

Abstract
Neuronal precursor cell migration in the developing mammalian brain is a complex process requiring the coordinated interaction of numerous proteins. We have recently shown that amyloid precursor protein (APP) plays a role in migration into the cortical plate through its interaction with two cytosolic signaling proteins, disabled 1 (DAB1) and disrupted in schizophrenia 1 (DISC1). In order to identify extracellular factors that may signal through APP to regulate migration, we performed an unbiased mass spectrometry-based screen for factors that bind to the extracellular domain of APP in the rodent brain. Through this screen, we identified an interaction between APP and pancortins, proteins expressed throughout the developing and mature cerebral cortex. Via co-immunoprecipitation, we show that APP interacts with all four of the mammalian pancortin isoforms (AMY, AMZ, BMY, BMZ). We demonstrate that the BMZ and BMY isoforms of pancortin can specifically reduce β-secretase- but not α-secretase-mediated cleavage of endogenous APP in cell culture, suggesting a biochemical consequence of the association between pancortins and APP. Using in utero electroporation to overexpress and knock down specific pancortin isoforms, we reveal a novel role for pancortins in migration into the cortical plate. Interestingly, we observe opposing roles for alternate pancortin isoforms, with AMY overexpression and BMZ knock down both preventing proper migration of neuronal precursor cells. Finally, we show that BMZ can partially rescue a loss of APP expression and that APP can rescue effects of AMY overexpression, suggesting that pancortins act in conjunction with APP to regulate entry into the cortical plate. Taken together, these results suggest a biochemical and functional interaction between APP and pancortins, and reveal a previously unidentified role for pancortins in mammalian cortical development.
Keywords: Pancortin, Olfactomedin, Noelin, APP, Migration, Mouse, Rat
INTRODUCTION
Migration of neuronal precursor cells in the developing cerebral cortex is a complex process that requires the coordinated interaction of many factors. Extracellular cues are relayed to intracellular signaling pathways via transmembrane receptors to mediate migration of formative neurons from the ventricular and subventricular zones through the intermediate zone and to the proper layer of the cortical plate. Although dozens of genes have been identified that play important roles in this process, large gaps remain in our understanding of the molecular mechanisms involved.
We have recently shown that amyloid precursor protein (APP) plays a role in migration of neuronal precursor cells into the cortical plate. APP is a type I single transmembrane glycoprotein centrally involved in the pathogenesis of Alzheimer's disease (AD) (reviewed by Hardy and Selkoe, 2002). APP undergoes sequential proteolytic processing by β- and γ-secretase to generate the amyloid β-peptide (Aβ) (reviewed by De Strooper and Annaert, 2000). Several therapeutic strategies for AD, some of which have already entered clinical trials, aim to chronically inhibit β- or γ-secretase in order to prevent Aβ generation (reviewed by Selkoe, 2011). To understand the potential effects of chronically reducing APP processing to treat AD, it is important to decipher the normal functions of APP and understand the molecular pathways through which APP executes these functions.
Recent studies from several labs have begun to clarify an essential role for APP in the development of the nervous system. APP is now believed to function in neurite outgrowth (Araki et al., 1991; Perez et al., 1997; Young-Pearse et al., 2008), cell adhesion (Ghiso et al., 1992; Soba et al., 2005), synapse formation (Priller et al., 2006; Wang et al., 2009) and migration of neuronal precursors (Pramatarova et al., 2006; Young-Pearse et al., 2007). The precise mechanisms regulating the proteolytic processing and function of APP are only partially understood. Although the cleaved extracellular domain of APP itself has biological activities in vivo (Ring et al., 2007), we have found that both the ectodomain and intracellular domain of APP are required to be expressed as a holoprotein in order to mediate proper neuronal precursor cell migration during cortical development (Young-Pearse et al., 2007), and that the cytoplasmic factors disabled 1 (DAB1) and disrupted in schizophrenia 1 (DISC1) biochemically and functionally interact with APP in this function (Pramatarova et al., 2008; Pramatarova et al., 2006; Young-Pearse et al., 2010). On the basis of these findings, we hypothesize that specific extracellular factors bind the ectodomain of holoAPP on the cell surface and transmit a signal to intracellular signaling cascades during development. Accordingly, identifying extracellular binding partners for APP will help reveal the molecular pathways that regulate APP processing and function.
Several candidate ligands for APP, including F-spondin (Ho and Südhof, 2004; Hoe et al., 2005), contactins (Ma et al., 2008; Osterfield et al., 2008) and reelin (Hoe et al., 2009; Hoe et al., 2006), have been reported to interact with the ectodomain of APP and modulate APP processing and/or APP function in neurodevelopment. However, despite these initial reports, an endogenous functional ligand for APP has yet to be widely confirmed in multiple laboratories. We initiated a search for additional factors that could interact with the APP ectodomain and modulate APP processing and/or function in vivo. Here, we have identified pancortin in an unbiased screen for proteins that interact with the ectodomain of APP in the mouse cerebral cortex.
Pancortin, also known as noelin (chicken and Xenopus), olfactomedin-related glycoprotein (rats) and OlfA (humans), is a secreted glycoprotein that is expressed at high levels in the developing and mature cortex (Danielson et al., 1994; Nagano et al., 2000). In the rodent, four isoforms of pancortin are expressed; these variants are named based upon the domains incorporated and are designated BMZ, BMY, AMZ and AMY (Danielson et al., 1994). Each isoform contains the common central M domain. Differential promoter use generates an N terminus composed of either the A or B domain, and alternative splicing produces a C terminus composed of either the Y or Z domain (Danielson et al., 1994) (Fig. 1A). Although the function of pancortin in the developing mammalian nervous system has not been elucidated, the BMZ and AMY isoforms have reported functions in neurodevelopment in Xenopus and chick. The BMZ isoform functions in the migration of neural crest cells in chick embryos (Barembaum et al., 2000), and BMZ and AMY isoforms have opposing roles in regulating the timing of neuronal differentiation in Xenopus (Moreno and Bronner-Fraser, 2001; Moreno and Bronner-Fraser, 2005).
Fig. 1.

Identification of pancortin as an APP-binding partner. (A) Schematic of pancortin isoforms and their domains. (B) Pancortin peptides identified by mass spectrometry in an unbiased screen for extracellular factors within murine cortical slices that interact with the APP ectodomain. (C) Western blot (WB) for pancortin (anti-FLAG) of the lysate and conditioned media (CM) from HEK293 cells transiently transfected with each of the pancortins or control vector both with and without PNGase F treatment. Red arrowheads highlight specific bands. (D) Western blot for pancortin isoforms (‘BMZ/AMZ’ and ‘BMY/AMY’), APP and GAPDH in E16 and adult brain homogenates from wild-type or APP knockout mice.
Here, we report the biochemical and functional interaction of APP and pancortins, and identify a novel function of pancortins in mammalian cortical development. We identify pancortin as an APP-binding partner and show that specific pancortin isoforms inhibit β-secretase cleavage of APP. We reveal that, similar to APP, pancortins play a role in migration of neuronal precursor cells into the cortical plate, with different variants of pancortin having opposing roles in this process. Together, our studies identify a novel function of pancortins in cortical cell migration and give further mechanistic insight into the physiological function of APP, a protein central to the pathogenesis of AD.
