

Abstract
Background
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder characterized by a decreased ability to repair DNA damaged by UV radiation and the early development of cutaneous and ocular malignant neoplasms. Approximately 20% of patients with XP also develop progressive neurologic degeneration.
Observations
We describe a boy who was found to have XP after a severe burn following minimal sun exposure. His maternal uncle, now age 20 years, had been diagnosed with XP after a similar sunburn in infancy. The uncle has the typical skin pigmentary findings of XP along with severe progressive neurologic involvement. Although the infant’s parents were not known to be blood relatives, the infant and his affected uncle proved to be compound heterozygotes for the same 2 frameshift mutations in the XPA DNA repair gene (c.288delT and c.349_353del). After the diagnosis of XP in the infant, genealogic investigation identified a common Dutch ancestor for both of his grandfathers 5 generations back.
Conclusions
Counseling families at risk for a rare inherited disease is not always straightforward. The socio-cultural and demographic backgrounds of the families must be considered for evaluation of risk assessment.
Xeroderma pigmentosum (XP) is a rare autosomal recessive disorder characterized by a decreased ability to repair DNA damaged by UV light and the subsequent early development of cutaneous and ocular malignant neoplasms.1,2 About 20% to 30% of patients with XP develop severe progressive neurologic deterioration characterized by loss of reflexes, sensorineural hearing loss, ataxia, dysphagia, and decreasing cognition.1,3,4 Disease-causing mutations have been identified in 8 different genes in patients with XP: 7 of these genes (XPA, XPB, XPC, XPD, XPE, XPF, and XPG) are involved in nucleotide excision repair, and the eighth gene, polymeraseeta, is involved in the replication of damaged DNA and is mutated in patients with the XP variant form.2,5–8
The overall frequency of XP is about 1 in 1 million people in Europe and the United States.2,6,9 In Japan, the frequency is about 1 in 22 000.7,10 Mutations in the XPA gene can cause a particularly severe form of XP with progressive neurodegeneration, often starting in the second decade of life, and significant UV sensitivity such that blistering burns result from minimal sun exposure.11 Xeroderma pigmentosum presents during infancy with or without sunburn but with the later development of often darkly pigmented lentigines. The occurrence of a blistering sunburn at such an early age should also lead one to consider porphyria (particularly congenital erythropoietic porphyria), neonatal lupus, Cockayne syndrome, and trichothiodystrophy in the differential diagnosis.
REPORT OF CASES
A 6-week-old boy born to presumably non-consanguineous parents developed a blistering burn after minimal sun exposure. The family history was significant for a 17-year-old maternal uncle who had been diagnosed with XP after a similar sunburn in infancy. Given the severe sunburn in the infant and the family history of XP, the parents were concerned about the possibility of XP in their son.
The family was evaluated under a National Institutes of Health (NIH), National Cancer Institute (NCI) natural history protocol. The present study was approved by the NCI institutional review board, and appropriate informed consent was obtained from the adult family members and parents of the proband and his uncle. The family members received genetic counseling that included a description of the research study; an explanation of the risks, limitations, and benefits of genetic testing; and education regarding the genetics and care of persons with XP.
Fibroblast cultures and lymphoblastoid cell lines were established from skin biopsy specimens and blood samples from the family members. Cell culture establishment and growth, UV-C treatment, host cell reactivation assays, and DNA sequencing assays were performed as previously described.12–14 The mutations were named according to GenBank accession Nos. NM_000380.3 for the XPA complementary DNA sequence and NP_000371.1 for the XPA protein sequence and confirmed by use of the Mutalyzer program (http://www.lovd.nl/mutalyzer). Single-nucleotide polymorphisms (SNPs) used to assess the haplotype on chromosome 9 in the region of the XPA gene (10 SNPs located up to 0.5 megabase [Mb] on one side of the XPA gene and 13 SNPs located up to 2.9 Mb on the other side of the XPA gene) were sequenced according to the format described by the International Hap-map Project (http://www.hapmap.org/).
PATIENT 1
Patient XP360BE was a healthy first-born boy delivered at term after an uncomplicated pregnancy. His neonatal period was complicated by hyperbilirubinemia that was treated for 36 hours with blue light without complications. At age 6 weeks, he developed a sunburned face after being outside in a stroller in northern Indiana for a total of 45 minutes during a 2-day period in early April (Figure 1A and Table). He was not wearing any type of sunscreen or hat. The erythema and crusting from the burn persisted for 1 week and cleared without scarring. After the initial sunburn, the infant’s mother kept him protected from UV (Figure 1B) and used a UV meter to assess the levels of UV in the infant’s environment.2,6,15 The mother’s concern was based on a known family history of XP in her brother.
