

Abstract
Viral vectors are emerging as potent basic research tools and gene therapy vehicles in many laboratory animal models. However, little information is available on the potential shedding of these vectors and the consequent exposure risk to investigators and animal care staff from animals over time. This study provides empirical information to Institutional Biosafety Committees and animal care programs, to enhance their ability to perform risk management of laboratory animals treated with viral vectors. Control experiments evaluated the limit of detection of third-generation lentivirus, recombinant adeno-associated virus, and E1-deleted adenovirus tested directly from stocks and after application onto cage plastic or bedding. After inoculation of ICR or NOD-SCID mice, we quantified the recovery of viral vector genomes directly from blood, urine, and fecal samples and assessed the persistence of infectious vector at the site of injection and from soiled bedding at different time points after inoculation. No differences were seen between ICR and NOD-SCID mice. We saw no evidence of vector amplification after in vivo inoculation. The most environmentally persistent vector was recombinant adeno-associated virus, which has no known pathogenicity in humans. In light of these data, we conclude that commonly used replication-deficient viral vectors pose minimal exposure risk by 72 h after inoculation. Prudent precautions at Animal Biosafety Level 2 are warranted during initial administration, but Level 1 safety measures may be sufficient after cage changing and biosafety evaluation.
Abbreviations: Ad, adenovirus; CMV, cytomegalovirus; EGFP, enhanced green fluorescent protein; LV, third-generation lentivirus; qPCR, qualitative PCR; rAAV, recombinant adeno-associated virus
The era of customizable transgenic mice and molecular manipulation of viruses to deliver targeted genes has revolutionized our ability to study gene-specific functions. A typical viral vector is engineered by separating the wildtype virus genome onto multiple nonoverlapping plasmids, with the minimal coding sequences necessary for replication provided in trans to the transgene construct, which contains only those cis-acting viral elements necessary to direct packaging into a noninfectious virion. Large portions of the genome necessary for pathogenicity are completely removed. The resulting engineered viral vectors offer the ability to specifically target tissues and genes and are emerging as potent gene therapy vehicles. With these tools for basic research, we are equipped to answer questions about the effects of genes and proteins on development, behavior, sensation, and organ function and in disease.
Each viral vector has unique properties and targets defined cell populations (Figure 1).1,18 For example, lentiviral (LV) vectors are used for in vivo gene delivery because they efficiently transduce both dividing and nondividing cells and stably integrate into the host genome, providing long-term transgene expression. Lentiviral vectors typically are based on HIV1 that has been substantially debilitated to provide multiple safeguards against the production of replication-competent lentivirus.6,32 Third-generation lentiviral vectors have deletions in the promoter region of the long terminal repeats, rendering these vectors self-inactivating after proviral integration. In addition, they include deletions in all 6 major genes involved in pathogenesis—including the genes for Env, Vpr, Vpu, Vif, Nef, Rev, and (in some versions) Tat. Typically, vesicular stomatitis virus glycoprotein is substituted for the native viral Env protein. The transient packaging system contains less than 30% of the original viral genome, and the produced replication-deficient self-inactivating vector particles integrate less than 8% of the HIV genome into infected cells. Such an HIV1-based vector, pseudotyped with the vesicular stomatitis virus glycoprotein as its major envelope protein, is the most commonly used lentiviral vector and is available commercially from most major life-science vendors and from core facilities at various scientific institutions.
Figure 1.

Viral vector characteristics.
Adenoviral vectors are small (90 nm) nonenveloped icosahedral vectors with a linear double-stranded DNA genome. They infect dividing and nondividing cells but do not actively integrate into the host cell genome; therefore, expression is transient in actively dividing cells and tissues. Long-term transgene expression in nondividing cells and tissues has been achieved as well. Adenoviral vectors are most commonly derived from adenoviral serotype 5 (Ad5).9 Helper-free, replication-defective recombinant Ad5 vectors often are generated through deletion of the essential E1a/b and nonessential E3 regions of the viral genome. Removal of these sequences allows the introduction of a gene of interest into the deleted region, with a packaging capacity of approximately 8 kb. Adenoviral replication is dependent on the E1a/b region of the viral genome; as such, recombinant Ad5 vectors are unable to replicate, and packaging of replication-defective Ad5 vector particles is achieved through the transfection of a linearized plasmid containing the recombinant Ad5 vector genome into HEK293/17 cells, or derivatives of this cell line, which stably express the Ad5 E1a gene. With E1a/b- and E3-deleted Ad5 vectors, 2 separate recombination events would need to occur during packaging to generate replication-competent wildtype virus. ‘Gutless’ (that is, helper-dependent) Ad5 viral vector systems have also been developed; in these systems, all viral genes have been removed from the recombinant vector genome. This manipulation greatly increases the packaging capacity of these vectors—from approximately 8 kb to 36 kb—and markedly reduces immune responses in vivo. Helper virus is required for the production of gutless adenoviral vector, and various strategies have been developed to remove this helper virus from the subsequent viral vector preparation.25
Recombinant adeno-associated viral (rAAV) vectors are derived from the AAV2 virus, a very small (20 nm) icosahedral nonenveloped virus with a linear single-stranded DNA genome that does not actively integrate into the host cell genome. Wildtype AAV2 is not a known human pathogen, and coinfection with a helper virus, such as adenovirus, is required for AAV2 to replicate within a host cell. All of the wildtype viral genome has been deleted from rAAV2 vectors, except for the 5′ and 3′ inverted terminal repeat regions, which are the only cis acting, noncoding viral sequences necessary for packaging of a recombinant vector genome into replication-defective particles. rAAV2 vectors typically are produced in a helper-virus-free system.34 The recombinant rAAV2 genome expressing the gene of interest, adenoviral helper genes, and the AAV coding sequences for the rep and cap proteins are provided on separate plasmids and are transiently transfected into producer cells, such as HEK293T/17. The packaging capacity of rAAV2 vectors is approximately 4.7 kb, although larger genomes have successfully been packaged or expressed in vivo. The produced, replication-defective rAAV2 vector particles can also be cross-packaged with capsid proteins from other AAV serotypes22 as well as engineered capsid proteins,15,31,33 offering attractive systems for altering the cell and tissue tropism of the produced vector.