MATERIALS AND METHODS
Screen for identification of APP binding partners
C57/BL6 mouse brains were vibratome sectioned (350 μm) and slices were aerated in artificial cerebrospinal fluid (aCSF), incubated with APPsα for 1 hour, followed by incubation in 250 μM DSS (Thermo Fisher, Rockford, IL, USA) for 30 minutes. DTT (1 mM) was added to terminate the reaction. Slices were then homogenized in TEVP buffer [10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 1 mM EGTA, protease inhibitors, 320 mM sucrose] and lysates spun at 1000 g to pellet nuclei. Membranes were then pelleted at 100,000 g for 1 hour and the pellet resuspended in 1% NP40 STEN buffer [150 mM NaCl, 50 mM Tris, 2 mM EDTA and 1.0% (v/v) NP-40]. Following lysis, APP was immunoprecipitated with 8E5 (a gift from Elan Pharmaceuticals) crosslinked to protein G agarose beads. Beads were washed in STEN buffer followed by 1% SDS-STEN buffer, and proteins eluted from the beads with ammonium hydroxide at pH 12 followed by neutralization. Eluted proteins were electrophoresed on a 10-20% Tricine gel, stained with colloidal blue, and bands excised and subjected to MALDI-TOF mass spectrometry.
Plasmids
Generation of APP and control shRNA constructs were described previously (Young-Pearse et al., 2007). Pancortin shRNA constructs were generated by cloning of oligonucleotides encoding the shRNA sequence into the pENTR-U6 plasmid (Invitrogen, Carlsbad, CA, USA). Pancortin shRNAs target the following sequences: shRNA 3 (targeting AMY) GGAGAAGATGGAGAACCAAAT; shRNA 4 (targeting BMZ) GCAACATTGTCATCAGCAAGC. Murine pancortin cDNAs were cloned by PCR into the pCAGGs expression vector for mammalian expression and the cDNAs confirmed to have the sequences corresponding to the following GenBank entries: NM_019498 (BMZ or transcript variant 1), NM_001038612 (BMY or transcript variant 2), NM_001038613 (AMZ or transcript variant 3) and NM_001038614 (AMY or transcript variant 4). In order to rescue shRNA 4 with a non-targetable BMZ construct (‘BMZmut’), four synonymous mutations (bold) were introduced at the shRNA 4 target site: GCAATATCGTGATAAGCAAGC.
Protein expression, immunoprecipitation and western blotting
HEK293 cells were transiently transfected using Lipofectamine 2000 (Invitrogen) or Fugene HD (Promega, Madison, WI, USA). Forty-eight hours after transfection, conditioned media (CM) were collected and cells lysed in 1% NP40 STEN buffer. E18 rat cortical neurons were nucleofected with the Amaxa 4D-Nucleofector Device using the P3 primary cell kit according to the manufacturer's protocol (Lonza, Basel, Switzerland). For deglycosylation of pancortin, lysates and CM from transfected HEK293 cells were treated with PNGase F (New England BioLabs, Ipswich, MA, USA) according to the manufacturer's protocol. For immunoprecipitation (IP), HEK293 cells were transfected with each of the FLAG-tagged pancortin isoforms or domains plus either empty vector alone, human APP (695 residue splice variant), the C-terminal 99 amino acid fragment of APP (C99) or the APP ectodomain E1 or E2 deletion constructs, APPΔ1 and APPΔ2, respectively (kindly provided by T. Sudhof, Stanford University, CA, USA). Lysates were immunoprecipitated with M2-agarose (Sigma, St Louis, MO, USA) or the APP antiserum C9 with protein A and G agarose resin (Sigma) overnight and washed three times with 1% NP40 STEN buffer. Lysates, CM and immunoprecipitations were electrophoresed on 10-20% tricine or 4-12% Bis-Tris gels (Invitrogen) and transferred to nitrocellulose. Western blotting was performed with primary antibodies anti-APP (C9, 1:1000; Selkoe Lab, Brigham and Women's Hospital), anti-APPsα (1736, 1:2000; Selkoe Lab), anti-rodent APP/APPsα (597, 1:200; Immuno-Biological Laboratories, Minneapolis, MN, USA), mouse anti-pancortin (K96/7, 1:1000; UC Davis/NIH NeuroMab Facility, Davis, CA, USA), rabbit anti-pancortin (ab3512, 1:1000; Abcam, Cambridge, MA, USA), anti-GAPDH (1:2000; Millipore, Billerica, MA, USA), anti-FLAG (M2, 1:1000; Sigma), anti-BACE1 (1:500; Millipore), anti-ADAM10 (1:1000; Millipore), anti-ADAM17 (1:500; Abcam), anti-nicastrin (1:1000; BD Biosciences, San Jose, CA, USA) and anti-presenilin 1 (1:1000; Millipore), followed by IRDye800- or IRDye680-conjugated secondary antibodies (1:10,000; Rockland Immunochemicals, Gilbertsville, PA, USA) and detection with the LICOR Odyssey detection system.
APP processing assay
HEK293 cells were plated in six-well plates at 1×106 cells/well and transiently transfected with pancortin isoforms or domains or empty vector (as control) using Fugene HD (Promega). Twenty-four hours post-transfection, media (DMEM + 10% FBS) were replaced, and at 48 hours post-transfection media were collected and centrifuged at 200 g for 5 minutes and cells were lysed in 1% NP-40 STEN buffer. APPsα, APPsβ and Aβ levels in the CM were quantified by ELISA (Meso Scale Discovery, Gaithersburg, MD, USA) and normalized to total intracellular protein. For each isoform, two to four independent experiments performed in triplicate were analyzed. For the individual domains, a representative experiment performed in triplicate is shown. One-way ANOVA tests were performed with the Bonferroni correction for multiple comparisons.
In utero electroporation
Sprague Dawley rats (Charles River Laboratories, Wilmington, MA, USA) were housed and cared for under the guidelines established by Harvard University's Institutional Animal Care and Use Committees in compliance with federal standards. Timed pregnant rats embryonic day 15.5 (E15.5) were anesthetized with ketamine/xylazine (100/10 mixture, 0.1 mg/g body weight, i.p.). The uterine horns were exposed, and a lateral ventricle of each embryo injected with DNA constructs and Fast Green (2 mg/ml; Sigma) via a microinjector (Picospritzer III; General Valve, Fairfield, NJ, USA) and pulled glass capillaries. For characterization of shRNA phenotypes, 1.0-1.5 mg/ml of shRNA was co-electroporated with 0.5 mg/ml pCAG-green fluorescent protein (GFP). For cDNA expression, 0.5 mg/ml pCAG-GFP was co-electroporated with 3.0 mg/ml cDNA constructs. For rescue experiments, 0.5 mg/ml shRNA was co-electroporated with 0.5 mg/ml pCAG-GFP and 3.0 mg/ml rescue constructs. All rescue constructs were expressed in the pCAGGs vector. Electroporation was accomplished by discharging a 500 mF capacitor charged to 50-100 V with a sequencing power supply or with a BTX square wave electroporator, at 50-75 V, for 50 mseconds on followed by 950 mseconds off for 5 pulses. The voltage was discharged across copper alloy oval plates placed on the uterine wall across the head of the embryo. Brains from rat embryos were harvested 3 or 6 days following electroporation in 4% paraformaldehyde by immersion. For each plasmid combination, at least three independent brains were analyzed. Ex vivo culture of electroporated cells were performed as described previously (Rice et al., 2010).
