

Abstract
Purpose
The purpose of this study is to identify the genetic defect in a Turkish family with autosomal recessive retinitis pigmentosa, nanophthalmos, and optic disc drusen.
Methods
Ophthalmological examinations consisted of measuring the best-corrected visual acuity and the refractive error, electroretinography, optical coherence tomography, B-mode ultrasonography, and fundus photography. The involvement of the membrane frizzled-related protein (MFRP) gene in this family was studied with direct DNA sequencing of the coding exons of MFRP and with linkage analysis with microsatellite markers. After MFRP was excluded, genome-wide homozygosity mapping was performed with 250 K single nucleotide polymorphism (SNP) microarrays. Mutation analysis of the crumbs homolog 1 (CRB1) gene was performed with direct sequencing.
Results
Ophthalmological evaluation of both affected individuals in the family revealed a decreased axial length (18–19 mm), retinal dystrophy, macular edema, and hyperopia of >+8.0 diopters. Sequencing of MFRP did not reveal any pathogenic changes, and microsatellite marker analysis showed that the chromosomal region did not segregate within the disease in this family. Genome-wide homozygosity mapping using single nucleotide polymorphism microarrays revealed a 28-Mb homozygous region encompassing the CRB1 gene, and direct sequencing disclosed a novel homozygous missense mutation (p.Gly833Asp) in CRB1.
Conclusions
Previous studies associated mutations in the MFRP gene with the syndrome nanophthalmos-retinitis pigmentosa-foveoschisis-optic disc drusen. In this study, we demonstrated that a similar disease complex can be caused by mutations in the CRB1 gene.
Introduction
Nanophthalmos-retinitis pigmentosa-foveoschisis-optic disc drusen disease complex has been described as a distinct recessive entity [1,2]. The disease can be described as characteristically having a short axial eye length (13.0–18.5 mm), high hyperopia (+8.00 to +25.00 diopters), retinal pigment epithelium atrophy, formation of optic disc drusen, and foveoschisis [3]. Mutations in the membrane frizzled related protein (MFRP) gene were described as responsible for causing the disease complex [1–3]. The MFRP gene is located on chromosome 11q13 and encodes a membrane receptor protein specifically expressed in the retinal pigment epithelium and ciliary epithelium of the eye [4]. MFRP is thought to play a role in eye development, as mutations in the gene that codes for this protein have been associated with nanophthalmos, retinitis pigmentosa (RP), and other degenerative disorders [5,6].
In this study, we describe the clinical and genetic features of a consanguineous Turkish family with two affected siblings with RP, nanophthalmos, and optic disc drusen. We excluded the involvement of the MFRP gene in the family, and report a novel mutation in CRB1, a gene previously associated with autosomal recessive RP and Leber congenital amaurosis [7].
Methods
Clinical analyses
Ophthalmological examinations of the affected siblings included measuring the best corrected visual acuity (BCVA) and refractive error, electroretinography (ERG) according to the International Society for Clinical Electrophysiology of Vision (ISCEV) protocol [8], B-mode ultrasonography, fundus photography, and spectral domain optical coherence tomography (SD-OCT). Because of the early-onset and severity of the disease, the unaffected parents who had no complaints were not subjected to the ophthalmological examinations.
Genetic analyses
We obtained blood samples and pedigree information after receiving informed consent from all individuals. Approval was obtained from the institutional review board. Genomic DNA was isolated from lymphocytes with automated DNA extraction (Hamilton ML Star, Hamiloton Bonaduz AG, Bonaduz, Switserland).
Primers were designed using Primer3 online software. All of the coding exons and the exon/intron boundaries of MFRP were amplified with polymerase chain reaction (PCR) using the primers contained in Table 1.