Institutional Biosafety Committees serve a critical function in providing oversight and balanced risk assessment of hazard use in research facilities, effectively managing the risk associated with viral vector use, both in the laboratory and in vivo after treatment of laboratory animals. Decisions related to the biosafety of viral vector use often are based on the known pathogenic properties of the parental wildtype viruses, in combination with case-by-case determination of additional risks related to the characteristics of the particular viral vector subtype and the biologic activity (if known) of the expressed transgene or regulatory nucleic acid to be packaged. Similar strategies are used to further classify these agents into 1 of 4 specific risk groups.3 Replication-deficient Ad and LV vectors currently are classified in section III-D-3-a as group 2 viruses, and experiments with these agents involving animals are categorized into section III-D-4-b.20 Although AAV vectors are classified as group 1 viruses, the biologic activity of the expressed transgene may lead an Institutional Biosafety Committee to mandate the use of Biosafety Level 2 or Animal Biosafety Level 2 precautions with rAAV vectors. To date, few published reports address direct measurement of the exposure risk from animals treated with different viral vectors have been available to Institutional Biosafety Committee members. Given this paucity of information, recommendations frequently are made based on the biologic properties of the parent (often virulent and replicative) strains of these vectors. These biologic properties most specifically relate to virulence, pathogenicity, infectious dose, environmental stability, route of spread, and communicability of the wildtype strain.30
Several potential risks are associated with these agents, including transmission of the viral vector itself, homologous recombination resulting in the generation of replication-competent vector or virulent wildtype-like virus, contamination by residual helper viruses (if used during vector preparation), and adverse effects of the inserted gene or regulatory nucleic acid (for example, oncogenesis).17,27 The wildtype, parental HIV and adenovirus strains can be stable for a month or longer in a laboratory setting.29 Although important in the context of traditional virus research, these guidelines are only broadly applicable to advanced, later-generation agents that have been specifically and rationally modified to deliver target genes without generating associated vector-induced disease or uncontrolled replication.
Managing these vector agents in rodents according to Animal Biosafety Level 2 guidelines can sometimes be problematic when the use of primary engineering controls (for example, a biologic safety cabinet) is impractical or impossible. The ability to incorporate suggested safety measures may be limited, in part, because of the experimental design, infrastructural limitation, and work processes. Examples of these procedures include behavioral testing, breeding, bioimaging, MRI and CT scanning, and other specialized research activities that require the use of large or immobile equipment. In addition, routine practices in vivaria may increase the potential for contamination thorough direct contact with infected animals, fomite contamination, or potential aerosolization through handling of soiled bedding.
A group of investigators at our institution are developing and using novel or modified viral vectors in various unconventional ways. These methodologies are expanding rapidly to other disciplines, and many institutions are witnessing the expansion of ‘gray areas’ in risk assessment and containment. Guidance in evaluating the safety and use of LV vectors is available,19 but few reports detailing empirical data are available to support these concepts. If the real threat of shedding or infection can be quantified, it then would be feasible to establish accurately the risk associated with treated animals and enhance laboratory and vivarium safety practices.
We hypothesized that risks associated with viral vectors in mice may be lower than those generally associated with the parent viruses. Once given to animals, the current state-of-the-art replication-deficient viral gene therapy and targeting vectors likely pose minimal risk of exposure to staff, and application of standard Animal Biosafety Level 2 biohazard precautions when working with rodents carrying viral vectors may not be necessary indefinitely. We report here our evaluation of the risk of the shedding of several classes of genetically modified viral vectors from rodents and subsequent potential for human exposure to these vectors.
Materials and Methods
Vectors.
The E1a/b-, E3-deleted Ad5, rAAV2/2, and third-generation self-inactivating LV vectors used in this study were produced in the Gene Transfer, Targeting, and Therapeutics (GT3) Core Facility (The Salk Institute, La Jolla, CA) under Biosafety Level 2 conditions by using helper-free transient production protocols (http://vectorcore.salk.edu/protocols.php) that are based on previously developed methods.8,10,28 These vectors all contain a cytomegalovirus (CMV)–enhanced green fluorescent protein (EGFP) reporter transgene cassette to allow the use of fluorescence detection and PCR in assaying for the presence of infectious vector particles in environmental and tissue samples. Unless otherwise stated, the titers of the stock viral vectors were: LV-CMV-EGFP, 2.14 × 1010 IU/mL; AAV2/2-CMV-EGFP, 3.01 × 1011 IU/mL; and Ad5-CMV-EGFP, 1.9 × 1010 IU/mL. Infectious titers (IU) were calculated by scoring EGFP fluorescence per cell in HEK293T/17 cells (ATCC, Manassas, VA) 72 h after inoculation by using serial log dilutions (10−2 to 10−5) of each viral vector.
Preparation of mouse biologic samples.