Quantitative analyses of cortical plate entry
For quantitative analyses of migration, all electroporations were performed targeting the same region of the developing rat cortex. This resulted in a reliable electroporation of the dorsal-lateral region of the neocortex adjacent to the lateral ventricle. After harvest, brains were vibratome-sectioned (150 μm) in the coronal plane and immunostained for microtubule-associated protein 2 (MAP2) to delineate the cortical plate. For each electroporation condition (i.e. each set of electroporated DNAs), greater than a total of 500 cells were counted and assessed for their location in either the MAP2+ cortical plate or the intermediate zone. To determine significant changes relative to control electroporations, at least three independent brains were electroporated and analyzed for each DNA condition. For each brain, the percentage of GFP-positive cells in the intermediate zone (IZ) and cortical plate (CP) were calculated. These values were then compared between electroporation conditions using GraphPad InStat (San Diego, CA, USA). Using this program, one-way ANOVA tests were performed with the Bonferroni multiple comparisons test. This analysis was used to determine whether the percentage of cells in the CP in the specified electroporation condition was significantly different from the percentage cells in the CP of control electroporations or, in the case of rescue experiments, of shRNA-receiving electroporations (similarly, these same conditions were assessed for statistical significance when comparing the percentage cells in the IZ for each condition). Data presented in Figs 3 and 4 were generated in concomitant experiments and are presented separately for clarity. Therefore, the combined data from the control electroporation condition with GFP alone for 3 and 6 days post-electroporation are presented twice in both figures for comparison with other conditions.
Fig. 3.

Specific pancortin isoforms reduce β-secretase- but not α-secretase-mediated cleavage of APP. HEK293 cells were transiently transfected with pancortin isoforms and individual domains or empty vector (ctrl). Twenty-four hours post-transfection, media were replaced, and after 24 additional hours media were collected and cells lysed. (A,D) Quantification of endogenous APPsα and APPsβ in the conditioned media (CM) via a multiplex ELISA, which allows for the simultaneous detection of each. (B) Quantification of Aβ40 in the CM by a separate ELISA. (C,E) Western blots of lysates (and conditioned media where noted by ‘CM’) showing expression of endogenous full-length APP, transfected pancortins and each of the secretases. Asterisk in C indicates a non-specific band. In A,B,D, each bar represents data from two to four independent experiments performed in triplicate for A and B, and a single representative experiment performed in triplicate for D. Error bars represent s.e.m. ***P<0.001.
Fig. 4.

Expression levels of different pancortin isoforms have opposing effects in migration of neural precursor cells into the cortical plate. E15.5 rat embryos were co-electroporated with GFP and constructs encoding pancortin cDNAs. (A,D) Quantification of the percent of GFP-positive cells that migrated into the cortical plate after 3 (A) and 6 (D) days post electroporation. Each bar represents the average of data acquired from at least three independent embryos electroporated with constructs listed. Error bars represent s.e.m. *P<0.05, ***P<0.005. Red and black asterisks indicate significance relative to AMY and control, respectively. (B) Representative images from control brains or those expressing the listed pancortin isoforms 3 days post-electroporation. MAP2 immunostaining is shown in red and TBR1 in blue. CP, cortical plate; IZ, intermediate zone. (C) Representative images showing the 6 days post-electroporation time point following electroporation of AMY with and without BMZ or APP co-electroporation. White lines indicate boundaries of the cortical plate. (E) Two panels of a representative image showing GFP, pancortin immunostaining (red) and TOPRO-3 staining (blue), or pancortin immunostaining alone of sections electroporated with AMY and BMZ.
Immunofluorescent staining and confocal microscopy
Coronal sections of electroporated brains were incubated in blocking buffer (2% donkey or goat serum; 0.1% Triton X-100 in PBS) for over 1 hour. Sections were then incubated in primary antibody [anti-MAP2, 1:10,000 (Abcam) and anti-Tbr1, 1:200 (Abcam)] between 6 hours and overnight at 4°C, followed by three washes in PBS. Sections were then incubated with Cy3- and Cy5-conjugated secondary antibodies (1:500 to 1:1000; Jackson ImmunoResearch, West Grove, PA, USA) for over 2 hours followed by four PBS washes. Sections were mounted on glass slides using GelMount (Biomeda, Foster City, CA, USA). Images were acquired using a Zeiss (Oberkochen, Germany) LSM 510 confocal microscope with Axiovert 100M system.
RESULTS
Identification of pancortin as an APP binding partner
In an unbiased screen for extracellular proteins that interact with the APP ectodomain, murine cortical slices were treated with human APPsα, crosslinked and homogenized. APP was immunoprecipitated with a monoclonal antibody to the APP ectodomain and eluted. The eluted proteins were run on SDS-PAGE, and mass spectrometry was performed on horizontal slices excised from the gel that contained any co-immunoprecipitated proteins. This method identified three dozen proteins, including APP. Of the published APP binding partners, only NCAM1 (Bai et al., 2008) was identified. Among the novel putative APP-interacting proteins identified, one of the top hits was pancortin, with recovery of two peptides within the Z domain and one within the B domain (Fig. 1B).
To examine the potential biochemical interactions between APP and pancortin, HEK293 cells were transiently transfected with empty vector (as control) or the four individually FLAG-tagged pancortin isoforms (Fig. 1C). Each of the pancortin isoforms was present in the HEK293 lysate, with BMY and AMY detected as multiple specific bands. In the conditioned media (CM), all of the isoforms with the exception of AMY (which often was not secreted at detectable levels) were detected as a band migrating at a slightly higher molecular weight than in the lysate. The immunoreactive bands in the lysate and the higher migration of pancortins in the CM appear to be due to N-linked glycosylation, as digestion of the respective lysates with Peptide: N-Glycosidase F (PNGase F) resulted in a single lower band now running at the same size in both the CM and lysate for each of the four isoforms.