Table 1. Primers used for amplification and sequence analysis of the MFRP, RBP3 and RP1 gene.
| Gene | Exon | Sequence (5′-3′) | |
|---|---|---|---|
|
MFRP |
1 |
F: CCCCCACACAGAGACAGAGT |
R: CTGGTGCTGGGTCTTAGGAG |
| |
2 and 3 |
F: CTCCTAAGACCCAGCACCAG |
R: TCATGGAGTTTCATTCCAAAGC |
| |
4 and 5 |
F: ACCCAGCTCCTCTGAACGC |
R: GATAGTGGTTCAGGACACGG |
| |
6 and 7 |
F: CTGACCCTGCTCTTGGAGC |
R: CTTGAACCCAGATCAGACGC |
| |
8 and 9 |
F: ATGGAGGCACAGATCCTAGC |
R: ACAGTGAGGATGGAGTTATCC |
| |
10 and 11 |
F: GTCAGCCAGGGCTGGTGC |
R: GCACCCAGCCTGCTCAGG |
| |
12 and 13 |
F: AGAGCCAGTGAGCAGTCCC |
R: GACCGGCAAAAGAGGACG |
| |
13 |
F: AGCTGACCTGGAAGCTTGTG |
R: GCAGAGAGATGAGGGTGGAG |
|
RBP3 |
1, fragment 1 |
F: CTTGCACACAGTCCAGGGAG |
R: AGATCCAGCACTAAGGCGG |
| |
1, fragment 2 |
F: TGGAGGGTAATGTGGGCTAC |
R: GTCCCCACACAGGGCAG |
| |
1, fragment 3 |
F: GCTGAGGATAGGCGAGTCTG |
R: CGGAGGCGTCAGCAAAAC |
| |
1, fragment 4 |
F: CTGAGGACGAGGCTATCCG |
R: TTGTCGATGAAGGTGAGGAC |
| |
1, fragment 5 |
F: CTTCCTTATGCAGTCGCTGG |
R: TCAAAACGCAGGTAGCCC |
| |
1, fragment 6 |
F: CGAGCTGGTGGTAGAGGAAG |
R: TGCATATAAGGGGCTGCTG |
| |
1, fragment 7 |
F: CTTTGCACACACCATGCAG |
R: CAATGGGTCAACTCACTCCC |
| |
2 |
F: CTGGGCTCTAAAACTGGCTG |
R: GCCCATAGCTTTGACTGTCC |
| |
3 |
F: GCACACAGGGCCTCACTG |
R: CTGTCTTTCCCTGGTTTCCC |
| |
4 |
F: GAGAAGACAGGTGCTCCAGG |
R: GGTGTGTGTCCCAGAGGTTC |
|
RP1 |
1 |
F: CCATGTATTCGCTATGGTGC |
R: TGTCCAGGTCTACAGGCTGC |
| |
2 |
F: GGCAGGCACAGCATCAC |
R: CACCATTCATATCCCACACG |
| |
3 |
F: TTCAAGCCTAGGAGGTTGTTG |
R: ATTGAAGCATGGATTTTGCC |
| |
4, fragment 1 |
F: GATATTTCTAACTTCTCTGCCTTCC |
R: CCCTGGATGATATCTGTGTCC |
| |
4, fragment 2 |
F: ATCAAGAGGGCAGTTTGGC |
R: TTGAAGTTCTTGATACCAGTTTTG |
| |
4, fragment 3 |
F: TCACATAATAATGGTTTGCCATC |
R: TTTCTATGGAAATTCTTGGAAATC |
| |
4, fragment 4 |
F: TCCCCTTAAAGGAGGGATAC |
R: AATTGAATGATGAGCAATAGCC |
| |
4, fragment 5 |
F: GAATGGCAAAGAAGAGTTTAGTTTC |
R: ACTGAAGCTTGCAATTGGTG |
| |
4, fragment 6 |
F: GCTTATTTGGTTCCCCTGC |
R: AGAGCAACCTCCATCCAAAG |
| |
4, fragment 7 |
F: ACTTGAAAGCTGCTGTTGCC |
R: GCTTAAATTACTGACATTTTGATGTG |
| |
4, fragment 8 |
F: CAATGTCTGCAATACCATTGAC |
R: TCCTTCATTGGTCTCCTTTTC |
| |
4, fragment 9 |
F: TTAATCCAAGAAGAGGTAGAGGC |
R: CCTGGAATTCCTGCAACATAG |
| |
4, fragment 10 |
F: TGGAATTTCAGTGTTCCAGG |
R: TGATGACTACCCTTCTCCTCTG |
| |
4, fragment 11 |
F: CATGGTAGTGACTCAGAACCTTTTC |
R: CCTTCTTCCTCTAACCCCAAG |
| 4, fragment 12 | F: GATAATGCCATTGGTGATATATTTG | R: CGTATTCGTCACATGTGCTTC | |
PCR products were purified with gel extraction (QIAquick Gel Extraction Kit; Qiagen, Venlo, the Netherlands) or with 96-well filter plates (MultiScreen HTS-PCR; Millipore, Bedford, MA). Bidirectional dideoxy sequencing was performed using the forward and reverse primers (BigDye Terminator, ver. Three on a 3730 or 3100 DNA Analyzer; Applied Biosystems, Inc., [ABI], Foster City, CA). Sequencing results were analyzed with Vector NTI (Invitrogen Life Technologies Europe BV, Bleiswijk, the Netherlands) software. The microsatellite markers used for linkage analysis are presented in Table 2.