Blood (100 μL per mouse per time point) was collected from the tail vein, snap-frozen on dry ice, and stored at −80 °C until use. Immediately prior to isolation of viral nucleic acids, each blood sample was diluted with 400 µL HBSS (Life Technologies, Carlsbad, CA) and mixed thoroughly by vortexing to dissolve any clotting. Urine samples were collected directly from each mouse at each time point by using a sterile cotton swab. Mice were restrained gently and the swab applied to the urethral opening; mice typically responded by urinating and defecating. The cotton swab containing mouse urine was snap-frozen on dry ice and stored at −80 °C until needed. Immediately prior to isolation of viral nucleic acids, the swab was submerged in 500 µL HBSS and vortexed to get the urine into solution. Feces (2 pellets per mouse per time point) were collected in a sterile tube as the animal defecated, snap-frozen on dry ice, and stored at −80 °C until needed. Immediately prior to nucleic acid isolation, each sample was mixed by vortexing with 500 µL HBSS until completely resuspended and centrifuged at 13,350 × g in a benchtop microfuge (model 5415R, Eppendorf, Hauppauge, NY) for 1 min to pellet debris.
Preparation of PCR and RT-PCR controls.
A log dilution series for each batch of viral vector (LV-CMV-EGFP, rAAV2/2-CMV-EGFP, and Ad5-CMV-EGFP) used in vivo was used as a positive control for qualitative PCR (qPCR) and RT–qPCR analysis. For each viral vector, 1 × 105 IU was added to 100 µL whole blood from an uninfected control mouse and the volume brought to 500 µL with HBSS. Samples were further diluted by using HBSS to generate a log dilution series ranging from 1 × 105 to 1 × 101 IU vector in a 500-µL volume. Positive-control samples were processed identically to the mouse samples.
Nucleic acid purification.
Both viral DNA and RNA were purified from mouse samples (blood, urine, and feces) and positive-control samples (High Pure Viral Nucleic Acid Large Volume Kit, Roche, Nutley, NJ) according to the manufacturer's instructions. All plasticware and reagents were RNase- and DNase-free. Purified nucleic acid was stored at −20 °C until use.
Detection of viral nucleic acids by RT-PCR and PCR.
Purified viral nucleic acids (RNA and DNA) were amplified (SensiFAST One-Step RT–PCR Kit, Bioline USA, Taunton, MA). For amplification of vector genomes from mice treated with DNA viral vectors (rAAV2/2 and Ad5), the RT step was omitted from the reaction. The reaction mix was made according to the manufacturer's instructions; 4 µL of each control or experimental sample was used in a final volume of 20 µL per reaction. For all samples, EGFP-specific primers were used, because this cDNA was present in each viral vector used and absent from the mouse genome. The primers used (EGFP fwd, 5′ GCT GAC CCT GAA GTT CAT CT 3′; EGFP rev, 5′ GAA GTC GTG CTG CTT CAT GT 3′) amplify a 127-bp amplicon, have been extensively validated for qPCR and RT-qPCR, and are used routinely for titration of EGFP-expressing viral vectors in the GT3 core facility. Cycling conditions for RT–PCR were 45 °C for 10 min; 95 °C for 2 min; 40 cycles of 95 °C for 5 s, 60 °C for 10 s, 72 °C for 5 s; and 72 °C for 5 min. Cycling conditions for PCR only were identical except for omission of the initial step (45 °C for 10 min) for RT. RT–PCR and PCR products were analyzed by using agarose gel electrophoresis, with a 2% agarose gel. DNA bands were stained by using GelSTAR DNA binding dye (Lonza, Basel, Switzerland). Bands were visualized by UV transillumination and images collected by using an imaging system (Gel-Logic, Bio-Rad, Hercules, CA).
Vector viability in the environment.
Control experiments evaluated the limit of detection for stock viral vectors that were serially diluted (10−1 to 10−9 in 1× HBSS) and incubated for 1 h at room temperature with 3 different types of clean and soiled (mixed with urine and feces) standard rodent contact bedding (3 mL of bedding incubated with 1mL of viral vector): corncob (Bed O’ Cobs, The Anderson's, Maumee, OH), careFRESH (Absorption, Ferndale, WA), and Pure O'cell (The Anderson's). In addition, undiluted vector stocks were spotted directly onto sterile, dry, animal-cage plastic. These experiments were designed to validate recovery protocols and determine vector viability over time under controlled conditions. Briefly, aliquots (10 µL) of each vector were spotted on sterile, polysulfone mouse caging (Allentown, Allentown, NJ) and swabbed at 0 h, 24 h, 72 h, 5 d, and 14 d after spotting. Finally, we tested the recovery of viral vectors that were premixed with soiled bedding samples and processed identically to the control bedding samples. Vector recovery and analysis proceeded as described.
Mice.