Developmental expression of pancortin isoforms
Previous studies have shown that multiple pancortin isoforms are expressed in both the embryonic and mature cortex (Danielson et al., 1994; Nagano et al., 2000). Here, multiple antibodies for pancortins were tested by western blot analysis of purified, baculovirus-expressed pancortin isoforms. We found that a rabbit polyclonal antibody (Abcam) preferentially recognizes the Z-domain-containing isoforms AMZ and BMZ and a monoclonal antibody (NeuroMab) that preferentially recognizes the Y-domain-containing isoforms AMY and BMY (supplementary material Fig. S1A). Using these antibodies, AMY/BMY and AMZ/BMZ expression were observed in rodent cortex at both E16 (during active cortical migration) and adult ages (Fig. 1D; supplementary material Fig. S1A). Levels of expression of the pancortins were not affected by genomic disruption (knock out) of APP (Fig. 1D). Immunostaining of wild-type E16 and adult mouse brain sections showed significant signal throughout the cerebral cortex with the pancortin polyclonal antibody (favoring the Z-domain) (supplementary material Fig. S1D,E,H,I). Immunostaining with the pancortin monoclonal antibody (favoring the Y-domain) also showed extensive signal in the E16 brain, but signal was strongly diminished in the cortical plate (supplementary material Fig. S1B,C).
Biochemical characterization of the interaction between APP and pancortins
In order to confirm a biochemical interaction between APP and pancortin, stable HEK293-APP695 cells were transiently transfected with the four individual FLAG-tagged pancortin isoforms or vector alone. Cell lysates were immunoprecipitated with M2 (anti-FLAG)-agarose beads and probed for both pancortin (M2) and APP (C-terminal APP antiserum, C9). APP was able to co-immunoprecipitate with each of the pancortin isoforms (Fig. 2A). Because each of the pancortin isoforms co-immunoprecipitates with APP, we predicted that the M domain common to each isoform was sufficient for an interaction with APP. FLAG-tagged B, M and Z domains of pancortin were expressed in stable HEK293-APP695 cells, and lysates were immunoprecipitated with C9 (APP). The Y and A domains were not examined as they encode only 1 and 22 amino acids, respectively. As predicted, the M domain was sufficient to immunoprecipitate with APP while the B domain was not (Fig. 2B; data not shown). Surprisingly, the Z domain also was capable of interacting with APP; however, this interaction was inconsistent between replicates (Fig. 2B).
Fig. 2.

APP and pancortins biochemically interact. Western blots for APP (anti-APP, C9) and FLAG-tagged pancortin (anti-FLAG, M2) are shown for both the input (lysates) and the immunoprecipitated products. Immunoprecipitations of lysates for FLAG (M2) are shown in A and for APP (C9) in B-F. Asterisks represent non-specific or cross-reactive IgG bands. (A) Western blots of lysates of HEK293-APP695 cells transiently transfected with FLAG-tagged pancortin isoforms or vector alone (ctrl). (B) Western blots of lysates and IPs of HEK293-APP695 cells transiently transfected with FLAG-tagged pancortin domains B, M and Z. (C) Western blots of lysates and IPs of HEK293-APP695 cells transiently transfected with BMZ and AMY isoforms alone or together. (D) Quantification of the percent of BMZ and AMY co-immunoprecipitated with APP relative to the total amount of each isoform in the lysate for BMZ and both the unglycosylated (lower band, ‘ungly’) and glycosylated (upper band, ‘gly’) forms of AMY when BMZ and AMY were expressed separately (−) or together (+). (E) Western blots of lysates and IPs of HEK293 cells transiently transfected with each of the pancortins and either full-length APP (APPfl) or a C-terminal fragment of APP (C99). (F) Western blots of lysates and IPs of HEK293 cells transiently transfected with the AMY isoform of pancortin and either full-length APP, APPΔ1 (lacking residues 36-289), APPΔ2 (lacking residues 288-493) or C99.
To investigate whether APP shows preferential interaction with specific isoforms of pancortin, BMZ and AMY were expressed alone or together in HEK293 cells and the percentage of BMZ and AMY that co-immunoprecipitated with APP relative to the total amount of each isoform in the lysate was quantified. AMY (both glycosylated and unglycosylated forms) had a higher co-immunoprecipitation efficiency than BMZ when each was expressed alone, but when expressed together the co-immunoprecipitation efficiency of AMY decreased and BMZ increased to reach a similar immunoprecipitation efficiency for each (Fig. 2C,D). Interestingly, the unglycosylated form of AMY (lower band) had a much greater binding efficiency than the glycosylated form (upper band) when expressed alone but not when co-expressed with BMZ (Fig. 2C,D). Furthermore, expression of BMZ stabilized the glycosylated form of AMY in the lysate of HEK293 cells (Fig. 2C).
To determine whether pancortin interacts with the intracellular or extracellular region of APP, each of the pancortins plus either full-length APP or the β-secretase-cleaved, membrane-tethered C-terminal fragment of APP (C99) were expressed in HEK293 cells. Each of the pancortins co-immunoprecipitated strongly with full-length APP but weakly or not at all with C99, suggesting a requirement of the APP ectodomain for interaction with pancortin (Fig. 2E). In order to narrow down this region of interaction, the AMY isoform of pancortin and either full-length APP, APPΔ1 (lacking residues 36-289), APPΔ2 (lacking residues 288-493) or C99 were expressed in HEK293 cells. Deletion of the E1 domain of APP led to failure to co-immunoprecipitate AMY, whereas deletion of the E2 domain did not disrupt the interaction with AMY (Fig. 2F). Taken together, these studies demonstrate that APP interacts with each of the pancortin isoforms and that the E1 domain of APP and both the M and Z domains of pancortin are important for the interaction.
Effects of pancortin isoforms on APP proteolytic processing
Pancortin may interact functionally with APP by altering the proteolytic processing of holoAPP, thus regulating potential downstream signaling activities of APP. Furthermore, cleavage of APP to generate Aβ is a process central to Alzheimer's disease pathogenesis, and understanding what factors trigger or inhibit this process may provide valuable insights into disease progression. The APP ectodomain is first shed by either α-secretase or β-secretase to release APPsα or APPsβ, respectively. When APP is cleaved by β-secretase followed by γ-secretase, Aβ is generated. To determine whether pancortins can modulate the processing of endogenous APP, HEK293 cells were transiently transfected with the pancortin isoforms or empty vector (as control). Endogenous APPsα, APPsβ and Aβ levels were measured in the conditioned media using ELISA.
Expression of the B domain-containing isoforms resulted in a dramatic reduction in the generation of β-secretase cleavage products (APPsβ and Aβ), but had no significant effect on the α-secretase cleavage product (APPsα). BMZ and BMY reduced APPsβ levels by 51.8±2.0% and 51.4±0.8%, respectively (Fig. 3A) and reduced Aβ levels by 57.5±3.4% and 65.6±6.1%, respectively (Fig. 3B). The inhibition of β-secretase cleavage of APP by BMZ and BMY was a robust effect seen consistently in each individual experiment. Although in these experiments slight changes in APPsα levels were seen in some experiments, overall there was no statistically significant effect of pancortins on APPsα levels (Fig. 3A). Expression of AMZ did not significantly modulate α-secretase or β-secretase cleavage of APP, and expression of AMY resulted in smaller reductions in β-secretase cleavage of APP when compared with BMZ and BMY (Fig. 3A,B).