Table 2. Microsatellite markers used for haplotype analysis at the MFRP locus.
| Chromosome | Position (hg18) | Name | D number |
|---|---|---|---|
| chr11 |
118,140,606–118,140,889 |
AFMA222XC5 |
D11S4104 |
| chr11 |
118,884,802–118,884,982 |
AFMB342ZE9 |
D11S4171 |
| chr11 | 120,333,420–120,333,756 | AFM220YB6 | D11S925 |
DNA samples of both affected individuals were genotyped with 250 K single nucleotide polymorphism (SNP) microarrays (GeneChip Mapping 250 K Nsp Array; Affymetrix, Santa Clara, CA). Array experiments were performed according to protocols provided by the manufacturer. Arrays were scanned, and genotypes were called as described [9]. The 250 K SNP data were analyzed with the software package CNAG [10], and chromosomal segments were accepted as homozygous if the loss of heterozygosity (LOH) score was ≥10. The LOH score measures the likelihood of a stretch of SNPs being homozygous based on the population SNP allele frequencies. An LOH score of ≥15 corresponds to regions of (on average) 4 Mb and larger [11]. Homozygous regions shared by both individuals were analyzed for the presence of known RP genes. Retinol-binding protein 3 (RBP3) and retinitis pigmentosa 1 (RP1) were screened for mutations as described above for MFRP. CRB1 amplification and sequencing were performed as described previously [7]. Turkish controls were screened for the novel mutation in CRB1, with restriction enzyme digestion with BccI.
Results
Clinical findings
Table 3 summarizes the ophthalmologic features of both affected individuals. Both patients demonstrated bilateral decreased axial length, retinal dystrophy, and macular edema (Figure 1). Patient IV:2 had optic disc drusen on funduscopy and confirmed by B-mode ultrasound, whereas patient IV:3 did not have optic disc drusen. The ERG for patient IV:3 showed an extinguished rod response and a subnormal cone photopic response. In patient IV:2, the rod and cone responses were extinguished on the ERG.
Table 3. Clinical characteristics of affected members of a family with retinitis pigmentosa, nanophthalmos and optic disc drusen.
| Patient | Age (years) | BCVA (Snellen) | Axial length (mm) | Refractive error (D) | Posterior Segment Findings | Ultrasound | OCT findings |
|---|---|---|---|---|---|---|---|
| IV:2 |
17 |
0.20 OD |
19.47 OD |
+8.25 OD |
Atrophy of the retina outside of the fovea, spots of hyperpigmentation |
Optic disc drusen |
Intraretinal macular edema |
| |
|
0.16 OS |
19.46 OD |
+7.75 OS |
|
|
|
| IV:3 |
7 |
0.10 OD |
18.86 OD |
+9.5 OD |
Atrophy of the retina outside of the fovea, spots of hyperpigmentation |
No optic disc drusen |
Intraretinal macular edema |
| 0.10 OS | 18.89 OS | +9.25 OS |
BCVA=best-corrected visual acuity; OD=right eye; OS=left eye; D=diopters; OCT=optical coherence tomography
Figure 1.
