAGG-Cre) MEFs. We cultured the MEFs under two conditions: in serum-free media for 24 hours (to induce cilia formation), followed by tamoxifen treatment to delete Arl13b, or in serum-containing media with tamoxifen, which permits the cells to proliferate as they would in vivo. Consistent with what we had seen in vivo, we saw a slight decrease of Arl13b 24 hours after tamoxifen addition and an absence of Arl13b between 36 and 42 hours after tamoxifen treatment in Arl13bΔCAGG-Cre MEFs (supplementary material Fig. S3A-F,I,J-O,R). These data establish that Arl13b protein was abolished 42 hours after tamoxifen injection, implying that Arl13b-dependent phenotypes could be analyzed in vivo 2 days post-injection (supplementary material Fig. S3G,H,P,Q).
The germline deletion of the conditional allele recapitulated the Arl13bhnn neural patterning phenotype, arguing that the Arl13b conditional deletion globally affected Shh activity, as expected (supplementary material Fig. S1C-F). At the cellular level, this is due to a lack of regulation of key components of the pathway; Smo requires Shh stimulation to localize to cilia in wild-type MEFs, but was found in the cilia of Arl13bhnn MEFs regardless of Shh stimulation (Larkins et al., 2011). Therefore, we tested whether the conditional deletion of Arl13b affected ciliary Smo localization like the null allele. We examined Smo localization in control and Arl13bΔCAGG-Cre MEFs after treating cells with tamoxifen and either control or Shh-conditioned media for 36 hours (when Arl13b still remains) or 48 hours (when we could no longer detect Arl13b protein). As expected, we found that when Arl13b protein was present in control or in Arl13bΔCAGG-Cre MEFs at 36 hours, Smo localized to cilia only in the presence of Shh (Fig. 1B-I). By 48 hours in the control, we no longer found Smo in cilia, consistent with the observation that continued Shh response requires progressively increasing stimulation (Fig. 1J,K,N,O) (Dessaud et al., 2007). By contrast, Smo localized to cilia in Arl13bΔCAGG-Cre at 48 hours with or without Shh stimulation (Fig. 1L,M,P,Q). This indicates that temporal deletion of Arl13b results in the same loss of Shh-dependent Smo regulation observed in Arl13bhnn MEFs (Larkins et al., 2011); thus, the temporal deletion of Arl13b with CAGG-CreER® in vivo results in a constitutive low-level of Shh activity via the same mechanism we saw in the null allele.
Neural progenitors are sensitive to changes in Shh activity at E9.5, but not at E10.5
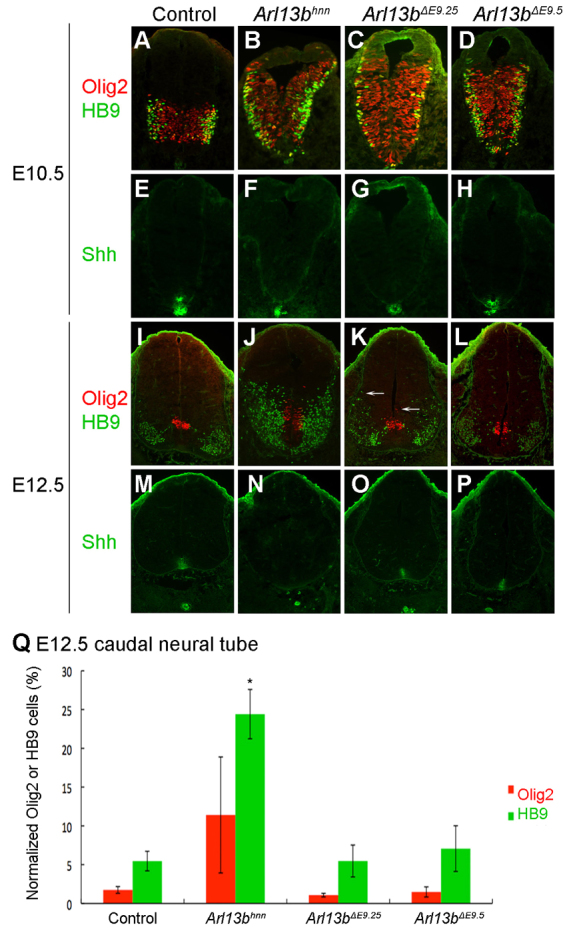
To investigate when cells are sensitive to changes in the level of GliA function, we first analyzed the consequences of removing Arl13b as neural patterning is established, at ~E9.5. By injecting tamoxifen at E7.75, we found a complete absence of Arl13b protein by E9.5, and refer to mice with this time of deletion as Arl13bΔ9.5 (Fig. 2Aii,H). In the caudal neural tube at E9.5, we found an expansion of Olig2 cells at the hindlimb level in the Arl13bΔE9.5 neural tube, although the expansion was not as extensive as in Arl13bhnn embryos (Fig. 2E,I). We also saw that, compared with control embryos, the progenitor marker Nkx6.1 was expanded dorsally in the Arl13bΔE9.5 neural tube, albeit not as far dorsally as in Arl13bhnn embryos (see Fig. 5A″,D″,J″). Our observation that the patterning defects were not as severe in Arl13bΔE9.5 embryos as in Arl13bhnn embryos suggests that the competence of the cells in the neural tube to ectopic GliA is progressively lost.
Fig. 2.

Distinct neural pattern when Arl13b is deleted at different time points. (A) Experimental design of tamoxifen injection to delete Arl13b. Tamoxifen is injected at E7.5 (i), E7.75 (ii) or E8.5 (iii), and embryos are collected at E9.5, E10.5 or E12.5 to examine neural tube patterning. Black neural tube indicates that ubiquitous Arl13b deletion has occurred, based on immunofluorescence with Arl13b antibody. (B,D,F,H) Arl13b expression can be observed in the ventricular zone of E9.5 control caudal neural tube (B), but not in E9.5 Arl13bhnn (D), Arl13bΔE9.25 (F) or Arl13bΔE9.5 (H). (C,E,G,I) Olig2 cells are specified in a restricted domain in E9.5 control (C), but are expanded in Arl13bhnn (E), Arl13bΔE9.25 (G) and Arl13bΔE9.5 (I) caudal neural tube. (C′,E′,G′,I′) Shh is expressed in the notochord and floor plate at E9.5 in both control and Arl13bΔE9.5 (C′,I′). Shh is only observed in the notochord in Arl13bhnn and Arl13bΔE9.25 (E′,G′). (J,L) Arl13b is expressed in the ventricular zone of E10.5 control caudal neural tube (J), but is absent in Arl13bΔE10.5 (L). (K,M) Olig2 cells are specified in their restricted domain in both control (K) and Arl13bΔE10.5 (M).