We next investigated the mechanism by which pancortin isoforms regulate β-secretase cleavage of APP. The effects of the pancortins on APP processing were not due to alterations in the expression of APP or its secretases, as transfection of the pancortins did not alter endogenous holoAPP or the principal secretases responsible for cleavage at the β-site, BACE-1 (β-site APP cleaving enzyme-1), or α-site, ADAM17 and ADAM 10 (members of the disintegrin and metalloproteinase domain-containing family of proteins) (Fig. 3C). Expression levels of nicastrin and presenilin 1, members of the γ-secretase complex, also were unchanged (Fig. 3C). Because B domain-containing isoforms of pancortin showed the most robust effects on APP processing but the B domain alone was not sufficient for binding APP, we tested whether a physical interaction with APP was necessary for the effects of BMZ and BMY on APP processing. As predicted, expression of the M and Z domains alone had no effect on APPsα or APPsβ (Fig. 3D,E) despite their ability to physically interact with APP (Fig. 2B). However, the B domain alone also was not sufficient to reduce β-secretase-mediated cleavage of APP (Fig. 3D,E), suggesting that BMZ and BMY require a physical interaction with APP via the M and/or Z domains to modulate APP processing.
Role of pancortin and APP in cortical cell migration
As APP has previously been shown to play an important role in cortical precursor cell migration (Young-Pearse et al., 2007; Young-Pearse et al., 2010), we asked whether pancortins also function in this capacity. To address this question, E15.5 rat embryos were electroporated with a cDNA construct encoding GFP alone or co-electroporated with constructs encoding GFP and each of the pancortin variants. Three days after electroporation, brains were harvested, sectioned coronally and immunostained for MAP2, which is expressed in differentiated neurons in the cortical plate, and for Tbr1, which is strongly expressed in cortical layer VI and in remnant subplate cells (and at lower levels in upper layers) at this developmental stage (Bulfone et al., 1995). At this time point (E18.5), electroporated cells are actively migrating from the ventricular zone through the intermediate zone and into the cortical plate. Using this experimental paradigm, cells electroporated with GFP alone were observed in both the cortical plate and below the cortical plate in the intermediate zone (Fig. 4A,B), as expected for E18.5. Expression of each of the pancortin isoforms had differential effects on location of the respective electroporated cells. Whereas expression of BMZ resulted in a distribution of GFP-positive (i.e. BMZ-expressing) cells qualitatively similar to that observed with the control GFP-only electroporation, expression of AMY had the consistent effect of preventing electroporated cells from entering the cortical plate (Fig. 4A,B). Expression of AMZ and BMY had intermediate effects on cell location (Fig. 4A,B).
Given the consistent qualitative effects of AMY overexpression on cell location, we aimed to evaluate these effects by quantifying migration into the cortical plate of the electroporated cells at two time points: 3 days after electroporation, when control (GFP-only) electroporated cells are actively migrating, and 6 days after electroporation when the control cells have completed migration. At least three brains per condition were analyzed, and the summary results of these analyses are shown in Fig. 4A,D. AMY expression had a significant retarding effect on cell migration into the cortical plate at both 3 and 6 days post electroporation. At 6 days, almost all of the cells of control- and BMZ-electroporated brains entered the cortical plate, whereas AMY-expressing brains show a major and significant impairment of cell movement past the IZ/CP boundary (Fig. 4C,D). Interestingly, co-expression of either BMZ or APP rescued this effect of AMY expression (Fig. 4C,D). To address whether expression of BMZ or APP prevented the overexpression of AMY, immunostaining for AMY was performed, and strong immunostaining for AMY was observed in sections from brains co-electroporated with AMY plus either BMZ or APP, at a level qualitatively similar to expression of AMY alone (Fig. 4E and data not shown).
To address whether a physical interaction between APP and pancortin is necessary for proper rescue of the AMY defect by APP, we examined whether deletion of the AMY binding site within APP (APPΔ1) affected its ability to rescue. Indeed, co-expression of APPΔ1 with AMY failed to rescue the migration defect of AMY, whereas no effect was observed upon deletion of a neighboring domain in the APP ectodomain (APPΔ2) that does not affect binding to AMY (Fig. 2F, Fig. 4C,D). These data support the hypothesis that a biochemical interaction between APP and AMY is required for APP to rescue the failure of neuronal precursors to enter the cortical plate with AMY overexpression.
It has been reported that the AMZ and AMY pancortin isoforms are more efficiently secreted than the BMZ and BMY isoforms (Barembaum et al., 2000; Moreno and Bronner-Fraser, 2005; Nagano et al., 2000). However, it also has been reported in multiple studies that the BMZ isoform is robustly secreted (Barembaum et al., 2000; Moreno and Bronner-Fraser, 2001). In order to examine whether pancortin variants are secreted and/or retained intracellularly when expressed in cells of the developing cerebral cortex, brains electroporated with each of the FLAG-tagged variants were sectioned coronally and immunostained with an anti-FLAG antibody. With AMY or AMZ electroporation, immunostaining for FLAG showed a primarily diffuse staining pattern in the cortical plate (Fig. 5A). By contrast, electroporation of BMY or BMZ resulted in an immunostaining pattern that was more strongly cell-associated and colocalized with cellular GFP fluorescence in the cortical plate, suggesting that these proteins are either primarily present intracellularly, or they are associated with the cell membrane of the cells in which they are expressed (Fig. 5B). In order to confirm and extend these observations, we performed ex vivo cultures of the cells electroporated with the individual pancortin isoforms. Total pancortin in the lysate and CM was determined by immunoprecipitation with M2 (anti-Flag)-agarose beads. Both BMZ and AMY were strongly detected in the CM, suggesting that both BMZ and AMY are secreted from electroporated neural cells. However, the level of secreted AMY was greater than BMZ relative to total expression in both the CM and the lysate (Fig. 5C). The levels of AMY in the lysate were below detection levels; therefore, to obtain higher expression levels and more accurately quantify the relative secretion of BMZ and AMY in neurons, we transfected E18 rat primary cortical neurons ex vivo using the Amaxa Nucleofector system. Although both BMZ and AMY were again highly secreted, the percentage of AMY in the CM relative to total AMY in both the lysate and CM was greater than that of BMZ (Fig. 5D,E). Co-expression of both BMZ and AMY had no effect on the secretion of either isoform (Fig. 5D,E). Thus, we conclude that all isoforms are secreted, but the A-domain isoforms are secreted more robustly than the B-domain isoforms in developing neurons.
Fig. 5.

Differential secretion of the pancortin isoforms from neuronal cells. (A,B) E15.5 rat embryos were co-electroporated in utero with GFP and constructs listed, and harvested 3 days later. Brains were sectioned and immunostained for FLAG and MAP2. Shown are representative images of GFP (green), anti-FLAG (red) and anti-MAP2 (blue). (C,D) Lysates and CM from ex vivo cultures of cortical neurons electroporated in utero (C) or transfected in vitro (D). BMZ or AMY were immunoprecipitated with M2 (anti-Flag)-agarose and western blots performed to detect FLAG-tagged pancortin (anti-FLAG) (C,D). (E) Quantification of the percent of BMZ and AMY detected in the CM relative to total (CM + lysate) following in vitro transfection of BMZ and AMY both together and separately.