We evaluated the selected viral vectors for shedding and transmission after inoculation of mice. Naïve SPF 8-wk-old female Hsd:ICR (CD1) and NOD.CB17-Prkdcscid/NCrHsd mice (Harlan Laboratories, Indianapolis, IN) were housed according to institutional Biosafety Level 2 housing standards in static microisolation shoebox-style cages (Allentown) on 1/4-in. corncob bedding with standard rodent diet (Laboratory Rodent Diet 5001, Lab Diet, PMI Nutrition International, Brentwood, MO), and reverse-osmosis–purified water. Mice were free from adventitial infection including viruses, bacteria, and parasites (internal and external). The CD1 strain was chosen to represent animals that react robustly to new antigen and infection, whereas the NOD/SCID strain lacks adaptive immunity and would therefore be more likely to support robust replication of infectious agents. This diversity is important because the immunologic status of many genetically altered mice, including those whose genome has been manipulated or altered (by selective breeding, by mutations, or artificially in the laboratory), is not well defined, and shedding of contaminated or recombined viral vectors may be pronounced. All animal procedures were reviewed and approved the Salk Institute Animal Care and Use Committee. All mice were treated in accordance with the Guide for the Care and Use of Laboratory Animals.12
Mice were anesthetized briefly by using isoflurane delivered in 100% O2 via precision vaporizer and a shielded nose cone and received an intravenous inoculation of 100 µL virus (dose per mouse: LV-CMV-EGFP, 9.51 × 108 IU; AAV2-CMV-EGFP, 1.03 × 109 IU; and Ad5-CMV-EGFP, 1.9 × 109 IU). On days −1, 0, 1, 2, 3, and 7 (relative to inoculation), we assessed levels of infectious virus at the site of injection (tail), in soiled cage bedding, and in tissues. Samples (5 mL each) of soiled bedding were collected, and swabs of the injection site were taken by using wetted, sterile cotton-tipped applicators. After sample collection, mice were transferred to clean cages. Mice were euthanized by using CO2, and tissues collected at 7 d after exposure. CO2 was delivered from a compressed gas source at a standardized 20% fill rate. Tissues were divided and either snap-frozen on dry ice for DNA extraction and qPCR analysis of viral genomes or fixed in 4% paraformaldehyde in PBS and processed for fluorescence microscopy. A cohort of 2 mice that received rAAV2/2 were euthanized at day 21 after exposure to assess cross-reactivity of vector antigens with NS1 murine parvoviral serology. Subsequently, to assess the presence and potential shedding of viral vectors directly, an additional cohort of 15 NOD.CB17-Prkdcscid/NCrHsd mice was inoculated as described earlier, and biologic samples (blood, urine and feces) were collected from each mouse on days −1, 1, 3, 7) and processed by using PCR and RT–PCR for the detection of viral nucleic acids.
Analysis of recovered infectious viral vectors.
All swabs and bedding samples for infectious assays were put immediately into 500 mL of 293T growth medium (DMEM high glucose with pirodoxone, L-glutamine, and 10% FBS; Mediatech, Manassas, VA) on ice, swirled, and incubated for 30 min at room temperature. The total inoculate from each sample was centrifuged for 5 min at 10,000 × g to remove debris, and the supernatant was filtered through a 0.22-µm filter (Millipore, Billerica, MA). The clarified filtrate was used to infect 5 × 104 HEK293T/17 cells per well in a 12-well tissue culture plate, with 1.5 mL of medium per well. The titers of recovered infectious virus were determined by assaying for EGFP expression by using epifluorescence microscopy or fluorescence-associated cell sorting at 48 h after infection.
Results
Using a cell-based assay for detection of replication-competent LV,7 we found no evidence of recombinant or replication-competent viruses in these vector stocks. rAAV2/2 vectors were generated by using a completely helper-free system with no known risk of generating replication-competent vector and therefore were not tested for replication competency. E1a/b-, E3-deleted Ad5 vector stocks also were generated by using a helper-virus–free system. These stocks were free of wildtype Ad5 according to results from a sensitive cell-based qPCR assay.13
Recovery of infectious viral vectors from the environment.
From control experiments testing the sensitivity of the cell-based infectivity assay, the lower limit of detection as assayed by EGFP expression in infected 293T cells was 1 × 103 IU/mL for LV, 6 × 104 IU/mL for rAAV2/2, and 2 × 104 IU/mL for Ad5. LV RT–qPCR titers were compared with biologic titers; 1 × 104 gene copies per mL in the RNA samples was equivalent to a biologic titer of 1 × 103 IU/mL.
Infectious LV was recoverable for 24 h after spotting onto dry plastic. In comparison with LV, Ad5 was more environmentally stable, and infectious Ad5 vector was recoverable for as long as 3 d after inoculation. rAAV2/2 was by far the most environmentally stable vector, with recoverable infectious vector present for as long as 14 d after inoculation onto plastic (Figure 2). In spiked soiled bedding, maximal LV recovery was 21.5% of inoculate after 30 min incubation, dropping to 5.2% after 24 h. No infectious LV was detected after a 72-h incubation. For rAAV2/2 and Ad5, infectious vector was detected at all time points assayed, with the following efficiencies of recovery: rAAV2/2 at 30 min, 95.2%; rAAV2/2 at 24 h, 92.6%: rAAV2/2 at 72 h, 84.4%; Ad5 at 30 min, 90.7%; Ad5 at 24 h, 71.2%; Ad5 at 72 h, 33.1% (Figure 3). Recovery did not differ significantly between bedding substrates but was significantly (P < 0.005) greater from soiled compared with dry bedding, regardless of bedding substrate (data not shown).
Figure 2.

Recovery of infectious viral vectors from cage plastic. In this experiment, 10-µL aliquots of vector were dried on cage plastic, recovered by swabbing at indicated intervals, and cultured on 293T cells for 48 h to assess infectivity. Beginning titers for each time point were 1.9 × 108 TU for the Ad5 construct, 3 × 109 TU for AAV2 construct, and 2.14 × 108 TU for the LV construct. For each sample, 3 wells of cells were infected, and the images shown are representative of each replicate. Scale bar, 50 µm.
Figure 3.