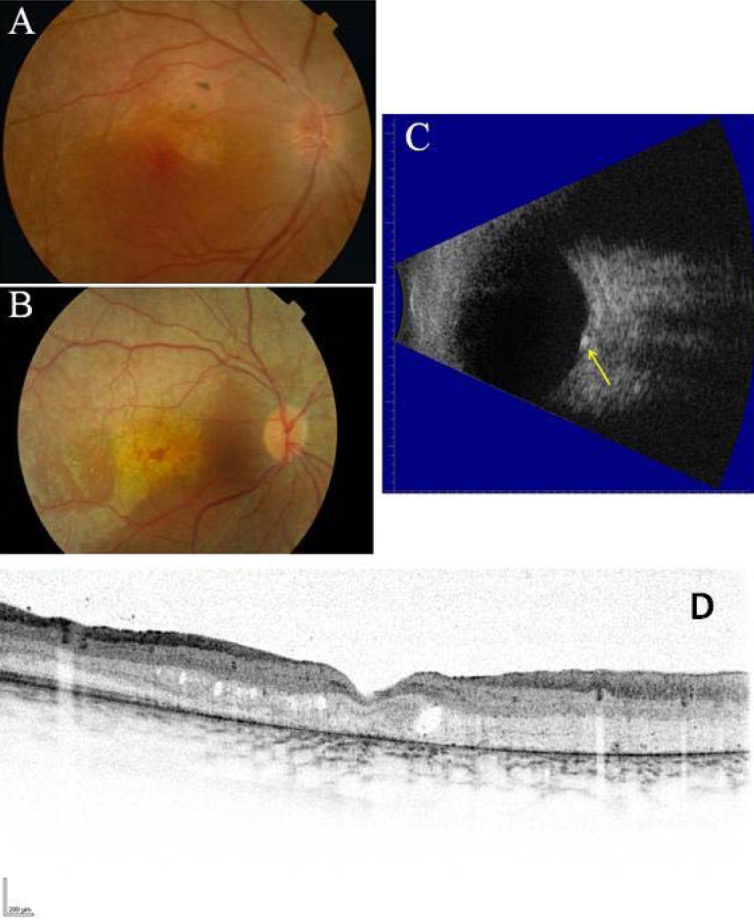
Ophthalmological images of two siblings affected by retinitis pigmentosa, nanophthalmus and optic disc drusen. Fundus photography of the right eye of patient IV:2 A: and of patient IV:3 B: showed atrophy of the retina outside the fovea and spots of hyperpigmentation. B-mode ultrasound of the left eye of patient IV:2 C: revealed optic disc drusen (indicated with the yellow arrow). Optical coherence tomography scan of the macula of patient IV:2 showed intraretinal edema and atrophy of the outer retinal layers D.
Genetic findings
Direct sequencing of the MFRP gene in the proband (patient IV:2) did not reveal a disease-causing mutation. The only detected variations were known SNPs in exons 1, 4, and 5 (Table 4). Haplotypes were constructed based on microsatellite markers and SNPs at the MFRP locus (Figure 2). Both affected individuals inherited different chromosomal haplotypes from their father at this locus, excluding involvement of this locus in this family.