Figure 1.
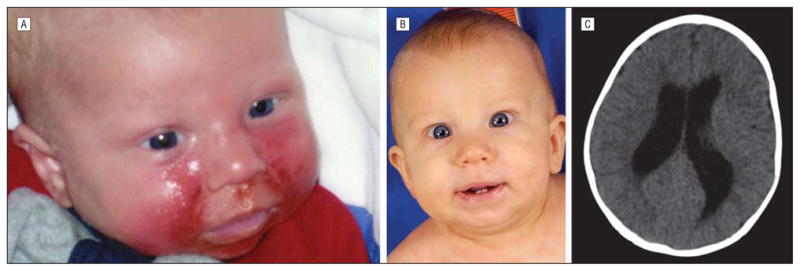
Proband XP360BE. A, Severe facial sunburn seen at age 6 weeks. B, At age 7 months, after beginning the regular use of effective sunlight protection, his skin shows no residual effects of the earlier sunburn. C, Computed tomographic scan of the brain at age 7 months shows moderately severe enlargement of both lateral ventricles.
Table.
Xeroderma Pigmentosum Progression in Patients Studieda
| Characteristic | Patient Age
|
|
|---|---|---|
| XP363BE | XP360BE | |
| Last visit | 20 y | 3 y |
|
| ||
| Skin Involvement | ||
| First sunburn with blistering | 6 wk | 6 wk |
| Freckling of sun-exposed skin | 1 y | None (sun protected) |
| Skin cancer | None | None |
|
| ||
| Progressive Neurologic Impairment | ||
| Dilated ventricles in the brain | 15 mo | 7 mo |
| Developmental delay first noted | 2 y | 3 y |
| Delayed speech | 25 words at 7 y | Sign language and single words at 3 y |
| Gait disturbance due to ataxia or spasticity | 9–14 y | NA |
| Loss of speech | 12 y | NA |
| Severe hearing loss documented | 12 y | None |
| Cannot walk or talk; confined to bed and wheelchair | 15–20 y | NA |
| Absent deep tendon reflexes | 17 y | Still present at 3 y |
| Massively dilated ventricles in the brain with cortical atrophy | 17 y | NA |
Abbreviation: NA, not applicable.
Both patients were born at term of uncomplicated pregnancies, patient XP363BE by cesarean delivery of a mother who had undergone cesarean delivery before.
Patient 1 was first seen at Children’s Memorial Hospital of Chicago at age 17 weeks. The findings of his skin and gross neurologic examinations at that time were unremarkable. Neither his teeth nor his urine showed pink fluorescence, and he had no hepatosplenomegaly. Although other photosensitivity disorders were considered, particularly erythropoietic porphyria and lupus, the infant had no other features, and evaluation for other disorders was not performed pending evaluation for XP. The parents had already begun vigorous UV protection. To consider the diagnosis of XP, blood was drawn for DNA testing; a biopsy was performed for fibroblast culture; and the patient was referred to the NIH for further evaluation.
An axial computed tomography (CT) scan of the brain at age 7 months showed moderately severe enlargement of both lateral ventricles (Figure 1C), indicative of neuronal loss. There were no focal brain abnormalities. His development through his first year appeared normal. However, at age 18 months, he was noted to have substantial speech delay and was enrolled in the First Steps early intervention program. At last follow-up at age 3 years, he spoke several words in response to questions and used some sign language to communicate his needs. He had an individualized education plan in place and received local services.
PATIENT 2
The maternal uncle (Figure 2) of patient 1, patient XP363BE, was born at term by cesarean delivery of a mother who had undergone cesarean delivery previously. Like patient 1, he also had mild hyperbilirubinemia neonatally that resolved spontaneously at age 2 weeks. He experienced a severe sunburn on his face and upper extremities at age 6 weeks after a 20-minute sun exposure (Figure 2A). He was hospitalized for 4 days to treat the burn. During his first year he experienced several other sunburns after minimal exposure and began developing freckling on sun-exposed skin surfaces of his face and arms.
Figure 2.