Fig. 5.

Floor-plate and p3 progenitor marker expression in different inducible Arl13b deletion caudal neural tubes. (A-C″) FoxA2 is expressed in the floor plate (A,B,C), Nkx2.2 is specified in the p3 domain (A′,B′,C′) and Nkx6.1 is in the ventral neural tube in control embryos at E9.5 (A-A″), E10.5 (B-B″) and E12.5 (C-C″). White arrow in C indicates FoxA2-expressing cells. (D-F″) FoxA2 is absent in Arl13bhnn at E9.5 (D), E10.5 (E) and E12.5 (F), whereas Nkx2.2 is expanded dorsally in all three developmental stages (D′,E′,F′). Nkx6.1 is specified in the whole neural tube at E9.5 (D″) and E10.5 (E″), and specified in the whole ventricular zone at E12.5 (F″). White arrow in F indicates the region that should be the floor plate. (G-L″) Both FoxA2 and Nkx2.2 cells are expanded dorsally in Arl13bΔE9.25 and Arl13bΔE9.5 at E9.5 (G,G′,J,J′) and E10.5 (H,H′,K,K′). FoxA2 is restored at E12.5 in Arl13bΔE9.25 and Arl13bΔE9.5 (I,L). Nkx2.2 is expressed normally at E12.5 in both Arl13bΔE9.25 and Arl13bΔE9.5 (I′,L′). Nkx6.1 is expanded to the dorsal quarter of the neural tube at E9.5 and E10.5 (G″,J″,H″,K″), but restricted to the ventral ventricular zone at E12.5 (I″,L″). Nkx6.1 expression in motoneurons is similar to control in E12.5 Arl13bΔE9.25 (I″), whereas fewer motoneurons express Nkx6.1 in Arl13bΔE9.5 (L″). White arrows in I and L show FoxA2-expressing cells.
Next, to examine whether cells are sensitive to changes in the level of GliA function at E10.5, we examined Arl13bΔE10.5 embryos, whose mothers were injected with tamoxifen at E8.5 (Fig. 2Aiii,J,L). We found Olig2, HB9, Shh and Nkx2.2 expression was normal at E10.5, indicating the cells were no longer sensitive to changes in Shh activity and arguing that the cells are committed at E10.5 (Fig. 2K,M; supplementary material Fig. S4; data not shown). Thus, cells in the mouse neural tube are progressively less sensitive to changes in the level of GliA function for a developmental window after E8.5 and prior to E10.5, implying that Shh via GliA is not a potent instructive signal in vivo after E10.5.
Rescue of neural tube patterning over time in Arl13bΔE9.5 embryos demonstrates an active role for low-level Shh activity
The loss of cell fates over time in the Shh conditional mice indicated that Shh expression must be maintained for patterning to persist (Dessaud et al., 2010). Without Shh ligand, Gli protein is no longer activated but is instead cleaved to its repressor form, so it is unclear whether this phenotype is due to lack of activation or active repression. Previously, it has been difficult to separate these two possibilities because Gli protein can be processed into either activator or repressor, making it difficult to perturb GliA function without affecting GliR, and vice-versa (Aza-Blanc et al., 2000; Ruiz i Altaba, 1998). Unlike mutations in Gli2 or in Gli3, mutations in Arl13b enable us to uncouple the regulation of GliA from that of GliR (Caspary et al., 2007; Ding et al., 1998; Matise et al., 1998; Persson et al., 2002). To test the consequences of altering the level of GliA function while leaving GliR intact on ventral cell fates over time, we examined the Arl13bΔE9.5 embryos at several time points. Differentiated motoneurons are normally restricted to the ventrolateral neural tube, but are expanded in Arl13bhnn-null embryos from E10.5 through E12.5 (Fig. 3A,B,I,J). In E10.5 Arl13bΔE9.5 embryos, we detected an expansion of Olig2 and HB9 cells similar to the E10.5 Arl13bhnn-null phenotype (Fig. 3B,D). However, by E12.5 we found the same number of Olig2- and HB9-positive cells in Arl13bΔE9.5 embryos as in control embryos (Fig. 3Q). Furthermore, their expression domain resembled the wild-type pattern, not the Arl13bhnn-null embryo pattern (Fig. 3I,J,L). This recovery of patterning from E10.5 to E12.5 was surprising because graded Shh does not direct different ventral neural cell fates after E10.5, according to the normal pattern we observed in Arl13bΔE10.5 embryos (Fig. 2K,M). Thus, whereas Shh activity must be maintained for normal neural cell fates to be specified, we find that the level of GliA function occurring in the absence of Arl13b, although abnormally regulated, permits initially mispatterned cells to be rescued to a wild-type fate over time.
Fig. 3.
The pMN expansion is restored over time when Arl13b is deleted at E9.25 and E9.5. (A-D) Olig2 (red) and HB9 (green) cells are expressed in the pMN domain of control at E10.5 (A), but are expanded in Arl13bhnn (B), Arl13bΔE9.25 (C) and Arl13bΔE9.5 (D). (E-H) Shh (green) is expressed in the notochord and the floor plate of E10.5 control (E) and Arl13bΔE9.5 (H), but only in the notochord of Arl13bhnn (F) and Arl13bΔE9.25 (G). (I-L) At E12.5, there is normal Olig2 (red) and HB9 (green) expression in control (I) and Arl13bΔE9.5 (L), whereas there is an expansion of cells in Arl13bhnn caudal neural tube (J). Olig2 and HB9 cells are mainly in their correct domains in Arl13bΔE9.25, except that there are some dorsally expressed Olig2 and HB9 cells (K and white arrows). (M-P) In control (M), Arl13bΔE9.25 (O) and Arl13bΔE9.5 (P) caudal neural tube, Shh is expressed in the notochord and floor plate at E12.5, whereas it is expressed in only the notochord of Arl13bhnn neural tube (N). (Q) Quantitative results showing that the percentage of normalized Olig2 (red) and HB9 (green) cells in E12.5 Arl13bΔE9.25 and Arl13bΔE9.5 caudal neural tube are similar to control, whereas there are excessive Olig2 and HB9 cells in Arl13bhnn. Data are mean±s.d. *P<0.05.