Next, we asked whether endogenous pancortin expression is necessary for proper cortical migration. shRNA constructs were generated to target different pancortin isoforms. The pancortin shRNAs were first tested via transient transfection of HEK cells with each FLAG-tagged pancortin isoform. Two days after this co-transfection, cells were lysed and western blots performed. Pancortin shRNA 3 targets the ‘M’ domain and was expected to target each of the pancortin isoforms. Interestingly, although all splice variants contain the target site for shRNA 3, it was only effective in knocking down expression of AMY (Fig. 6A). Pancortin shRNA 4 targets domain ‘Z’, and it effectively knocked down BMZ but not other pancortin isoforms (Fig. 6A).
Fig. 6.

Pancortins are required for proper migration in the developing cerebral cortex. (A) shRNA constructs targeting pancortin isoforms were generated and tested for their ability to knock down each of the FLAG-tagged pancortin isoforms. HEK293 cells were transfected with construct combinations as labeled. GFP was co-transfected in all cases to monitor transfection efficiency. Forty-eight hours following transfection, cells were lysed and western blots performed to examine GFP and pancortin levels, using M2 (α-FLAG). (B-F) E15.5 rat embryos were electroporated in utero with GFP and constructs listed, and harvested 3 or 6 days later. Brains were dissected, fixed, sectioned coronally and immunostained for MAP2 and TBR1. (B) Representative images of a 3-day harvest following introduction of shRNA 3 (which knocks down AMY) and shRNA 4 (which knocks down BMZ). MAP2 immunostaining is shown in red and TBR1 in blue. (C) Representative images of brains electroporated with constructs listed and harvested 6 days later. White lines delineate the boundaries of the cortical plate as determined by MAP2 and TBR1 immunostaining. (D,E) Quantification of the percentage of GFP-positive cells present in the cortical plate 3 (D) and 6 (E) days post electroporation. Each bar presents data from the average of at least three independent embryos electroporated with constructs listed. Error bars represent s.e.m. *P<0.05, **P<0.01, ***P<0.005. Black asterisks represent significance relative to GFP vector control; red asterisks represent significance relative to AMY; green asterisks represent significance relative to pancortin shRNA 4 (BMZ); blue asterisks represent significance relative to APP shRNA. Quantifications from Fig. 3A,D for GFP, AMY, BMZ, AMY+BMZ and AMY+APP are re-shown here for direct comparison with knock down data. (F) Representative images of brains electroporated with shRNA targeting AMY with and without co-electroporation of AMY, APP or BMZ and harvested at 6 days post electroporation. MAP2 immunostaining is shown in red.
With harvesting at 3 days post-electroporation, pancortin shRNA 3 (targeting AMY) had no qualitative effect on cell location, whereas pancortin shRNA 4 (targeting BMZ) resulted in a failure of electroporated cells to enter the cortical plate (Fig. 6B). The effects of knock down of BMZ with shRNA 4 persisted at 6 days post-electroporation, producing a phenotype strikingly similar to that observed with APP knock down or AMY overexpression (Fig. 6C) (Young-Pearse et al., 2007). Quantification of these effects was performed at both 3 and 6 days, with analysis of at least three brains per condition (Fig. 6D,E). Knock down of AMY (by shRNA 3) only modestly decreased migration into the cortical plate at 3 days, and by 6 days, no significant effect on entry into the cortical plate was observed. However, those cells that entered the cortical plate were not localized to a single layer but rather were found in a ‘wavy’ pattern throughout the cortical plate, which could be rescued by either APP or BMZ overexpression (Fig. 6F).
As expected, electroporation of shRNA 3, which knocks down AMY, rescued the AMY overexpression phenotype (Fig. 6D,E). By contrast, knock down of BMZ (with shRNA 4) did not rescue the AMY overexpression phenotype at 3 or 6 days (Fig. 6D,E). Similarly, knock down of APP had no additional effect on AMY overexpression. Overexpression of a mutated BMZ cDNA construct, which contains 4 synonymous mutations in the shRNA target site (‘BMZmut’), rescued the knock down phenotype of pancortin shRNA 4 (BMZ) at 3 and 6 days, suggesting that the effects observed with this shRNA are specifically due to a loss of pancortin expression (Fig. 6E). Interestingly, overexpression of BMZ partially rescued the APP knock down phenotype at 6 days (Fig. 6E). Taken together, these data demonstrate a role of the pancortins in proper migration of neuronal precursor cells into the cortical plate. Furthermore, our data suggest that APP and pancortin may act in concert to regulate cortical precursor cell migration, as APP overexpression rescues both the defect in cortical plate entry of AMY overexpression and the ‘wavy’ phenotype of AMY knock down.
DISCUSSION
A role for pancortins in mammalian cortical development
Pancortin, originally named for its high expression in the cerebral cortex (Nagano et al., 1998), has been implicated in migration of neural crest cells and neuronal differentiation in chick embryos and Xenopus, respectively (Barembaum et al., 2000; Moreno and Bronner-Fraser, 2001; Moreno and Bronner-Fraser, 2005). However, no studies have yet identified a role for pancortin in the development of the mammalian brain. In a single report of a pancortin knockout mouse, it was proposed that pancortins are mediators of ischemia-induced neuronal cell death in the adult brain (Cheng et al., 2007). However, it remains unclear what the normal function of pancortin may be during development, as the loss of pancortin only protected adult but not embryonic neurons against ischemic death, and other putative developmental phenotypes were not presented. Here, using the technique of in utero electroporation to both overexpress pancortin and knock down endogenous pancortin in a subset of neural cells, we reveal a novel function of pancortin in the migration of neuronal precursors into the cortical plate of the rat cortex. AMY knock down produces a phenotype in which neuronal precursors enter the cortical plate but exhibit abnormal positioning, a unique phenotype that we have not observed with in utero electroporation of over a dozen different genes thought to have roles in neuronal development. Overexpression of the AMY isoform or knock down of the BMZ isoform results in failure of neuronal precursors to enter the cortical plate. Phenotypes of abnormal cell positioning can result from a number of different types of primary insult, including a decline in cell survival, defects in the cytoskeletal network that are crucial for the mechanics of cell movement, and disruptions of signaling pathways that instruct cells when to migrate and when to stop and differentiate. Here, we believe that the primary defect is not in cell survival, as there is no dramatic loss in electroporated cells between 3 and 6 days post electroporation. Furthermore, electroporated cells are able to migrate out of the ventricular zone and through the intermediate zone with AMY overexpression and BMZ knock down, suggesting that these cells have the necessary machinery for cell movement. Rather, cells stop just below the cortical plate boundary, similar to the defect observed with knock down of APP. We hypothesize that this is due to a defect in signaling within the cells that either fails to direct the cells to continue migrating or else directs the cells to prematurely halt migration.