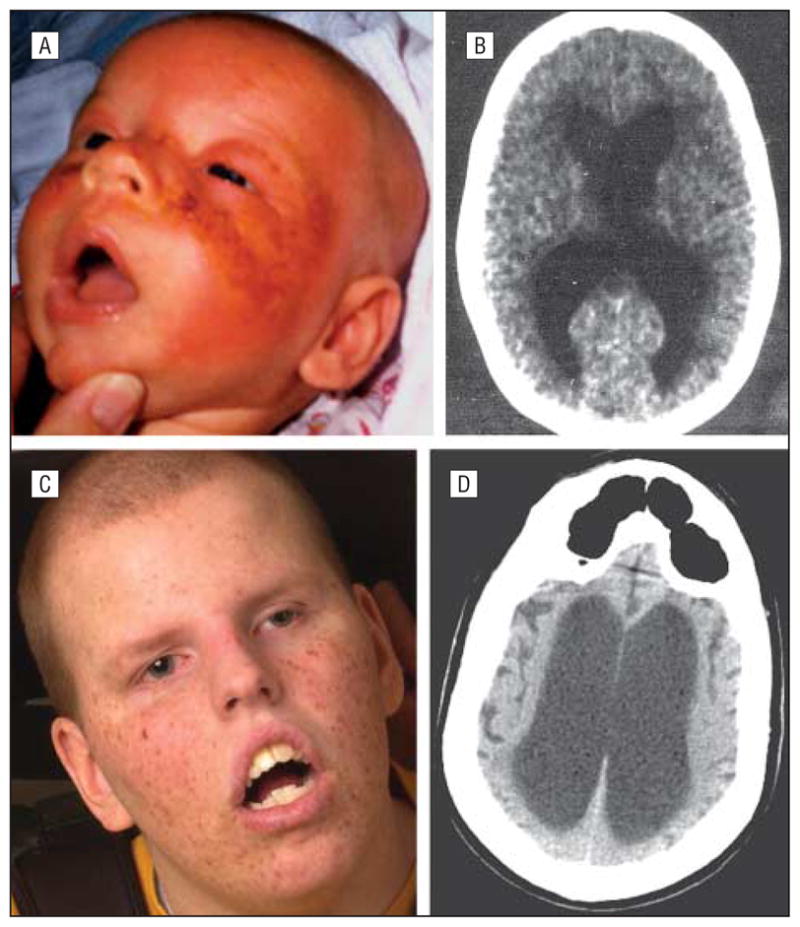
Affected uncle, patient XP363BE. A, Severe facial sunburn seen at age 6 weeks. B, Computed tomographic scan of the brain at age 15 months shows slight enlargement of the lateral ventricles. C, At age 17 years, despite total avoidance of UV exposure after xeroderma pigmentosum diagnosis at age 1 year, he has numerous facial freckles and lentigines. D, Computed tomographic scan of the brain at age 17 years shows markedly enlarged ventricles indicative of progressive neuronal loss.
He was first seen at Children’s Memorial Hospital of Chicago at age 1 year, and the diagnosis of XP was suspected owing to his substantial sun sensitivity, unusual freckling, and developmental delay. At that time, testing of his cultured fibroblasts revealed severe UV hypersensitivity. His development progressed at a much slower rate than that of his 2 older siblings, and he did not walk unassisted until age 18 months (Table). An axial CT scan of the brain obtained at age 15 months showed slight enlargement of the lateral ventricles (Figure 2B).
After diagnosis he had impeccable skin protection throughout his life. His parents had the windows in their home and vehicles treated with sunblocking film and used a UV meter to measure the levels of UV in the environment.2,6,15 In addition, the parents applied sun-blocking skin creams and made sure the patient wore long-sleeved shirts, long pants, and gloves when he needed to go outside. The mother designed a UV protective hood with a face shield to provide UV protection to his face, ears, and neck. Since the face shield was made of UV-blocking material, this also provided good protection for his eyes. The protective clothing enabled him to be UV protected when he was transported to school, church, or family activities.
While he never had skin cancers or premalignant lesions (Figure 2C), patient 2 developed severe, progressive neurologic deterioration (Table). He had marked speech delay and at age 7 years was able to use only about 25 words. An axial CT scan of the brain at age 17 years performed at the NIH (Figure 2D) showed markedly enlarged ventricles indicative of progressive neuronal loss. There was also bilateral enlargement of cortical sulci of the cerebral hemispheres. Additional features of brain atrophy included increased thickness of the skull and overpneumatization of the frontal sinuses. At last follow-up at age 20 years, he was profoundly deaf, unable to speak, confined to a wheelchair, incontinent, and fed by gastrostomy tube.