This recovery of patterning was unexpected, so we investigated three potential artifacts that could stem from inducing deletion of Arl13b. A trivial explanation for the rescue of patterning we see is that Cre-induced deletion might not be complete. In such a scenario, wild-type cells might out-compete mutant cells over time. We ruled out this possibility by examining Arl13b expression via immunofluorescence (Fig. 4C,D,G,H) and western blotting (Fig. 4K), as well as by confirming Arl13b deletion via PCR of the deleted Arl13bfloxed allele (Fig. 4L). In all cases, we could detect no protein or unrecombined DNA allele, indicating that the deletion was ubiquitous and no wild-type cells remained.
Fig. 4.

Rescue of patterning is not due to reactivation of Shh activity or incomplete Arl13b deletion. (A,B) Ptch1-lacZ shows a ventral-to-dorsal gradient in E10.5 control (A), but is expressed ubiquitously in the whole neural tube of Arl13bΔE9.25 (B). (C,D,G,H) Arl13b (red) is expressed in cilia of controls at E10.5 (C) and E12.5 (G), but is absent from Arl13bΔE9.25 at E10.5 (D), as well as from Arl13bΔE9.5 at E12.5 (H). Hoechst (blue) stains nuclei. (E,F) Ptch1-lacZ is uniformly expressed in E12.5 control (E) and Arl13bΔE9.5 (F) ventral neural tube. (I,J) Ptch1 mRNA is expressed along the ventricular zone of E12.5 control (I) and Arl13bΔE9.5 (J) ventral neural tubes. White bracket indicates strong expression of Ptch1. (K) Western blot shows the absence of a 60 kDa Arl13b band in Arl13bΔE9.5. (L) PCR shows there is no conditional Arl13b allele DNA (679 bp), whereas a deleted band (109 bp) can be detected in Arl13bΔE9.5. A 526 bp band indicates the endogenous Arl13bhnn allele. Controls used were either Cre-positive Arl13bfloxed/+, Cre-negative Arl13bfloxed/floxed or Cre-negative Arl13bfloxed/hnn.
Alternatively, one could argue that the recovery is due to cells in the dorsal and ventral Arl13bΔE9.5 neural tube proliferating more than those within in the Olig2 domain. We eliminated this model by staining Arl13bΔE9.25 neural tubes with markers of cell proliferation: phospho-histone H3 and BrdU (supplementary material Fig. S5; data not shown). We found no proliferative differences from wild-type controls, indicating that differential proliferation cannot explain the patterning recovery. This is consistent with our previous data showing that loss of Arl13b does not impact cell proliferation (Caspary et al., 2007; Horner and Caspary, 2011).
Another possibility to account for the recovery of patterning is that, in contrast to Arl13bhnn embryos that never expressed Shh in the floor plate, Shh was expressed in the floor plate in Arl13bΔE9.5 embryos at E9.5, E10.5 and E12.5 (Fig. 2E′,I′; Fig. 3F,H,N,P). Over time, perhaps Shh from the floor plate re-established the pattern. We thought this model unlikely for several reasons: Arl13bΔE10.5 embryos had shown ventral neural cells are insensitive to shifts in Shh activity after E10.5 (Fig. 2M), Shh Arl13bhnn double mutant analysis had revealed that the absence of Arl13b results in ligand-independent constitutive Shh activity (Caspary et al., 2007), and in vitro data had indicated that cells lacking Arl13b do not show a transcriptional response to ligand stimulation (Fig. 1A). Nevertheless, we reasoned that we could eliminate this possibility if we could identify a time point for Arl13b deletion that would allow the initial Shh activity gradient to be established, but would eliminate Shh expression in the floor plate. We achieved this by injecting tamoxifen at E7.5, 6 hours before the injection that generated the Arl13bΔE9.5embryos, and called these embryos Arl13bΔE9.25 (Fig. 2Ai). Shh was absent in the floor plate in the Arl13bΔE9.25 embryos at E9.5, as in Arl13bhnn embryos (Fig. 2E′,G′). We did find an expansion of the Olig2-positive domain at E9.5 and E10.5 in the Arl13bΔE9.25 embryos (Fig. 2G; Fig. 3C); however, by E12.5, Olig2 was restricted to its normal wild-type domain (Fig. 3K). The equivalent recovery of patterning by E12.5 in Arl13bΔE9.25 as in Arl13bΔE9.5 embryos indicates that, as long as the initial gradient of Shh activity is induced, Shh expression in the floor plate of Arl13bΔE9.5 embryos cannot explain the recovery.
Recovery of patterning is complete
To further characterize the patterning phenotypes and the extent of recovery in Arl13bΔE9.25 and Arl13bΔE9.5 embryos, we examined other markers affected in the Arl13bhnn-null neural tube. Nkx6.1 marks all ventral progenitors, which are divided into subdomains demarcated by FoxA2 in the floor plate, Nkx2.2 in the p3 cells and Olig2 in the pMN cells (Fig. 5A-C″). In Arl13bhnn embryos, we rarely saw FoxA2-expressing cells; Nkx2.2-expressing cells were specified across the ventral midline, intermingled with Olig2 cells; and Nkx6.1 cells were expanded into dorsal neural tube (Fig. 5D-F″). In both Arl13bΔE9.25 and Arl13bΔE9.5 caudal neural tubes at E9.5, FoxA2 was present, consistent with the establishment of the Shh activity gradient inducing FoxA2 expression (Fig. 5G,J); however, by E10.5, there were FoxA2-positive cells in more dorsal positions than normal, which is likely to reflect the shift in the level of GliA function due to the loss of Arl13b (Fig. 5H,K). Nkx2.2 and Nkx6.1 expression expanded further dorsally in both the Arl13bΔE9.25 and Arl13bΔE9.5 caudal neural tube, albeit not as far dorsally as in Arl13bhnn (Fig. 5G′,G″,H′,H″,J′,J″,K′,K″). When we examined these markers in E12.5 Arl13bΔE9.25 and Arl13bΔE9.5 embryos, we found they were similar to control embryos, evidence that the pattern was rescued (Fig. 5I-I″,L-L″).