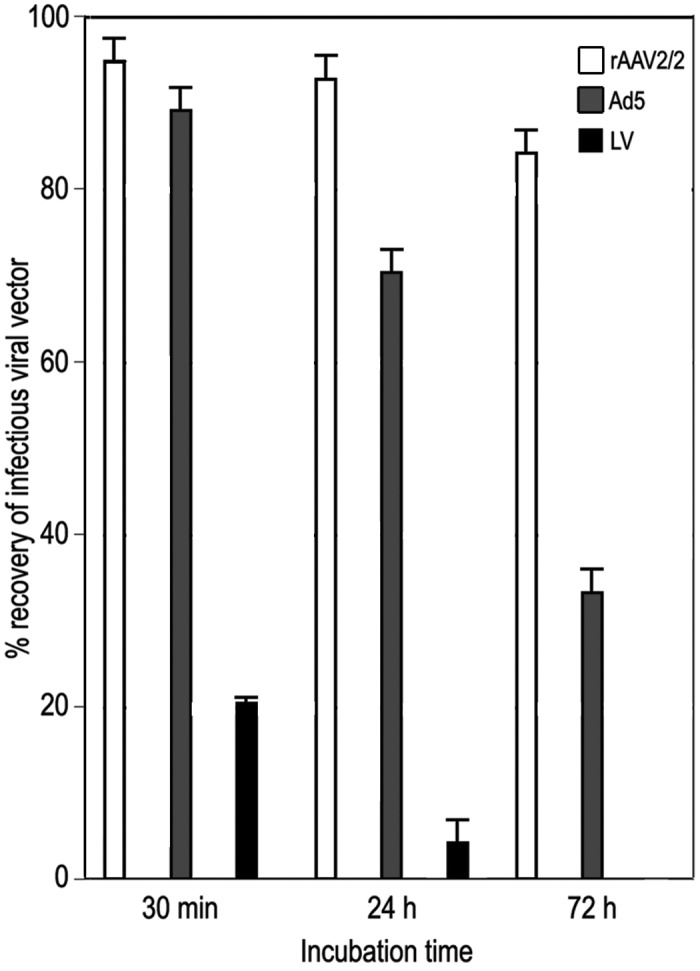
Recovery of infectious viral vectors from spiked bedding. Viral vectors were recovered from soiled rodent bedding at 30 min, 24 h, or 72 h after inoculation and used to infect 293T indicator cells (3 replicates for each time point). The histogram shows the percentage recovery of infectious viral vector compared with that of cells treated with stock vectors in parallel.
Recovery of infectious viral vectors from treated animals.
After administration to experimental mice, infectious virus was delectable on swabs of the injection site (tail) for as long as 24 h for LV, 72 h for rAAV2/2, and 24 h for Ad5 (Figure 4). We further analyzed the data from the LV swabs, because this viral vector typically is considered to have a higher risk profile than either rAAV2/2 or Ad5 given that LV can integrate stably into host cell genomes. The concentration of infectious LV recovered on swabs of the injection site was between 1 to 3 × 103 IU/mL, or about 0.00005% of initial inoculate. For LV at time 0, 9 of 10 mice were positive on tail swab, whereas only 2 of 10 swabs taken at 24 h after injection showed minimal but detectable virus at the site of injection (Figure 5). No detectable infectious LV, rAAV2/2, or Ad5 was found at any time in soiled corncob bedding samples from experimental cages despite group housing of infected mice.
Figure 4.

Recovery of infectious viral vectors from injection sites. Inoculum isolated from a direct swab of the injection site of mice treated with LV, Ad5, or rAAV2/2 constructs was cultured on 293T cells for the indicated times. Data shown are GFP expression at 48 h after infection of cells. All swabs were negative by 7 d after injection of mice. Scale bar, 50 µm.
Figure 5.

Recovery of lentiviral genomes from injection site. qRT-PCR quantification of LV RNA genomes isolated from inoculum recovered from direct swabs of the injection site at various time points after injection. Bar, SEM of qRT-PCR replicates (n = 3 per sample).
Shedding of viral vectors from treated mice.
On the basis of these initial results, we tested for the presence of potentially infectious viral vector directly from biologic samples isolated from inoculated mice (n = 4 per vector). Blood, urine, and fecal samples were collected on days −1, 1, 3, and 7 relative to inoculation. In addition, an untreated naïve, sentinel mouse was housed in each cage with the vector-treated mice and samples were collected in parallel with those from treated mice. Viral nucleic acids were isolated from each sample, and the presence of viral vector was assessed by PCR (Ad5 and rAAV2/2) or RT–PCR (LV) using EGFP-specific primers. For all viral vectors tested, the lower limit of detection was 200 IU. For LV- and Ad5-treated mice, no viral nucleic acids were detected before or at any time point after inoculation. For rAAV2/2-treated mice, viral nucleic acids were detected at 24 h after inoculation in the blood samples from 2 (mice 6 and 7) of the 4 treated mice and the fecal samples from 1 (mouse 7) of the 4 treated mice (Figure 6 A). Urine samples from the same mice were negative at 24 h after inoculation. By 72 h, the rAAV2/2-inoculated mice were negative for the presence of viral nucleic acids in blood, urine, and feces. All samples from untreated sentinel mice were negative at all points tested. Control experiments tested the sensitivity of the EGFP-specific PCR and RT–PCR assays for detection of viral nucleic acids in biologic samples (blood, urine, and feces) isolated from each experimental mouse over time. For each viral vector, the lowest amount of control vector that gave a positive band by PCR or RT–PCR was 2 × 102 IU per reaction (Figure 6 B).
Figure 6.

Detection of viral vector genomes in biologic samples by using PCR and RT-PCR. PCR assay of mice inoculated with rAAV2/2-CMV-EGFP shows (A) virus-positive bands in 24-h blood samples from 2 animals and a virus-positive band in the 24-h fecal sample from 1 animal. All urine samples from the same mice at this time point were negative. No LV or AD5 nucleic acids were detected at any time point after inoculation. The right panel shows positive-control PCR and RT-PCR bands after amplification directly from the vector stocks (2 µL vector per reaction). (B) Control PCR and RT-PCR assays of isolated nucleic acids from each viral vector (LV-CMV-EGFP, rAAV2/2-CMV-EGFP, Ad5-CMV-EGFP) after being spiked into whole blood from an untreated mouse. Very low concentrations of virus were used deliberately in the spiking experiments to define the lower limit of detection and demonstrate sensitivity and specificity above background.