LABORATORY INVESTIGATIONS
Cultured fibroblasts from patient XP360BE were hypersensitive to the killing effects of UV. A dose of UV-C (5 J/m2) that did not affect the growth of the normal fibro-blasts resulted in marked growth inhibition of the XP360BE cells (data not shown). Host cell reactivation studies indicated that cells from both patients had a defect in the XPA gene (data not shown). Analysis of genomic DNA from both patients showed them to be compound heterozygotes with the same 2 mutations in the XPA gene: c.288delT and c.349_353del (Figure 3). The proband’s mother and maternal grandmother carried the same mutation (c.288delT), a deletion of T at position 288 in the XPA gene (Figure 3A and B). This mutation abolishes an Eco0109I restriction site in the XPA gene and can be detected using a polymerase chain reaction (PCR)-based restriction fragment length polymorphism (RFLP) assay (Figure 3B). Surprisingly, the proband’s father and both grandfathers carried the same second mutation (c.349_353del), a deletion of 5 bases (CTTAT) in the XPA gene (Figure 3A and C). This mutation abolishes a BasB1 restriction site in the XPA gene and can also be detected using a PCR-based RFLP assay (Figure 3C). Both of these mutations are in exon 3 of the XPA gene. The findings of haplotype analysis of the XPA gene and the surrounding region on chromosome 9 were consistent with both grandfathers and the father of the pro-band having a common haplotype over a region of about 0.25 Mb on one side of the mutation and about 2.9 Mb on the other side. These haplotype data were not informative for calculation of the most recent common ancestor using the method previously reported for XPC families in Turkey and Italy.16
Figure 3.

Familial and genetic analysis of our 2 patients. A, Pedigree of the proband, XP360BE (arrow), and his affected uncle, XP363BE. Both affected patients have the same 2 frameshift mutations: c.288delT (red) and c.349_353del (blue) in the XPA gene. The proband’s mother and maternal grandmother are heterozygous carriers of the c.288delT mutation in the XPA gene, and the proband’s father and both grandfathers are heterozygous for the c.349_353del mutation. B, The deletion of thymine at base pair (bp) 288 destroys an Eco0109I restriction site, as seen in genomic DNA from the proband and his uncle as well as his mother and maternal grandmother, but not his father and maternal grandfather. C, The CTTAT deletion at bp 349 to 353 destroys a BsaB1 restriction site in the genomic DNA from the proband and his uncle as well as the proband’s father and both grandfathers but not in the proband’s paternal grandmother.
Although the proband’s father and maternal grandfather were not known to be related at the time of initial counseling, the laboratory findings were highly suggestive of a common ancestor. The families of both parents, although unrelated, belonged to a large, close-knit community and are of Dutch ancestry. After they discussed with us and among themselves the ramifications of the diagnosis of XP in the proband and the surprising molecular findings of common shared alleles, the proband’s relatives conducted a detailed genealogic investigation using records of the church denomination to which they all belonged. They discovered that the maternal and paternal grandfathers of the proband had a common relative 5 generations before him. Presumably, this individual was also a carrier of the c.349_353del mutation. Our molecular findings of a possible common haplotype among the grandparents and father of the proband are consistent with the hypothesis that this mutation was passed through all of the intervening relatives by direct descent.
COMMENT
XPA GENE
The human XPA DNA repair gene was first described in 1990 and reported to be localized to chromosome 9q34.1.17,18 The XPA gene is 25 kb long and contains 6 exons that code for a 273–amino acid protein. Exon 1 is important for nuclear localization but not for DNA repair; exon 2 encodes a domain for binding to ERCC 1; exon 3 encodes the zinc finger protein that binds replication protein A; exons 4 and 5 make up a DNA binding domain; and exon 6 interacts with the TFIIH complex.19 Most mutations in XPA are deletions and splice site mutations in exon 3, intron 3, or exon 4 that result in frameshift mutations within DNA binding regions.5,14 However, mutations have been found throughout the XPA gene with the exception of exon 1, and the sites of mutations are more randomly distributed in non-Japanese patients.5,11,13,14,20–23 In European and American cases, mutations have been detected at a variety of sites on the XPA gene. (For lists of additional XPA mutations, see Allelic Variations of the XP Genes at http://xpmutations.org/ and The Human Gene Mutation Database at the Institute of Medical Genetics in Cardiff at http://www.hgmd.cf.ac.uk/ac/index.php.)