Our data support an opposing role for BMZ and AMY in regulating developmental processes. Although BMZ promotes migration into the cortical plate, AMY appears to inhibit normal cortical plate entry. Furthermore, overexpression of BMZ rescues the phenotype observed with AMY overexpression. Interestingly, the observation of functional antagonism between the BMZ and AMY isoforms in neurodevelopment is consistent with previous studies in Xenopus in which BMZ promoted neuronal differentiation and AMY rescued this effect (Moreno and Bronner-Fraser, 2001; Moreno and Bronner-Fraser, 2005), perhaps through direct interaction of the BMZ and AMY isoforms (Moreno and Bronner-Fraser, 2005). In addition, our observation that both isoforms are able to bind to APP but differ in their co-immunoprecipitation efficiency with APP when each are expressed separately, but reach similar co-immunoprecipitation efficiencies with APP when expressed together, lend support to an additional model of competition between these isoforms for binding to putative receptors.
Biochemical and functional interaction of pancortin and APP
Here, we report a novel biochemical and functional interaction between pancortins and APP. In an unbiased screen, we identified pancortins as extracellular binding partners for APP, and confirmed that each of the pancortin isoforms binds either directly or indirectly to the APP ectodomain. The E1 region of the APP ectodomain and the M and Z domains of pancortin were crucial for a physical interaction. Furthermore, expression of the B domain-containing isoforms strongly inhibits β-secretase-mediated cleavage of APP, suggesting a biochemical consequence of the association between pancortins and APP. However, the B domain, which confers activity but cannot physically interact with APP, has no effect on β-secretase processing when expressed alone, suggesting that a physical interaction between APP and pancortin is required to modulate APP processing.
Knock down of BMZ or overexpression of AMY phenocopies a loss of APP expression in the developing rodent brain, each showing a specific defect in migration of neural precursor cells into the cortical plate. We hypothesize that for this function in cortical development, pancortins act in part through binding to APP, but also through APP-independent mechanisms. Based upon the data presented herein (summarized in Fig. 7), we propose that BMZ binding to the extracellular domain of APP promotes proper migration into the cortical plate while binding of AMY to APP inhibits proper migration. Thus, loss of APP or BMZ, or else overexpression of AMY inhibits cortical plate entry (Figs 4, 6). We posited that if AMY overexpression inhibits the functions of BMZ and APP in normal migration, then overexpression of either BMZ or APP should rescue the AMY defect, which was the effect that was observed (Fig. 4D). Furthermore, we found that APP rescues the AMY defect through a physical interaction, as blockade of the interaction through deletion of the E1 domain of APP prevents rescue of the AMY defect (Fig. 4D). In HEK293 cells, co-expression of BMZ and AMY reduces binding of AMY to APP, suggesting that BMZ may rescue the AMY defect by preventing AMY from binding APP (Fig. 2C,D). Interestingly, overexpression of APP does not rescue the loss of BMZ (Fig. 4D,E), indicating that the migration-promoting effect of APP is dependent upon expression of BMZ.
Fig. 7.
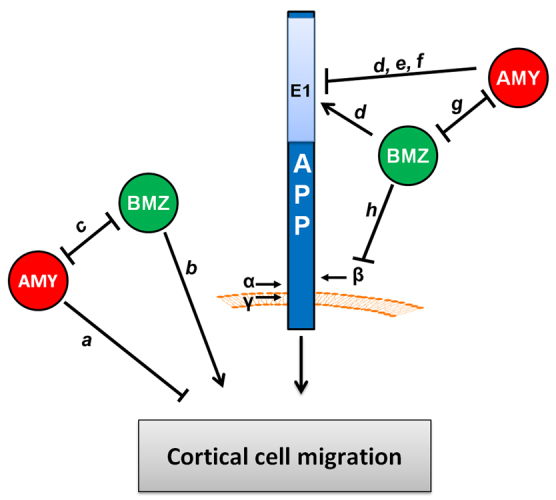
Summary of the effects observed of pancortins and APP on migration in the developing cerebral cortex. In utero electroporation of pancortin constructs in the developing rat brain revealed novel and opposing roles of the AMY and BMZ isoforms in cortical cell migration. (a) Cells overexpressing AMY fail to enter the cortical plate, suggesting that AMY inhibits cortical plate entry. (b) Knock down of BMZ results in failure to enter the cortical plate, suggesting that endogenous BMZ promotes cortical plate entry. (c) BMZ overexpression rescues the defect observed with AMY overexpression. Taken together with previous studies of neuronal differentiation in Xenopus (Moreno and Bronner-Fraser, 2005), the data support a model where AMY and BMZ mutually inhibit the activity of the other. (d) In support of an APP-dependent mechanism, peptides to the B and Z domain of pancortin were identified in an unbiased mass spectrometry screen for APP interacting proteins and both BMZ and AMY co-immunoprecipitate with APP, an interaction disrupted by deletion of the E1 region of the APP ectodomain. (e,f) Overexpression of APP rescues AMY overexpression (e), but blockade of the interaction through deletion of the AMY binding site within APP (the E1 domain) (f) prevents rescue of the AMY defect by APP, supporting a model whereby AMY inhibits the function of APP in cortical cell migration through a physical interaction. (g) Expression of BMZ can decrease the co-immunoprecipitation efficiency of AMY with APP, which suggest that BMZ may rescue the AMY defect by preventing AMY from binding to APP. (h) BMZ inhibits β-secretase cleavage of APP; however, whether this activity is mechanistically involved in regulating migration has yet to be determined.
Taken together, the data presented suggest that pancortins functionally and biochemically interact with APP in cortical development. However, our data showing that BMZ overexpression can partially rescue the phenotype observed with knock down of APP suggest that pancortins also may act through APP-independent mechanisms, perhaps through interaction with the highly homologous APP family members (APLP1 and APLP2) or possibly through other type I transmembrane domain proteins. Future studies are warranted to examine how pancortins interact with other known signaling pathways to regulate proper migration and differentiation of neuronal precursor cells in the mammalian cerebral cortex.
Supplementary Material
Acknowledgements
We thank Marion Tilearcio and Sophie Ran Wang for technical assistance, and Matt LaVoie and members of the Selkoe and Young-Pearse laboratories for helpful discussions.