Table 4. Sequence variants identified by sequence analysis of the MFRP and RP1 gene.
| Gene | Exon | cDNA | Protein | SNP number |
|---|---|---|---|---|
|
MFRP |
1 |
c.-88C>T |
- |
rs883245 |
| |
1 |
c.-65G>A |
- |
rs883246 |
| |
1 |
c.-31G>A |
- |
rs883247 |
| |
4 |
c.406G>A |
p.Val136Met |
rs3814762 |
| |
5 |
c.540T>C |
p.His180= |
rs2510143 |
| |
5 |
c.492C>T |
p.Tyr164= |
rs36015759 |
|
RP1 |
4 |
c.5175A>G |
p.Gln1725= |
rs441800 |
| |
4 |
c.2615G>A |
p.Arg872His |
rs444772 |
| 4 | c.5071T>C | p.Ser1691Pro | rs414352 |
Figure 2.
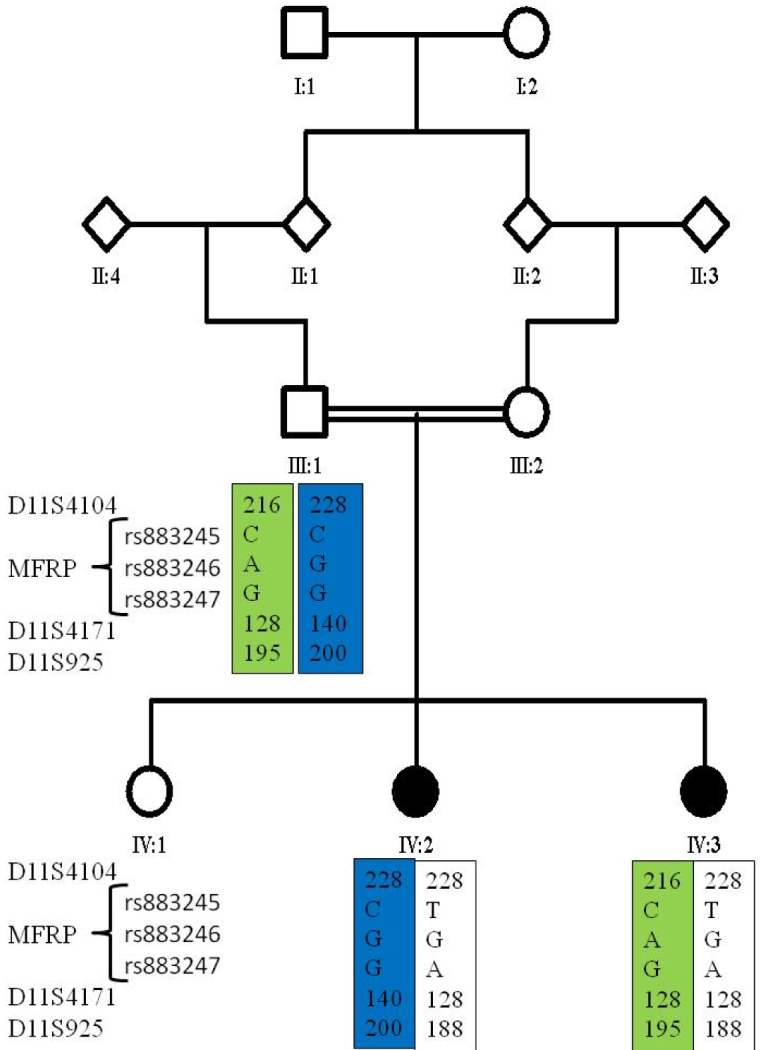
Exclusion of the membrane frizzled-related protein gene in a family with retinitis pigmentosa, nanophthalmos, and optic disc drusen with haplotype analysis. Haplotypes were constructed using microsatellite markers and single nucleotide polymorphisms detected in exon 1 of the membrane frizzled-related protein gene.
Genome-wide homozygosity mapping using SNP microarrays revealed several homozygous regions in both patients. Thirteen regions were shared in both, with the largest region spanning 28.8 Mb on chromosome 1. Analysis of the shared homozygous regions for the known RP genes revealed that the CRB1 gene resided in the largest homozygous segment, and two other known RP genes (RP1 and RBP3) were present in smaller homozygous regions (Table 5). Sequence analysis of the RP1 and RBP3 genes in the proband (patient IV:2) revealed only nonpathogenic SNPs in exon 4 of RP1 (Table 4).