In Japan, mutations in the XPA gene are responsible for the vast majority of XP cases. More than 90% of the mutant XPA alleles in Japanese patients have an identical G→C base change mutation at the 3′ splice acceptor site of intron 3 that results in no detectable protein production.7,11,21 This is a founder mutation that is present in about 1% of the general population in Japan.10 In the United States and Europe, the frequency of XP is about 1 per 1 million.2,9 About 13% of the XP families from the United States tested at NIH have defects in the XPA gene (data not shown). Spontaneous disease-causing mutations in XP genes have not yet been reported in XP.
In our patients, XP360BE and XP363BE, both of the mutations were in exon 3 of the XPA gene, a region involved in the zinc finger domain affecting binding of the protein to DNA.13,18,21 The paternal mutation (c.349_353del, p.Leu117Glufs*4) results in a truncated 119–amino acid protein that changes the leucine at position 117 to a glutamic acid and then would be predicted to insert a stop codon after inserting 2 abnormal amino acids. This mutation was described previously in 2 European families.13 The maternal allele, c.288delT, p.Val97Leufs*7, results in a truncated 102–amino acid protein that changes the valine at position 97 to leucine and then would be predicted to insert a stop codon after inserting 5 abnormal amino acids. To our knowledge, this mutation has not been previously reported.
There is marked clinical heterogeneity among the XP groups in the frequency of skin cancer and the severity of the neurologic involvement.1–3,6 The neurologic features (developmental delay, sensorineural deafness, abnormal motor function, spasticity, and reduced or absent deep tendon reflexes in association with a primary progressive neuronal degeneration) are most severe in patients in the XP-A complementation group but have also been described in patients in the XP-B, XP-D, and XP-G complementation groups.14 Even within a complementation group, there is marked phenotypic variability.
The pathogenesis of the neurologic abnormalities in patients with XP-A is not understood, in part because mouse homologs of XP-A have no detectable nervous system abnormalities.3 The XPA gene is required for repair of oxidative damage in mitochondrial DNA24 and of nuclear DNA damage related to reactive oxygen species.25 The neurologic defects seen in patients with XP-A appear to arise predominantly from mutations in the DNA binding region and may therefore be a direct consequence of the failure to recognize DNA damage and initiate nucleotide excision repair.14 It has been proposed that faulty repair by the nucleotide excision repair system of certain types of oxidative DNA lesions, such as cyclopurines, may lead to progressive neurologic degeneration in patients with XP.26,27
GENETIC COUNSELING AND RISK ESTIMATION
Given the autosomal recessive nature of XP, and the presumed low probability that the baby’s mother (XPH366BE) had married a carrier of a defect in the same XP gene, the family was initially counseled that there was a low likelihood that she would bear an infant with XP. The accuracy of such risk estimates of an affected child depends to a great degree on the available information and can be estimated by the equation Risk=A×B×C. In this equation, A is the probability of the mother being a carrier of a mutated allele; B is the probability of the father being a carrier of a mutated allele in the same gene; and C is the probability of 2 carriers of mutated alleles in the same gene giving birth to an affected child. Assuming that neither parent has had any DNA testing to determine genotype (complementation group not known), an estimate of the risk for this family can be made as follows:
Subject XPH366BE is the unaffected sister of an affected patient, XP363BE (Figure 3A). Since both of her parents are obligate carriers and she is clinically normal, the probability of her being a carrier is 2 out of 3, and so A=⅔.
The probability of the father (XPH367BE) (Figure 3A) being a carrier of a mutant allele can be calculated by the Hardy-Weinberg equilibrium equation,28 assuming that the parents are unrelated. This equation is X2+2Xy+y2=1, where X is the frequency of the normal allele, y is the frequency of the mutated allele, and 2Xy is the calculated risk of being a heterozygote for the mutant allele (B). In the United States and Europe, the frequency of XP (y2) is about 1 per million (10−6),2,9 so y=10−3. Since the population has either the mutant or normal allele (X+y=1), and y is very small, then X can be estimated as about 1 and X2= 1. Using the Hardy-Weinberg equilibrium equation (X2+2Xy+y2=1), we can calculate that the frequency of heterozygotes who carry 1 mutant allele for any of the XP genes (2Xy or B) is 1 in 500, and so B=1/500.
The probability of 2 carriers of mutant alleles of a recessive gene having an affected child is 1 out of 4, and so C=¼.
Thus if the genotype of XPH366BE were not known, her risk of having an affected child could be estimated as A×B×C=about 1 in 3000.