All mice that received viral vectors showed robust EGFP expression in the liver, confirming delivery and transduction (Figure 7). At 5 d after injection, qPCR of organs demonstrated high LV proviral integration in the liver (4.83 × 107 ± 1.8 × 106 IU/mL), moderate integration in kidney (7.69 × 106 ± 3.95 × 105 IU/mL), and minimal integration in lung tissue (3.15 × 105 ± 1.98 × 104 IU/mL; Figure 8). At 21 d after injection, Ad5 and AAV2 generated robust levels of EGFP expression in liver by fluorescence microscopy and by qPCR (Ad5, 2.03 × 107 ± 6.33 × 106 IU/mL; rAAV2/2, 1.69 × 108 ±1.64 × 107 IU/mL). Levels were below the limit of detection for other organs by both fluorescence microscopy and qPCR. For all vectors, no differences in vector recovery, infection, or shedding were seen between ICR and NOD-SCID mice.
Figure 7.

Transduction of target tissue by viral vectors. These micrographs demonstrate virus-mediated GFP fluorescence in whole-mount mouse livers at 7 d after tail-vein injection. Scale bar, 50 µm.
Figure 8.

Vector genomes in tissue. qPCR analysis of viral vector concentrations (TU/mL) in liver, kidney, and lung at 7 d after tail-vein injection of LV-CMV-EGFP (LV), Ad5-CMV-EGFP (Ad5), or rAAV2/2-CMV-EGFP (AAV2). Bar, SEM of replicates (n = 3 per sample).
Discussion
Infectious virus was not found in bedding samples from cages housing LV-, rAAV2/2-, or Ad5-treated mice at any time point after injection but was detectable at the site of injection for several days, most likely representing leakage after withdrawal of the needle. We saw no evidence of viral vector amplification or environmental persistence from leakage after in vivo inoculation. We intentionally group-housed mice after inoculation to exaggerate the potential for environmental recovery. Persistence in the laboratory environment of virulent wildtype parent strains of these vectors can be prolonged for a month or longer for HIV and adenovirus.29 In comparison to that of the parental strains, we noted much shorter persistence of modified viral vectors on animal caging and bedding. In pilot studies, recovery of all vectors tested was enhanced in soiled bedding as compared with dry bedding. Soiled bedding has higher water content and may have helped to preserve the biologic integrity of these enveloped and nonenveloped vectors, especially the enveloped LV vectors, which are the least environmentally stable vector that we tested and which are sensitive to uncontrolled desiccation.
Direct assessment of the presence of viral vector genomes (DNA or RNA) in blood, urine, and fecal samples from treated mice demonstrated that no viral vectors were present in these samples by 72 h after intravenous inoculation. rAAV2/2 was the only viral vector detected in biologic samples isolated directly from treated mice (blood and feces at 24 h after inoculation only). In addition, rAAV2/2 was the most environmentally persistent vector (longer than 14 d on dried cage plastic) we tested. rAAV2/2 vectors have no known pathogenicity in humans and are currently in phase I to III human clinical trials for the treatment of ocular diseases.4 Importantly, the current study also demonstrated that naive virus-free sentinel mice that were group-housed with each cohort of vector-treated mice tested negative for all 3 viral vectors assayed. These findings present robust evidence of the lack of shedding and transmission of viral vectors between animals housed within the same cage after intravenous inoculation. The lower detection limit of this assay was less than 2 × 102 IU of viral vector per reaction, which is equivalent to a circulating concentration of each viral vector of less than 10 IU/mL of blood. Given this sensitivity, it is unlikely that mice with undetectable viral loads at or below this threshold would present a significant exposure risk. Infectious doses for the wildtype parental viruses in humans are either not known (Ad5 and rAAV2/2) or vary across a large range (HIV1). For example, minimum infectious doses of HIV1 reportedly vary from 2 to 65,000 genome copies in human blood samples.23 Minimum infectious doses for these wildtype viruses cannot be applied accurately to the viral vectors derived from them, because the vectors are replication-defective and as such cannot amplify an initial infectious event, nor are these vectors pathogenic.
No differences were observed between ICR and NOD SCID mice. Immunocompromised NOD SCID mice were included as a ‘worst-case scenario’ for shedding. These results from NOD SCID mice are important because the immunologic status of many genetically altered mice is not well defined, and shedding of contaminated viral vectors may be pronounced. The use of these strains also provides a sensitive model for assessing the risks of shedding, because these mice are robust and susceptible to many viruses. Although C57Bl/6 mice are a common background strain for genetically altered mice, this strain is known to have innate resistance to many viruses.2,26,36
Our study was designed to specifically test the actual presence of LV, rAAV2/2, and Ad5 vectors on the animal, in body fluids and feces, and in the environment (either from contamination, leakage, or shedding) after intravenous administration of these vectors. The plasma half-life of LV after intravenous administration was evaluated in a rat model,14 which showed that LV is virtually undetectable in plasma by 24 h after injection. Our current study confirms the absence of LV after 24 h by using a sensitive RT–PCR based assay and extends the information to urine and feces, both of which are alternative routes for viral vector shedding. Furthermore, Ad5, a nonenveloped viral vector that was shown to be more environmentally persistent that were the LV vectors tested, was undetectable in all samples of blood, urine, and feces by 24 h after injection. The risk of environmental shedding or contamination appears to follow the in vivo time course. It is likely that shedding profiles will differ for alternative delivery routes, such as intranasal aerosol delivery or direct injection into solid tissues such as intramuscular inoculation and stereotactic delivery to the CNS. Although beyond the scope of the current study, testing these alternative delivery routes potentially would provide additional important information to aid in risk management of laboratory animals after vector administration.