Table 5. Homozygous regions shared by patients IV:2 and IV:3 identified by genome-wide SNP microarray analysis.
| Chromosome | Size (Mb) | Start position (hg18) | End position (hg18) | Number of homozygous SNPs | RP Gene |
|---|---|---|---|---|---|
| 1 |
28.8 |
167,580,132 |
196,437,697 |
2788 |
CRB1 |
| 12 |
20.2 |
12,966,664 |
33,172,827 |
2440 |
|
| 8 |
20.0 |
20,080,993 |
40,179,228 |
1901 |
|
| 10 |
18.3 |
29,819,314 |
48,158,305 |
1334 |
RBP3 |
| 4 |
15.1 |
147,053,197 |
162,160,056 |
1516 |
|
| 8 |
13.7 |
42,830,763 |
56,575,870 |
855 |
RP1 |
| 4 |
12.1 |
84,301,279 |
96,462,585 |
1123 |
|
| 11 |
8.1 |
47,323,947 |
55,496,802 |
307 |
|
| 19 |
6.3 |
49,894,480 |
56,209,610 |
207 |
|
| 12 | 5.2 | 5,127,842 | 10,400,609 | 450 |
Sequence analysis of CRB1 revealed a novel homozygous missense mutation in exon 7 (c.2498G>A; p.Gly833Asp), which affects a highly conserved amino acid residue (Figure 3). The mutation was found homozygously in both affected siblings, and heterozygously in the unaffected father. Restriction enzyme digestion did not reveal the mutation in 100 Turkish controls. Bioinformatic analyses confirmed pathogenicity of the mutation (Grantham score: 94, Sorting Intolerant From Tolerant [SIFT]: deleterious, Polymorphism Phenotyping v2 [PolyPHen-2]: probably damaging with a score of 1.000, PhyloP: 5.3).
Figure 3.
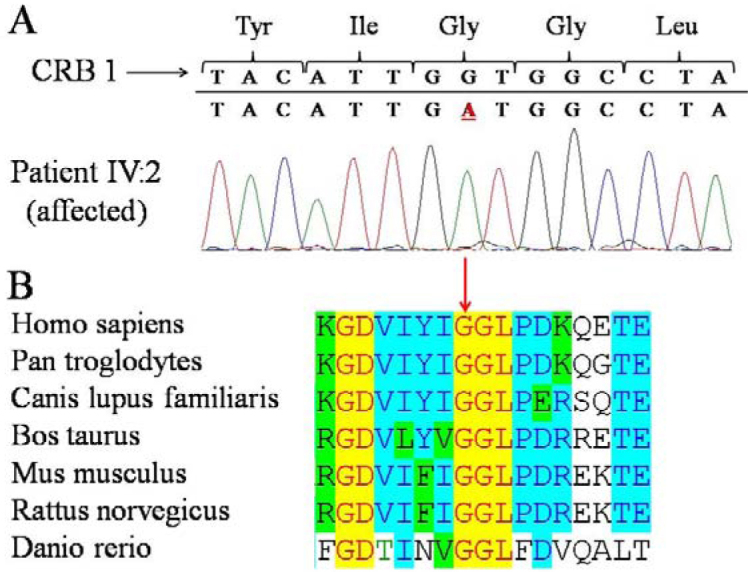
Identification of a novel crumbs homolog 1 mutation in a family with retinitis pigmentosa, nanophthalmos, and optic disc drusen. A: Crumbs homolog 1 sequence analysis demonstrated a homozygous mutation (c.2498G>A) in exon 7. B: At the protein level, the mutation (p.Gly833Asp) alters a highly conserved amino acid residue.
Discussion
Several research groups have described mutations in the MFRP gene, leading to an autosomal recessive disease characterized by nanophthalmos, RP, foveoschisis, and optic disc drusen [1–3]. In this study, we demonstrate that a similar disease complex can be caused by a novel missense mutation in the CRB1 gene. This is in agreement with a recent study that identified a homozygous CRB1 mutation in a Mexican family with similar features [12].