Footnotes
Funding
This work was supported by the National Institutes of Health (NIH) [R00 MH085004 to T.L.Y.-P., R01 AG06173 to D.J.S.] and a Jerome L. Rappaport Fellowship (to H.C.R.). The authors acknowledge the donors of ADR, a program of the American Health Assistance Foundation, for support of this research (T.L.Y.-P.). Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.082909/-/DC1
References
- Araki W., Kitaguchi N., Tokushima Y., Ishii K., Aratake H., Shimohama S., Nakamura S., Kimura J. (1991). Trophic effect of β-amyloid precursor protein on cerebral cortical neurons in culture. Biochem. Biophys. Res. Commun. 181, 265-271 [DOI] [PubMed] [Google Scholar]
- Bai Y., Markham K., Chen F., Weerasekera R., Watts J., Horne P., Wakutani Y., Bagshaw R., Mathews P. M., Fraser P. E., et al. (2008). The in vivo brain interactome of the amyloid precursor protein. Mol. Cell. Proteomics 7, 15-34 [DOI] [PubMed] [Google Scholar]
- Barembaum M., Moreno T. A., LaBonne C., Sechrist J., Bronner-Fraser M. (2000). Noelin-1 is a secreted glycoprotein involved in generation of the neural crest. Nat. Cell Biol. 2, 219-225 [DOI] [PubMed] [Google Scholar]
- Bulfone A., Smiga S. M., Shimamura K., Peterson A., Puelles L., Rubenstein J. L. (1995). T-brain-1: a homolog of Brachyury whose expression defines molecularly distinct domains within the cerebral cortex. Neuron 15, 63-78 [DOI] [PubMed] [Google Scholar]
- Cheng A., Arumugam T. V., Liu D., Khatri R. G., Mustafa K., Kwak S., Ling H. P., Gonzales C., Xin O., Jo D. G., et al. (2007). Pancortin-2 interacts with WAVE1 and Bcl-xL in a mitochondria-associated protein complex that mediates ischemic neuronal death. J. Neurosci. 27, 1519-1528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson P. E., Forss-Petter S., Battenberg E. L., deLecea L., Bloom F. E., Sutcliffe J. G. (1994). Four structurally distinct neuron-specific olfactomedin-related glycoproteins produced by differential promoter utilization and alternative mRNA splicing from a single gene. J. Neurosci. Res. 38, 468-478 [DOI] [PubMed] [Google Scholar]
- De Strooper B., Annaert W. (2000). Proteolytic processing and cell biological functions of the amyloid precursor protein. J. Cell Sci. 113, 1857-1870 [DOI] [PubMed] [Google Scholar]
- Ghiso J., Rostagno A., Gardella J. E., Liem L., Gorevic P. D., Frangione B. (1992). A 109-amino-acid C-terminal fragment of Alzheimer's-disease amyloid precursor protein contains a sequence, -RHDS-, that promotes cell adhesion. Biochem. J. 288, 1053-1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J., Selkoe D. J. (2002). The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297, 353-356 [DOI] [PubMed] [Google Scholar]
- Ho A., Südhof T. C. (2004). Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc. Natl. Acad. Sci. USA 101, 2548-2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe H. S., Wessner D., Beffert U., Becker A. G., Matsuoka Y., Rebeck G. W. (2005). F-spondin interaction with the apolipoprotein E receptor ApoEr2 affects processing of amyloid precursor protein. Mol. Cell. Biol. 25, 9259-9268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoe H. S., Tran T. S., Matsuoka Y., Howell B. W., Rebeck G. W. (2006). DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. J. Biol. Chem. 281, 35176-35185 [DOI] [PubMed] [Google Scholar]
- Hoe H. S., Lee K. J., Carney R. S., Lee J., Markova A., Lee J. Y., Howell B. W., Hyman B. T., Pak D. T., Bu G., et al. (2009). Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 29, 7459-7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q. H., Futagawa T., Yang W. L., Jiang X. D., Zeng L., Takeda Y., Xu R. X., Bagnard D., Schachner M., Furley A. J., et al. (2008). A TAG1-APP signalling pathway through Fe65 negatively modulates neurogenesis. Nat. Cell Biol. 10, 283-294 [DOI] [PubMed] [Google Scholar]
- Moreno T. A., Bronner-Fraser M. (2001). The secreted glycoprotein Noelin-1 promotes neurogenesis in Xenopus. Dev. Biol. 240, 340-360 [DOI] [PubMed] [Google Scholar]
- Moreno T. A., Bronner-Fraser M. (2005). Noelins modulate the timing of neuronal differentiation during development. Dev. Biol. 288, 434-447 [DOI] [PubMed] [Google Scholar]
- Nagano T., Nakamura A., Mori Y., Maeda M., Takami T., Shiosaka S., Takagi H., Sato M. (1998). Differentially expressed olfactomedin-related glycoproteins (Pancortins) in the brain. Brain Res. Mol. Brain Res. 53, 13-23 [DOI] [PubMed] [Google Scholar]
- Nagano T., Nakamura A., Konno D., Kurata M., Yagi H., Sato M. (2000). A2-Pancortins (Pancortin-3 and -4) are the dominant pancortins during neocortical development. J. Neurochem. 75, 1-8 [DOI] [PubMed] [Google Scholar]
- Osterfield M., Egelund R., Young L. M., Flanagan J. G. (2008). Interaction of amyloid precursor protein with contactins and NgCAM in the retinotectal system. Development 135, 1189-1199 [DOI] [PubMed] [Google Scholar]
- Perez R. G., Zheng H., Van der Ploeg L. H., Koo E. H. (1997). The beta-amyloid precursor protein of Alzheimer's disease enhances neuron viability and modulates neuronal polarity. J. Neurosci. 17, 9407-9414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A., Ochalski P. G., Lee C. H., Howell B. W. (2006). Mouse disabled 1 regulates the nuclear position of neurons in a Drosophila eye model. Mol. Cell. Biol. 26, 1510-1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pramatarova A., Chen K., Howell B. W. (2008). A genetic interaction between the APP and Dab1 genes influences brain development. Mol. Cell. Neurosci. 37, 178-186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priller C., Bauer T., Mitteregger G., Krebs B., Kretzschmar H. A., Herms J. (2006). Synapse formation and function is modulated by the amyloid precursor protein. J. Neurosci. 26, 7212-7221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice H., Suth S., Cavanaugh W., Bai J., Young-Pearse T. L. (2010). In utero electroporation followed by primary neuronal culture for studying gene function in subset of cortical neurons. J. Vis. Exp. 44, pii: 2103 doi:10.3791/2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ring S., Weyer S. W., Kilian S. B., Waldron E., Pietrzik C. U., Filippov M. A., Herms J., Buchholz C., Eckman C. B., Korte M., et al. (2007). The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 27, 7817-7826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe D. J. (2011). Resolving controversies on the path to Alzheimer's therapeutics. Nat. Med. 17, 1060-1065 [DOI] [PubMed] [Google Scholar]
- Soba P., Eggert S., Wagner K., Zentgraf H., Siehl K., Kreger S., Löwer A., Langer A., Merdes G., Paro R., et al. (2005). Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 24, 3624-3634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wang B., Yang L., Guo Q., Aithmitti N., Songyang Z., Zheng H. (2009). Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J. Neurosci. 29, 10788-10801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Pearse T. L., Bai J., Chang R., Zheng J. B., LoTurco J. J., Selkoe D. J. (2007). A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 27, 14459-14469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Pearse T. L., Chen A. C., Chang R., Marquez C., Selkoe D. J. (2008). Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 3, 15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young-Pearse T. L., Suth S., Luth E. S., Sawa A., Selkoe D. J. (2010). Biochemical and functional interaction of disrupted-in-schizophrenia 1 and amyloid precursor protein regulates neuronal migration during mammalian cortical development. J. Neurosci. 30, 10431-10440 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.