One additional concern when administering viral vectors to laboratory animals is the existing viral load, if any, of the animal or laboratory and animal care staff. Although direct testing of viral vector mobilization and shedding from animals with preexisting viral infections was beyond the scope of the current study, it is worth commenting on any potential increases in exposure risk that such evaluation may provide. We were able to confirm that the LV vectors used in this study were replication-incompetent, consistent with data generated by the National Center for Gene Therapy at Indiana University, which tested 60 third-generation self-inactivating vector preps and found no replication-competent LV.19 LV vectors are associated with a low but statistically relevant probability of mobilization, encaspidation, and shedding of integrated vector genomes in animals and humans with preexisting HIV1 infections.16 For animals falling into this category that are capable of supporting HIV1 infection, including rodents engrafted with human cells or tissues and nonhuman primates, Institutional Biosafety Committees typically perform additional risk assessment on a case-by-case basis. We also recommend notifying laboratory staff regarding the theoretical risk of viral vector mobilization in HIV1-positive personnel.
rAAV2-based viral vectors theoretically could be packaged and shed from animals with preexisting infections of helper viruses, such as adenovirus, vaccinia, and herpes simplex virus.11,24 Given that replication-competent wildtype rAAV is not associated with any known human pathology, risk assessment in these cases should primarily be based on the biologic properties of the vector-packaged transgene. Recombinant Ad5-based vectors with E1a/b and E3 deletions would not be mobilized in virally infected animals other than those already infected by wildtype adenovirus virus. Most immunocompetent persons have robust neutralizing antibody responses to adenoviruses,4 which effectively prevents infection. Interestingly, replication of Ad5 vectors can occur in human cells with dysfunctional p53 pathways,35 so there is a theoretical risk of mobilization and shedding of these vectors from mice harboring mutations in the p53 tumor suppressor pathway. Overlap between the tumor cell program and adenoviral replication is being harnessed to develop the next generation of oncolytic viral therapies for cancer treatment.21
We present compelling data by using standard vector constructs for LV, rAAV2/2, and Ad5. Published guidelines provide few data to support the recommendations.19 As suggested previously,9 there is no standardized approach to managing viral vector use, ranging from reduction in containment requirements after as little as 24 h to maintenance at Animal Biosafety Level 2 levels for the life of the animal. We sought to provide empirical data to prove our hypothesis and to provide Institutional Biosafety Committees and animal care programs with temporally quantified data. Essential for adequate risk assessment is knowledge of viral vector integrity (sequence, source, and validation). Source data should be evaluated for each case and appropriate biosafety containment assigned after evaluation of the host, inserted gene transcripts, and anticipated biologic outcome from activation. The potential for viral vector shedding from animal models after administration is dependent on the route of vector administration, vector titer, vector detection methods, and sample collection techniques. In the present study, we administered high titers of vector (108 to 109 IU) to mice by using a common route of delivery (intravenous) that has a high likelihood of generalized dissemination and increased risk of shedding compared with localized injection to target organs, such as the brain and eye.
In light of our current data, novel replication-deficient LV, rAAV2/2, and Ad5 vectors pose minimal exposure risk to staff and colony animals after intravenous inoculation. Animal Biosafety Level 2 precautions are warranted during the initial exposure stage, but levels of virus resulting from leakage or shedding dissipate quickly under typical housing conditions. Because of the potential for the presence of viral vectors in the environment, caging and equipment that can serve as potential fomites should be thoroughly disinfected between uses. After thorough evaluation by the Institutional Biosafety Committee, Animal Biosafety Level 1 precautions may be appropriate after a routine cage-change at 72 h after vector administration.
Acknowledgment
This work was supported by a grant from the American College of Laboratory Animal Medicine (ACLAM) Foundation.
References
- 1.Boeckle S, Wagner E. 2006. Optimizing targeted gene delivery: chemical modification of viral vectors and synthesis of artificial virus vector systems. AAPS J 8:E731–E742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brownstein DG, Smith AL, Johnson EA. 1981. Sendai virus infection in genetically resistant and susceptible mice. Am J Pathol 105:156–163 [PMC free article] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention and National Institutes of Health. 2009. Biosafety in microbiological and biomedical laboratories, 5th ed. Bethesda (MD): Department of Health and Human Services.
- 4.Chirmule M, Propert K, Magosin S, Qian Y, Wilson J. 1999. Immune response to adenovirus and adeno-associated virus in humans. Gene Ther 6:1574–1583 [DOI] [PubMed] [Google Scholar]
- 5.Cideciyan AV. 2010. Leber congenital amaurosis due to RPE65 mutations and its treatment with gene therapy. Prog Retin Eye Res 29:398–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. 1998. A third-generation lentivirus vector with a conditional packaging system. J Virol 72:8463–8471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Escarpe P, Zayek N, Chin P, Borellini F, Zufferey R, Veres G, Kiermer V. 2003. Development of a sensitive assay for detection of replication competent lentivirus in large-scale HIV-based vector preparations. Mol Ther 8:332–341 [DOI] [PubMed] [Google Scholar]
- 8.Grieger JC, Choi VW, Samulski RJ. 2006. Production and characterization of adeno-associated viral vectors. Nat Protoc 1:1412–1428 [DOI] [PubMed] [Google Scholar]
- 9.Hackney RW, Myatt TA, Gilbert KM, Caruso RR, Simon SL. 2012. Current trends in institutional biosafety committee practices. Appl Biosafety 17:11–18 [Google Scholar]
- 10. Haj-Ahmad Y, Graham FL. 1986. Characterization of an adenovirus type 5 mutant carrying embedded inverted terminal repeats. Virology 153:22–34 [DOI] [PubMed] [Google Scholar]
- 11.Handa H, Carter BJ. 1979. Adeno-associated virus DNA replication complexes in herpes simplex virus or adenovirus-infected cells. J Biol Chem 254:6603–6610 [PubMed] [Google Scholar]
- 12. Institute for Laboratory Animal Research. 2011. Guide for the care and use of laboratory animals, 8th ed. Washington (DC): National Academies Press.