In addition to the cell fates, we monitored the Shh activity gradient in Arl13bΔE9.25 and Arl13bΔE9.5 caudal neural tubes by examining Ptch1 expression. Normally, a steep Shh activity gradient is visible starting at E8.5, and it is maintained at E9.5 and E10.5; we previously showed that Ptch1 expression in Arl13bhnn is dorsally expanded and not in a gradient (Caspary et al., 2007). We found Ptch1-lacZ reporter expression at E10.5 in the Arl13bΔE9.25 caudal neural tube resembled that of Arl13bhnn at E10.5, consistent with the cell fates we observed (Fig. 4B). Similarly, by E12.5, when the recovery appeared complete, we found no difference in Ptch1 expression between the wild-type and Arl13bΔE9.5 caudal neural tubes using either the Ptch1-lacZ allele or Ptch1 in situ hybridization (Fig. 4E,F,I,J). This suggests that in Arl13bΔE9.25 and Arl13bΔE9.5 caudal neural tubes by E12.5, the gradient of Shh target gene transcriptional activity in the neural tube is normal. Taken together, the recovery of the cell fates and the recovery of the Shh activity gradient in the Arl13bΔE9.25 and Arl13bΔE9.5 caudal neural tubes indicate that, as long as the Shh activity gradient was initially established, improperly patterned cells along the dorsal-ventral axis were rescued in the absence of Arl13b.
The recovery of patterning in Arl13bΔE9.25 and Arl13bΔE9.5 neural tubes is Gli3 dependent
By conditionally deleting Arl13b, we were able to modulate the level of GliA function while leaving GliR function intact, raising the possibility that GliR could be responsible for the recovery of patterning we observed. To test this directly, we conditionally deleted Gli3, which acts predominantly as a repressor in the neural tube, along with Arl13b at E9.25. Gli3 mutants display normal patterning of ventral markers, such as FoxA2, Nkx2.2, Olig2 and HB9, and only disrupt a few cell fates in the p0-p2 domains (Persson et al., 2002). In contrast to the relatively normal pattern we observed in single mutant Arl13bΔE9.25 neural tubes at E12.5, in double mutant Arl13b Gli3ΔE9.25 neural tubes, we saw abnormal patterning: Nkx2.2 cells were concentrated in the p3 domain, but some were expressed dorsal to Olig2 cells (Fig. 6G,H); Olig2 and Nkx6.1 cells were scattered to more dorsal positions compared with the domain in E12.5 Arl13bΔE9.25 neural tubes (Fig. 6H,J,L; Fig. 3K; Fig. 5I″); and HB9 motoneurons were expanded dorsally (Fig. 6I,J). The overall patterning was less severe than in Arl13bhnn-null embryos, consistent with the initial Shh activity gradient being established in Arl13b Gli3ΔE9.25 double mutants as in Arl13bΔE9.25 and Arl13bΔE9.5 embryos.
Fig. 6.

Ventral neural tube patterning is impaired in E12.5 Arl13b Gli3ΔE9.25. (A-F) Olig2 (A), Nkx2.2 (C) and Nkx6.1 (E) cells are expressed in pMN, p3 and ventral neural tube of Cre negative Arl13bfloxed/floxed Gli3floxed/+ (control) embryos at E9.5, whereas those ventral progenitors are expanded in Arl13b Gli3ΔE9.25 (B,D,F). (G,H) Olig2 (red) and Nkx2.2 (green) cells are in their restricted domains in E12.5 Cre negative Arl13bfloxed/floxed Gli3floxed/floxed (control) neural tube (G). Although most Olig2 and Nkx2.2 cells are localized normally in Arl13b Gli3ΔE9.25, some cells are scattered to a more dorsal domain (H). (I,J) In E12.5 Arl13b Gli3ΔE9.25 embryo, some Olig2 (red) and HB9 (green) cells are localized in more dorsal domains than their normal restricted domains (J), but expressed normally in control embryo (I). (K,L) Nkx6.1 cells are mostly restricted to the ventricular zone of both control (K) and Arl13b Gli3ΔE9.25 (L) at E12.5, except some Nkx6.1 cells are localized more dorsally in Arl13b Gli3ΔE9.25 (L).
One explanation for the abnormal patterning at E12.5 in Arl13b Gli3ΔE9.25 double mutant embryos is that, at deletion, the removal of Gli3 resulted in a relatively higher burst of ectopic GliA in the Arl13b Gli3ΔE9.25 double mutants than in the Arl13bΔE9.25 single mutant embryos. To address this, we examined Arl13b Gli3ΔE9.25 double mutant embryos at E9.5 and observed an expansion of Nkx2.2-, Olig2- and Nkx6.1-positive cells (Fig. 6A-F). However, this expansion was indistinguishable from what we saw in the Arl13bΔE9.25 single mutant embryos, arguing that the levels of GliA upon Arl13b and Gli3 deletion are not higher at a functional level than the level of GliA upon Arl13b deletion alone. The abnormal patterning at E12.5 in Arl13b Gli3ΔE9.25 double mutant embryos suggested that the recovery of patterning we saw in Arl13bΔE9.25 and Arl13bΔE9.5 caudal neural tubes depended on the proper regulation of Gli3 repressor. Thus, our data reveal a previously unrecognized function of Gli3 in the maintenance of neural patterning (Fig. 7).
Fig. 7.

Summary of phenotypic analyses. In wild-type neural tube, Shh activity (blue) establishes a ventral-to-dorsal gradient, while a Gli3 repressor activity gradient (orange) is established dorsally at E9.0, and normal patterning is observed at E10.5 and E12.5. In Arl13bhnn, there is a constitutive low level of Shh activity, but normal Gli3 repressor activity, resulting in an expansion of pMN cells and motoneurons that persist to E12.5. In Arl13bΔE9.25 and Arl13bΔE9.5, a normal Shh activity gradient is established initially, but disrupted at E9.25 and E9.5, respectively; however, Gli3 repressor activity is intact. An expansion of pMN cells and motoneurons is detected in E10.5 Arl13bΔE9.25 and Arl13bΔE9.5, but both pMN cells and motoneurons are expressed normally at E12.5. In Arl13bΔE10.5, Shh activity gradient is disrupted at E10.5, but patterning is normal, suggesting that cells are no longer sensitive to changes in Shh activity after E10.5. In Arl13b Gli3ΔE9.25, Gli3 repressor is absent and there is a constitutive low level of Shh activity at E9.0, resulting in an expansion of Olig2 at E9.5 and a slight dorsal expansion of Olig2 and HB9 cells at E12.5. Lines on the top indicate Shh plays an instructive role at E9.0, and switches to become a persistent signal after E9.25. Asterisks represent the time points when tamoxifen is injected into pregnant females. Blue and orange shading demonstrate Shh and Gli3 repressor activity, respectively. In E9.5, E10.5 and E12.5 neural tube, red and green circles represent cell fates that are resulting from Shh activity: red circles are pMN cells; green circles are motoneurons. Black trapezoid is the floor plate. T.I., tamoxifen injection.