In both individuals of the family described in this study, we observed a decreased axial length consistent with nanophthalmos, resulting in high hyperopia. High hyperopia is commonly seen in patients with CRB1 mutations [13,14]. Optic disc drusen were observed in patient IV:2, but not in patient IV:3, which may be due to her young age (7 years) at examination [15]. On OCT, we noted a similar cystic appearance as observed in patients with MFRP mutations in previous studies [1–3], but in our opinion, this does not resemble former publications of classical foveoschisis [16]. More likely, the patients developed macular edema secondary to RP, resulting in a split appearance of the macula on OCT.
The involvement of MFRP was excluded in this family, and homozygosity mapping revealed a novel missense mutation in the CRB1 gene. The mutation resides in the second laminin A G-like domain, where the mutation affects a residue in a highly conserved region and localizes near several other missense mutations previously identified in CRB1 [17]. Our results demonstrate that mutations in not only MFRP but also CRB1 are associated with small eye size. The combination of features observed in this family closely resembles the nanophthalmos- retinitis pigmentosa-foveoschisis-optic disc drusen disease complex previously associated with MFRP mutations.
Acknowledgments
This study was supported by the Foundation Fighting Blindness USA (grants BR-GE-0510–04890RAD and C-GE-0811–0545-RAD01 to A.I.d.H and F.P.M.C.), and the Netherlands Organization for Scientific Research (TOP-grant 40–00812–98–09047 to A.I.d.H. and F.P.M.C.).
References
- 1.Ayala-Ramirez R, Graue-Wiechers F, Robredo V, Amato-Almanza M, Horta-Diez I, Zenteno JC. A new autosomal recessive syndrome consisting of posterior microphthalmos, retinitis pigmentosa, foveoschisis, and optic disc drusen is caused by a MFRP gene mutation. Mol Vis. 2006;12:1483–9. [PubMed] [Google Scholar]
- 2.Crespí J, Buil JA, Bassaganyas F, Vela-Segarra JI, Díaz-Cascajosa J, Ayala-Ramírez R, Zenteno JC. A novel mutation confirms MFRP as the gene causing the syndrome of nanophthalmos-renititis pigmentosa-foveoschisis-optic disk drusen. Am J Ophthalmol. 2008;146:323–8. doi: 10.1016/j.ajo.2008.04.029. [DOI] [PubMed] [Google Scholar]
- 3.Zenteno JC, Buentello-Volante B, Quiroz-Gonzalez MA, Quiroz-Reyes MA. Compound heterozygosity for a novel and a recurrent MFRP gene mutation in a family with the nanophthalmos-retinitis pigmentosa complex. Mol Vis. 2009;15:1794–8. [PMC free article] [PubMed] [Google Scholar]
- 4.Mandal MN, Vasireddy V, Jablonski MM, Wang X, Heckenlively JR, Hughes BA, Reddy GB, Ayyagari R. Spatial and temporal expression of MFRP an its interaction with CTRP5. Invest Ophthalmol Vis Sci. 2006;47:5514–21. doi: 10.1167/iovs.06-0449. [DOI] [PubMed] [Google Scholar]
- 5.Sundin OH, Leppert GS, Silva ED, Yang J-M, Dharmaraj S, Maumenee IH, Santos LC, Parsa CF, Traboulsi EI, Broman KW, DiBernardo C, Sunness JS, Toy J, Weinberg EM. Extreme hyperopia is the result of null mutations in MFRP, which encodes a frizzled-related protein. Proc Natl Acad Sci USA. 2005;102:9553–8. doi: 10.1073/pnas.0501451102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kameya S, Hawes NL, Chang B, Heckenlively JR, Naggert JK, Nishina PM. Mfrp, a gene encoding a frizzled related protein, is mutated in the mouse retinal degeneration 6. Hum Mol Genet. 2002;11:1879–86. doi: 10.1093/hmg/11.16.1879. [DOI] [PubMed] [Google Scholar]