- 13.Ishii-Watabe A, Uchida E, Iwata A, Nagata R, Satoh K, Fan K, Murata M, Mizuguchi H, Kawasaki N, Kawanishi T, Yamaguchi T, Hayakawa T. 2003. Detection of replication-competent adenoviruses spiked into recombinant adenovirus vector products by infectivity PCR. Mol Ther 8:1009–1016 [DOI] [PubMed] [Google Scholar]
- 14.Karlen S, Zufferey R. 2007. Declassification of rodents exposed to third-generation HIV-based vectors into class 1 animals. Appl Biosaf 12:93–99 [Google Scholar]
- 15.Kwon I, Schaffer DV. 2008. Designer gene-delivery vectors: molecular engineering and evolution of adeno-associated viral vectors for enhanced gene transfer. Pharm Res 25:489–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Logan AC, Haas DL, Kafri T, Kohn DB. 2004. Integrated self-inactivating lentiviral vectors produce full-length genomic transcripts competent for encapsidation and integration. J Virol 78:8421–8436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moens W. 2003. Preface [hot topic: biosafety of virus-derived vectors (guest editor: William Moens)]. Curr Gene Ther 3:i–ii [Google Scholar]
- 18.Moullier P. 2005. Editorial [hot topic: recombinant adeno-associated virus: current achievements and limitations (guest editor: Philippe Moullier)]. Curr Gene Ther 5:263 [Google Scholar]
- 19. National Institutes of Health. [Internet] 2006. Biosafety considerations for research with lentiviral vectors. [Cited 11 May 2011]. Available at: http://oba.od.nih.gov/rdna_rac/rac_guidance_lentivirus.html.
- 20. National Institutes of Health . [Internet] 2011. NIH guidelines for research involving recombinant DNA molecules (NIH guidelines). [Cited11 May 2011]. Available at: http://oba.od.nih.gov/oba/rac/Guidelines/NIH_Guidelines.htm.
- 21.O'Shea CC. 2005. Viruses—seeking and destroying the tumor program. Oncogene 24:7640–7655 [DOI] [PubMed] [Google Scholar]
- 22.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, Samulski RJ. 2002. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol 76:791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reid S, Juma OA. 2009. Minimum infective dose of HIV for parenteral dosimetry. Int J STD AIDS 20:828–833 [DOI] [PubMed] [Google Scholar]
- 24.Schlehofer JR, Ehrbar M, zur Hausen H. 1986. Vaccinia virus, herpes simplex virus, and carcinogens induce DNA amplification in a human cell line and support replication of a helper-virus-dependent parvovirus. Virology 152:110–117 [DOI] [PubMed] [Google Scholar]
- 25.Segura MM, Alba R, Bosch A, Chillon M. 2008. Advances in helper-dependent adenoviral vector research. Curr Gene Ther 8:222–235 [DOI] [PubMed] [Google Scholar]
- 26.Selin LK, Santolucito PA, Pinto AK, Szomolanyi-Tsuda E, Welsh RM. 2001. Innate immunity to viruses: control of vaccinia virus infection by γδ T cells. J Immunol 166:6784–6794 [DOI] [PubMed] [Google Scholar]
- 27.Tenenbaum L, Lehtonen E, Monahan PE. 2003. Evaluation of risks related to the use of adeno-associated virus-based vectors. Curr Gene Ther 3:545–565 [DOI] [PubMed] [Google Scholar]
- 28.Tiscornia G, Singer O, Verma IM. 2006. Production and purification of lentiviral vectors. Nat Protoc 1:241–245 [DOI] [PubMed] [Google Scholar]
- 29.Valtierra HN. 2008. Stability of viral pathogens in the laboratory environment. Appl Biosaf 13:21–26 [Google Scholar]
- 30.Van Regenmortel MHV. 2003. Viruses are real, virus species are man-made: taxonomic constructions. Arch Virol 148:2481–2488 [DOI] [PubMed] [Google Scholar]
- 31.Vandenberghe LH, Wilson JM, Gao G. 2009. Tailoring the AAV vector capsid for gene therapy. Gene Ther 16:311–319 [DOI] [PubMed] [Google Scholar]
- 32.Verma IM, Weitzman MD. 2005. Gene therapy: 21st-century medicine. Annu Rev Biochem 74:711–738 [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Faust SM, Rabinowitz JE. 2011. The next step in gene delivery: molecular engineering of adeno-associated virus serotypes. J Mol Cell Cardiol 50:793–802 [DOI] [PubMed] [Google Scholar]
- 34.Xiao X, Li J, Samulski RJ. 1998. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J Virol 72:2224–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeh P, Perricaudet M. 1997. Advances in adenoviral vectors: from genetic engineering to their biology. FASEB J 11:615–623 [DOI] [PubMed] [Google Scholar]
- 36.Zawatzky R, Gresser I, DeMaeyer E, Kirchner H. 1982. The role of interferon in the resistance of C57BL/6 mice to various doses of herpes simplex virus type 1. J Infect Dis 146:405–410 [DOI] [PubMed] [Google Scholar]