DISCUSSION
Although the requirement of Shh morphogen for patterning the neural tube is clear, its precise mechanism of action has been more difficult to dissect. Shh acts as an extrinsic signal by establishing the GliA/GliR ratio to induce the expression of specific transcription factors (Briscoe et al., 2000; Briscoe et al., 1999; Chiang et al., 1996; Ericson et al., 1997a; Ericson et al., 1997b). Subsequently, the transcription factors regulated by Shh form a gene regulatory network (GRN) with both positive feed-forward and negative-feedback outputs (Briscoe et al., 1999; Ericson et al., 1997b; Novitch et al., 2001; Sander et al., 2000; Vallstedt et al., 2001). Recent work has proposed this GRN buffers the ultimate cell fate decision against temporary fluctuations in the extrinsic signal (Balaskas et al., 2012). Our previous genetic work established that Arl13b regulates only GliA and that loss of Arl13b leaves GliR intact (Caspary et al., 2007). Here, we took advantage of this unique regulatory role to perturb Shh signaling by temporally deleting Arl13b during neural patterning. Our data show that in vivo, the initial Shh activity gradient is ultimately instructive in neural tube patterning and defines the window of time during which cells progressively lose their ability to respond to shifts in the GliA/GliR ratio. Our analysis of Arl13b Gli3 double mutants also reveals an essential role for GliR in sustaining neural patterning.
The initial role of GliA as an instructive signal
We define the in vivo window during which neural cell fates can respond to shifts in GliA to be prior to E10.5. Indeed, misregulation of GliA in Arl13bΔE9.25 and Arl13bΔE9.5 embryos results in Olig2 expansion in the neural tube similar to Arl13bhnn mutants, although the change in other ventral markers is not as dramatic. Later deletion of Arl13b results in less severe patterning defects, however (compare Fig. 3B-D with Fig. 5), indicating that the neural tube becomes progressively less sensitive to shifts in GliA over time, consistent with previous work showing the system requires increasing amounts of GliA inputs over time (Dessaud et al., 2007). Nonetheless, comparison of the germline null Arl13bhnn embryos with the Arl13bΔE9.25 and Arl13bΔE9.5 embryos underscores the crucial role of the initial Shh activity gradient in the mammalian neural tube (Fig. 7). At E12.5, abnormal patterning persists in Arl13bhnn mutants. In Arl13bΔE9.25 and Arl13bΔE9.5 embryos, however, the neural tube recovers its wild-type pattern by E12.5, suggesting that the initial establishment of the Shh activity gradient in Arl13bΔE9.25 and Arl13bΔE9.5 embryos provides the instructive signals that guide the ultimate fate of each cell (Fig. 7). This is in keeping with the prevailing view of Shh as an instructive morphogen.
Recovery of patterning
The recovery of patterning we see in the Arl13bΔE9.25 and Arl13bΔE9.5 embryos is striking; the cells outside the normal pMN domain that expressed Olig2 at E9.5 switch to expressing the markers of the adjacent dorsal and ventral domains by E12.5. Two distinct models might explain this result. In the first model, the recovery may reflect the response of the cells to GliA over time. In this scenario, the cells would initially be mispatterned owing to the shift in GliA caused by loss of Arl13b, but the cells would remain sufficiently plastic to be repatterned over time, provided Gli3 regulation is intact. Alternatively, the normal Shh gradient prior to Arl13b deletion might have turned on the appropriate target genes so that the downstream GRN is consolidated, but the cells remain sufficiently responsive to short-term fluctuations in GliA to induce the expression of Olig2; thus, the cells that ectopically express Olig2 at E9.5 would be respecified over time by the GRN established earlier in development. The latter is termed hysteresis, a memory of the initial signal, which has been argued for by recent work in neural patterning (Balaskas et al., 2012).
It remains unclear which model accounts for the recovery of patterning we observe or if both mechanisms are involved. The expansion of the Olig2 domain we observed in Arl13bΔE9.25 embryos was more pronounced than in Arl13bΔE9.5 embryos. The fact that earlier perturbation of the system results in more severe patterning defects could mean that GliA requires sufficient time to truly activate target genes and induce the downstream GRN, consistent with the first model. However, our analysis of the Arl13b Gli3ΔE9.25 double mutants at E9.5 might point towards the second model (hysteresis). In the double mutants, loss of Gli3 effectively increases GliA function by removing the primary GliR in the neural tube, altering the GliA/GliR ratio. If the GRN is not consolidated by E9.5, we would have expected to see greater expansion of Olig2 expression in double mutants compared with single mutants owing to this shift in inputs. Instead, we saw that the Arl13b Gli3ΔE9.25 double mutants resemble Arl13bΔE9.25 single mutants at E9.5, arguing that the functional level of ectopic GliA can be increased with no functional consequence to cell fate. It is possible, however, that increased ectopic GliA over a greater duration of time is what precludes the recovery of patterning. Because we cannot measure levels of GliA directly, only the downstream transcriptional response, we cannot experimentally distinguish between these models. Regardless of whether the GliA inputs or the downstream GRN are responsible for the recovery, this capacity for recovery shows the robustness of the system that patterns the neural tube.
The role of Gli3 in neural patterning
Although Gli3 acts predominantly as a repressor of Shh target genes in the neural tube, its exact function remains enigmatic because Gli3 mutants display a relatively mild patterning phenotype (Persson et al., 2002). We saw mispatterning in the Arl13bΔE9.25 and Arl13bΔE9.5 embryos, indicating that GliR could not initially compensate for the shift in GliA activity. By E12.5, however, we saw normal patterning only in the Arl13bΔE9.25 and Arl13bΔE9.5 single mutant embryos, and not in Arl13b Gli3ΔE9.25 double mutant embryos, demonstrating that the recovery depends on Gli3. This might be because the absence of Gli3 lowers the level of GliR, and thus alters the balance of activation and repression such that the downstream transcriptional profile of cells is altered. Alternatively, our data raise the possibility that there might be temporal distinctions in the roles of GliA and GliR in neural patterning, whereby neural patterning is first most responsive to the positive inputs of Shh signaling, mediated by GliA, and subsequently requires the negative inputs, via GliR. Finally, we cannot rule out the possibility of other signaling pathways active in the neural tube being responsible for the recovery of patterning in a Gli3-dependent manner.