- 7.den Hollander AI, ten Brink JB, de Kok YJ, van Soest S, van den Born LI, van Driel MA, van de Pol DJ, Payne AM, Bhattacharya SS, Kellner U, Hoyng CB, Westerveld A, Brunner HG, Bleeker-Wagemakers EM, Deutman AF, Heckenlively JR, Cremers FP, Bergen AA. Mutations in a human homologue of Drosophila crumbs cause retinitis pigmentosa (RP12). Nat Genet. 1999;23:217–21. doi: 10.1038/13848. [DOI] [PubMed] [Google Scholar]
- 8.Marmor MF, Fulton AB, Holder GE, Miyake Y, Brigell M, Bach M. Standard for clinical electroretinography (2008 update). Doc Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 9.Leung YF, Cavalieri D. Fundamentals of cDNA microarray data analysis. Trends Genet. 2003;19:649–59. doi: 10.1016/j.tig.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 10.Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, Ogawa S. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–9. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 11.den Hollander AI, Lopez I, Yzer S, Zonneveld MN, Janssen IM, Strom TM, Hehir-Kwa JY, Veltman JA, Arends ML, Meitinger T, Musarella MA, van den Born LI, Fishman GA, Maumenee IH, Rohrschneider K, Cremers FP, Koenekoop RK. Identification of novel mutations in patients with Leber congenital amaurosis and juvenile RP by genome-wide homozygosity mapping with SNP microarrays. Invest Ophthalmol Vis Sci. 2007;48:5690–8. doi: 10.1167/iovs.07-0610. [DOI] [PubMed] [Google Scholar]
- 12.Zenteno JC, Buentello-Volante B, Ayala-Ramirez R. Villanueva-Mendoza. Homozygosity mapping identifies the Crumbs homologue (Crb1) gene as responsible for a recessive syndrome of retinitis pigmentosa and nanophthalmos. Am J Med Genet. 2011;155A:1001–6. doi: 10.1002/ajmg.a.33862. [DOI] [PubMed] [Google Scholar]
- 13.den Hollander AI, Heckenlively JR, van den Born LI, de Kok YJ, van der Velde-Visser SD, Kellner U, Jurklies B, van Schooneveld MJ, Blankenagel A, Rohrschneider K, Wissinger B, Cruysberg JR, Deutman AF, Brunner HG, Apfelstedt-Sylla E, Hoyng CB, Cremers FPM. Leber congenital amaurosis and retinitis pigmentosa with Coats-like exudative vasculopathy are associated with mutations in the Crumbs homolog 1 (CRB1) gene. Am J Hum Genet. 2001;69:198–203. doi: 10.1086/321263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abouzeid H, Li Y, Maumenee IH, Dharmaraj S, Sundion OA. G1103R mutations in CRB1 is co-inherited with high hyperopia and Leber congenital amaurosis. Ophthalmic Genet. 2006;27:15–20. doi: 10.1080/13816810500481840. [DOI] [PubMed] [Google Scholar]
- 15.Auw-Haedrich C, Staubach F, Witschel H. Optic disc drusen. Surv Ophthalmol. 2002;47:515–32. doi: 10.1016/s0039-6257(02)00357-0. [DOI] [PubMed] [Google Scholar]
- 16.Sayanagi K, Morimoto Y, Ikuno Y, Tano Y. Spectral-domain optical coherence tomographic findings in myopic foveoschisis. Retina. 2010;30:623–8. doi: 10.1097/iae.0b013e3181ca4e7c. [DOI] [PubMed] [Google Scholar]
- 17.den Hollander AI, Davis J, van der Velde-Visser SD, Zonneveld MN, Pierrottet CO, Koenekoop RK, Kellner U, van den Born LI, Heckenlively JR, Hoyng CB, Hanford PA, Roepman R, Cremers FPM. CRB1 mutation spectrum in inherited retinal dystrophies. Hum Mutat. 2004;24:355–69. doi: 10.1002/humu.20093. [DOI] [PubMed] [Google Scholar]