Most of the pathways known to play patterning roles in the neural tube, including bone morphogenetic protein (BMP), Wingless (Wnt) and retinoic acid, interact with Shh signaling (Nishi et al., 2009). Retinoic acid signaling from the paraxial mesoderm works with Shh signaling to specify ventral progenitors (Novitch et al., 2003; Pierani et al., 1999; Wichterle et al., 2002). BMP antagonists in the ventral neural tube mediate a BMP activity gradient reciprocal to that of Shh (Liem et al., 2000; McMahon et al., 1998; Patten and Placzek, 2002). BMPs and Wnts are expressed at the dorsal midline of the neural tube and are needed for specifying dorsal progenitors (Chesnutt et al., 2004; Lee et al., 2000; Lee et al., 1998; Liem et al., 1997; Liem et al., 1995; Muroyama et al., 2002; Parr et al., 1993; Timmer et al., 2002; Wine-Lee et al., 2004; Zechner et al., 2007). Shh signaling interacts with both the Wnt and BMP pathways; Wnts directly regulate Gli3 repressor, and Gli3 activity can regulate canonical Wnt signaling (Alvarez-Medina et al., 2008; Ulloa et al., 2007; Yu et al., 2008). Furthermore, Wnt signaling regulates ventral neural fates through Gli3 in a time-dependent manner (Yu et al., 2008). Effectors of both BMP and Wnt signaling interact with Gli3 (Liu et al., 1998; Meyer and Roelink, 2003; Ulloa et al., 2007). Our finding that the recovery of patterning in Arl13bΔE9.25 and Arl13bΔE9.5 embryos is Gli3 dependent raises the possibility that one of these pathways could control the Gli3 repressor activity that is crucial to the recovery we saw.
Our findings further define the critical time points when Shh signaling is required in neural patterning during development. Our data show that in the neural tube, the initial Shh activity gradient is ultimately instructive, yet inputs to the transcriptional circuit can be manipulated and perturbed in ways that can temporarily disrupt any cell fate decision. Our observation that initially mispatterned neural progenitors can recover a wild-type pattern over time highlights the functional transition from an activity gradient of Shh signaling to the role of the downstream transcriptional circuit in regulating cell fate decisions. In addition, our data reveal a crucial role for Gli3 in maintaining the balance of the feed-forward and negative-feedback signals that determine neural cell fate. Thus, our data provide in vivo evidence to support a model of a fundamental mechanism through which positive and negative effectors of Shh signaling function to properly specify and maintain cell fates in the mammalian spinal cord.
Supplementary Material
Acknowledgements
We are grateful to Dr K. V. Anderson for the Smo antibody. The FoxA2, HB9, Nkx2.2, Nkx6.1 and Shh antibodies developed by Dr Thomas Jessell were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242, USA. We thank Kathryn Anderson, James Briscoe, Vanessa Ribes and members of the Caspary Lab for helpful suggestions on the manuscript. This research project was aided by the Transgenic Mouse and Gene Targeting Core of the Emory University School of Medicine.
Footnotes
Funding
This work was supported by the National Institutes of Health [R01NS056380] and the Burroughs Wellcome Fund (Hitchings Elion Career Development Award). Additional support was via an American Heart Association predoctoral fellowship [11PRE7200011] to L.E.M., predoctoral support from the National Institute of General Medical Sciences [T32GM008490] to S.N.B., and the Microscopy Core of the Emory Neuroscience National Institute of Neurological Disorders and Stroke Core Facilities [P30NS055077]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.082321/-/DC1
References
- Ahn K., Mishina Y., Hanks M. C., Behringer R. R., Crenshaw E. B., 3rd (2001). BMPR-IA signaling is required for the formation of the apical ectodermal ridge and dorsal-ventral patterning of the limb. Development 128, 4449-4461 [DOI] [PubMed] [Google Scholar]
- Alvarez-Medina R., Cayuso J., Okubo T., Takada S., Martí E. (2008). Wnt canonical pathway restricts graded Shh/Gli patterning activity through the regulation of Gli3 expression. Development 135, 237-247 [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P., Lin H. Y., Ruiz i Altaba A., Kornberg T. B. (2000). Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development 127, 4293-4301 [DOI] [PubMed] [Google Scholar]
- Balaskas N., Ribeiro A., Panovska J., Dessaud E., Sasai N., Page K. M., Briscoe J., Ribes V. (2012). Gene regulatory logic for reading the Sonic Hedgehog signaling gradient in the vertebrate neural tube. Cell 148, 273-284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belo J. A., Bouwmeester T., Leyns L., Kertesz N., Gallo M., Follettie M., De Robertis E. M. (1997). Cerberus-like is a secreted factor with neutralizing activity expressed in the anterior primitive endoderm of the mouse gastrula. Mech. Dev. 68, 45-57 [DOI] [PubMed] [Google Scholar]
- Briscoe J., Sussel L., Serup P., Hartigan-O'Connor D., Jessell T. M., Rubenstein J. L., Ericson J. (1999). Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature 398, 622-627 [DOI] [PubMed] [Google Scholar]
- Briscoe J., Pierani A., Jessell T. M., Ericson J. (2000). A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell 101, 435-445 [DOI] [PubMed] [Google Scholar]
- Caspary T., Larkins C. E., Anderson K. V. (2007). The graded response to Sonic Hedgehog depends on cilia architecture. Dev. Cell 12, 767-778 [DOI] [PubMed] [Google Scholar]
- Chen M. H., Wilson C. W., Li Y. J., Law K. K., Lu C. S., Gacayan R., Zhang X., Hui C. C., Chuang P. T. (2009). Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 23, 1910-1928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Struhl G. (1996). Dual roles for patched in sequestering and transducing Hedgehog. Cell 87, 553-563 [DOI] [PubMed] [Google Scholar]
- Chesnutt C., Burrus L. W., Brown A. M., Niswander L. (2004). Coordinate regulation of neural tube patterning and proliferation by TGFbeta and WNT activity. Dev. Biol. 274, 334-347 [DOI] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H., Beachy P. A. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407-413 [DOI] [PubMed] [Google Scholar]
- Corbit K. C., Aanstad P., Singla V., Norman A. R., Stainier D. Y., Reiter J. F. (2005). Vertebrate Smoothened functions at the primary cilium. Nature 437, 1018-1021 [DOI] [PubMed] [Google Scholar]
- Dessaud E., Yang L. L., Hill K., Cox B., Ulloa F., Ribeiro A., Mynett A., Novitch B. G., Briscoe J. (2007). Interpretation of the sonic hedgehog morphogen gradient by a temporal adaptation mechanism. Nature 450, 717-720 [DOI] [PubMed] [Google Scholar]
- Dessaud E., Ribes V., Balaskas N., Yang L. L., Pierani A., Kicheva A., Novitch B. G., Briscoe J., Sasai N. (2010). Dynamic assignment and maintenance of positional identity in the ventral neural tube by the morphogen sonic hedgehog. PLoS Biol. 8, e1000382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q., Motoyama J., Gasca S., Mo R., Sasaki H., Rossant J., Hui C. C. (1998). Diminished Sonic hedgehog signaling and lack of floor plate differentiation in Gli2 mutant mice. Development 125, 2533-2543 [DOI] [PubMed] [Google Scholar]
- Dubois N. C., Hofmann D., Kaloulis K., Bishop J. M., Trumpp A. (2006). Nestin-Cre transgenic mouse line Nes-Cre1 mediates highly efficient Cre/loxP mediated recombination in the nervous system, kidney, and somite-derived tissues. Genesis 44, 355-360 [DOI] [PubMed] [Google Scholar]
- Ericson J., Briscoe J., Rashbass P., van Heyningen V., Jessell T. M. (1997a). Graded sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harb. Symp. Quant. Biol. 62, 451-466 [PubMed] [Google Scholar]
- Ericson J., Rashbass P., Schedl A., Brenner-Morton S., Kawakami A., van Heyningen V., Jessell T. M., Briscoe J. (1997b). Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell 90, 169-180 [DOI] [PubMed] [Google Scholar]
- Goodrich L. V., Johnson R. L., Milenković L., McMahon J. A., Scott M. P. (1996). Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev. 10, 301-312 [DOI] [PubMed] [Google Scholar]
- Goodrich L. V., Milenković L., Higgins K. M., Scott M. P. (1997). Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 277, 1109-1113 [DOI] [PubMed] [Google Scholar]
- Han Y. G., Kim H. J., Dlugosz A. A., Ellison D. W., Gilbertson R. J., Alvarez-Buylla A. (2009). Dual and opposing roles of primary cilia in medulloblastoma development. Nat. Med. 15, 1062-1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S., McMahon A. P. (2002). Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev. Biol. 244, 305-318 [DOI] [PubMed] [Google Scholar]
- Haycraft C. J., Banizs B., Aydin-Son Y., Zhang Q., Michaud E. J., Yoder B. K. (2005). Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 1, e53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horner V. L., Caspary T. (2011). Disrupted dorsal neural tube BMP signaling in the cilia mutant Arl13b hnn stems from abnormal Shh signaling. Dev. Biol. 355, 43-54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde C., Dickinson R. J., Houtzager V. M., Cullum R., Montpetit R., Metzler M., Simpson E. M., Roy S., Hayden M. R., Hoodless P. A., et al. (2006). Hippi is essential for node cilia assembly and Sonic hedgehog signaling. Dev. Biol. 300, 523-533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D., Liu A., Rakeman A. S., Murcia N. S., Niswander L., Anderson K. V. (2003). Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 426, 83-87 [DOI] [PubMed] [Google Scholar]
- Jeong J., McMahon A. P. (2005). Growth and pattern of the mammalian neural tube are governed by partially overlapping feedback activities of the hedgehog antagonists patched 1 and Hhip1. Development 132, 143-154 [DOI] [PubMed] [Google Scholar]
- Lakso M., Pichel J. G., Gorman J. R., Sauer B., Okamoto Y., Lee E., Alt F. W., Westphal H. (1996). Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc. Natl. Acad. Sci. USA 93, 5860-5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkins C. E., Aviles G. D., East M. P., Kahn R. A., Caspary T. (2011). Arl13b regulates ciliogenesis and the dynamic localization of Shh signaling proteins. Mol. Biol. Cell 22, 4694-4703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. J., Mendelsohn M., Jessell T. M. (1998). Neuronal patterning by BMPs: a requirement for GDF7 in the generation of a discrete class of commissural interneurons in the mouse spinal cord. Genes Dev. 12, 3394-3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. J., Dietrich P., Jessell T. M. (2000). Genetic ablation reveals that the roof plate is essential for dorsal interneuron specification. Nature 403, 734-740 [DOI] [PubMed] [Google Scholar]
- Liem K. F., Jr, Tremml G., Roelink H., Jessell T. M. (1995). Dorsal differentiation of neural plate cells induced by BMP-mediated signals from epidermal ectoderm. Cell 82, 969-979 [DOI] [PubMed] [Google Scholar]
- Liem K. F., Jr, Tremml G., Jessell T. M. (1997). A role for the roof plate and its resident TGFbeta-related proteins in neuronal patterning in the dorsal spinal cord. Cell 91, 127-138 [DOI] [PubMed] [Google Scholar]
- Liem K. F., Jr, Jessell T. M., Briscoe J. (2000). Regulation of the neural patterning activity of sonic hedgehog by secreted BMP inhibitors expressed by notochord and somites. Development 127, 4855-4866 [DOI] [PubMed] [Google Scholar]
- Liu A., Wang B., Niswander L. A. (2005). Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 132, 3103-3111 [DOI] [PubMed] [Google Scholar]
- Liu F., Massagué J., Ruiz i Altaba A. (1998). Carboxy-terminally truncated Gli3 proteins associate with Smads. Nat. Genet. 20, 325-326 [DOI] [PubMed] [Google Scholar]
- Marigo V., Tabin C. J. (1996). Regulation of patched by sonic hedgehog in the developing neural tube. Proc. Natl. Acad. Sci. USA 93, 9346-9351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matise M. P., Epstein D. J., Park H. L., Platt K. A., Joyner A. L. (1998). Gli2 is required for induction of floor plate and adjacent cells, but not most ventral neurons in the mouse central nervous system. Development 125, 2759-2770 [DOI] [PubMed] [Google Scholar]
- May S. R., Ashique A. M., Karlen M., Wang B., Shen Y., Zarbalis K., Reiter J., Ericson J., Peterson A. S. (2005). Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev. Biol. 287, 378-389 [DOI] [PubMed] [Google Scholar]
- McMahon J. A., Takada S., Zimmerman L. B., Fan C. M., Harland R. M., McMahon A. P. (1998). Noggin-mediated antagonism of BMP signaling is required for growth and patterning of the neural tube and somite. Genes Dev. 12, 1438-1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N. P., Roelink H. (2003). The amino-terminal region of Gli3 antagonizes the Shh response and acts in dorsoventral fate specification in the developing spinal cord. Dev. Biol. 257, 343-355 [DOI] [PubMed] [Google Scholar]
- Muroyama Y., Fujihara M., Ikeya M., Kondoh H., Takada S. (2002). Wnt signaling plays an essential role in neuronal specification of the dorsal spinal cord. Genes Dev. 16, 548-553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi Y., Ji H., Wong W. H., McMahon A. P., Vokes S. A. (2009). Modeling the spatio-temporal network that drives patterning in the vertebrate central nervous system. Biochim. Biophys. Acta 1789, 299-305 [DOI] [PubMed] [Google Scholar]
- Novitch B. G., Chen A. I., Jessell T. M. (2001). Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron 31, 773-789 [DOI] [PubMed] [Google Scholar]
- Novitch B. G., Wichterle H., Jessell T. M., Sockanathan S. (2003). A requirement for retinoic acid-mediated transcriptional activation in ventral neural patterning and motor neuron specification. Neuron 40, 81-95 [DOI] [PubMed] [Google Scholar]
- Parr B. A., Shea M. J., Vassileva G., McMahon A. P. (1993). Mouse Wnt genes exhibit discrete domains of expression in the early embryonic CNS and limb buds. Development 119, 247-261 [DOI] [PubMed] [Google Scholar]
- Patten I., Placzek M. (2002). Opponent activities of Shh and BMP signaling during floor plate induction in vivo. Curr. Biol. 12, 47-52 [DOI] [PubMed] [Google Scholar]
- Persson M., Stamataki D., te Welscher P., Andersson E., Böse J., Rüther U., Ericson J., Briscoe J. (2002). Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. 16, 2865-2878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierani A., Brenner-Morton S., Chiang C., Jessell T. M. (1999). A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell 97, 903-915 [DOI] [PubMed] [Google Scholar]
- Ribes V., Balaskas N., Sasai N., Cruz C., Dessaud E., Cayuso J., Tozer S., Yang L. L., Novitch B., Marti E., et al. (2010). Distinct Sonic Hedgehog signaling dynamics specify floor plate and ventral neuronal progenitors in the vertebrate neural tube. Genes Dev. 24, 1186-1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohatgi R., Milenkovic L., Scott M. P. (2007). Patched1 regulates hedgehog signaling at the primary cilium. Science 317, 372-376 [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A. (1998). Combinatorial Gli gene function in floor plate and neuronal inductions by Sonic hedgehog. Development 125, 2203-2212 [DOI] [PubMed] [Google Scholar]
- Sander M., Paydar S., Ericson J., Briscoe J., Berber E., German M., Jessell T. M., Rubenstein J. L. (2000). Ventral neural patterning by Nkx homeobox genes: Nkx6.1 controls somatic motor neuron and ventral interneuron fates. Genes Dev. 14, 2134-2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmer J. R., Wang C., Niswander L. (2002). BMP signaling patterns the dorsal and intermediate neural tube via regulation of homeobox and helix-loop-helix transcription factors. Development 129, 2459-2472 [DOI] [PubMed] [Google Scholar]
- Tran P. V., Haycraft C. J., Besschetnova T. Y., Turbe-Doan A., Stottmann R. W., Herron B. J., Chesebro A. L., Qiu H., Scherz P. J., Shah J. V., et al. (2008). THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat. Genet. 40, 403-410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukachinsky H., Lopez L. V., Salic A. (2010). A mechanism for vertebrate Hedgehog signaling: recruitment to cilia and dissociation of SuFu-Gli protein complexes. J. Cell Biol. 191, 415-428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulloa F., Itasaki N., Briscoe J. (2007). Inhibitory Gli3 activity negatively regulates Wnt/beta-catenin signaling. Curr. Biol. 17, 545-550 [DOI] [PubMed] [Google Scholar]
- Vallstedt A., Muhr J., Pattyn A., Pierani A., Mendelsohn M., Sander M., Jessell T. M., Ericson J. (2001). Different levels of repressor activity assign redundant and specific roles to Nkx6 genes in motor neuron and interneuron specification. Neuron 31, 743-755 [DOI] [PubMed] [Google Scholar]
- van der Weyden L., Adams D. J., Harris L. W., Tannahill D., Arends M. J., Bradley A. (2005). Null and conditional semaphorin 3B alleles using a flexible puroDeltatk loxP/FRT vector. Genesis 41, 171-178 [DOI] [PubMed] [Google Scholar]
- Wichterle H., Lieberam I., Porter J. A., Jessell T. M. (2002). Directed differentiation of embryonic stem cells into motor neurons. Cell 110, 385-397 [DOI] [PubMed] [Google Scholar]
- Wine-Lee L., Ahn K. J., Richardson R. D., Mishina Y., Lyons K. M., Crenshaw E. B., 3rd (2004). Signaling through BMP type 1 receptors is required for development of interneuron cell types in the dorsal spinal cord. Development 131, 5393-5403 [DOI] [PubMed] [Google Scholar]
- Yamada T., Placzek M., Tanaka H., Dodd J., Jessell T. M. (1991). Control of cell pattern in the developing nervous system: polarizing activity of the floor plate and notochord. Cell 64, 635-647 [DOI] [PubMed] [Google Scholar]
- Yu W., McDonnell K., Taketo M. M., Bai C. B. (2008). Wnt signaling determines ventral spinal cord cell fates in a time-dependent manner. Development 135, 3687-3696 [DOI] [PubMed] [Google Scholar]
- Zechner D., Müller T., Wende H., Walther I., Taketo M. M., Crenshaw E. B., 3rd, Treier M., Birchmeier W., Birchmeier C. (2007). Bmp and Wnt/beta-catenin signals control expression of the transcription factor Olig3 and the specification of spinal cord neurons. Dev. Biol. 303, 181-190 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.