Alternative risk estimates could be generated if genetic information were known and would vary depending on the specific XP gene involved. For example, if XPH366BE is known to be a carrier of an XPA mutation, then A=1 rather than ⅔, and B=1/1400 for being a carrier of a mutation in the XPA gene in the US population. Then her risk of having an affected child would be about 1 in 5500. If the genotype of the father (XPH367BE) were unknown but the family is aware of the common Dutch ancestor 5 generations ago, then B=1/32, since the probability of a carrier passing the defective gene to the next generation is ½ for each generation. In that case the estimate of risk without laboratory testing of the father or mother would be about 1 in 200. If both the mother’s status as a carrier (A=1) and the common ancestor (B=1/32) were known, then the risk would be about 1 in 100. We now know that the father (XPH367BE) is also a carrier of a mutated allele in the same gene (Figure 3A), thus A=1 and B=1. The risk of any future pregnancies resulting in an affected child is 1 in 4.
The present cases illustrate the importance of informative counseling in the dermatology clinic. When a clinician discusses with a family the possible risk for transmitting an inherited disease, he or she must consider at least 2 factors. First, to determine an accurate risk assessment, the clinician must obtain a detailed multigeneration pedigree and discuss with the family ethnicity and sociocultural practices. Although the family in the present study was not part of a commonly recognized endogamous population, such as the Amish,29 they were members of a large Dutch community with very close religious affiliations who tended to marry members of their large religious group. While other diseases have been reported to have founder mutations in the Dutch population,30 we do not have sufficient population data to determine the frequency of these mutations in the XPA gene.
Second, risk assessment may also depend on obtaining detailed laboratory information and comparing the frequency of mutated alleles in the general population with their frequency in the family.
GENETIC TESTING
The clinician must be aware of the patterns of inheritance of a given disease to effectively discuss with a family the possibility of transmitting a genetic condition to offspring.28 The family also should be informed of the various signs of the disorder and approaches to either prevent or ameliorate the signs in the offspring.
Methods available for laboratory testing before, during, or after a pregnancy also should be addressed.28 There are many considerations relating to genetic testing for inherited diseases, including the ethical issues outlined in “ACOG Committee Opinion No. 410: Ethical Issues in Genetic Testing.”31 For XP we would consider testing of the other parent if (1) the mutations are known in the proband; (2) the relative of the proband has been tested and determined to be a carrier of 1 mutation; and (3) after appropriate genetic counseling, the parents desire genetic testing of their DNA. Families who are members of close communities may be at greater risk than the general population. The ability to identify mutations is limited by the extent of DNA testing performed. The most limited DNA test would exclude only the mutations present in the presenting individual. More extensive DNA testing might include sequencing of the entire XP gene. However, relatively few mutations have been identified in these genes, and sequencing might identify changes of unknown significance.
In conclusion, the present cases illustrate important concepts not only in the presentation of genotype-phenotype correlations of XPA mutations, but also in the surprising occurrence of the same mutation in presumably unrelated individuals. Also illustrated is the notion that informed counseling of the family should include a discussion of their sociocultural background to provide more accurate risk information to help the physician initiate prompt medical intervention. Management of patients with XP involves extensive sun protection of the skin and eyes along with early detection and removal of skin cancers.2,6,15 Early diagnosis and initiation of protective measures can minimize skin cancer morbidity and may extend life.32 Monitoring for neurologic involvement can enable early institution of supportive measures.
Acknowledgments
Funding/Support: This research was supported in part by the Intramural Research Program of the NIH, Center for Cancer Research, NCI.
Footnotes
Financial Disclosure: None reported.
Author Contributions: Dr Paller had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Drs Christen-Zaech and Imoto contributed equally to this project. Study concept and design: Christen-Zaech, Di-Giovanna, Kraemer, and Paller. Acquisition of data: Christen-Zaech, Imoto, Khan, Oh, Tamura, DiGiovanna, Boyle, Patronas, Schiffmann, Kraemer, and Paller. Analysis and interpretation of data: Christen-Zaech, Imoto, Khan, Oh, Tamura, DiGiovanna, Boyle, Patronas, Schiffmann, Kraemer, and Paller. Drafting of the manuscript: Christen-Zaech, Khan, Oh, Tamura, DiGiovanna, Patronas, Schiffmann, Kraemer, and Paller. Critical revision of the manuscript for important intellectual content: Imoto, Di-Giovanna, Boyle, and Paller. Administrative, technical, and material support: DiGiovanna, Boyle, and Paller. Study supervision: Kraemer and Paller.
References
- 1.Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum: cutaneous, ocular, and neurologic abnormalities in 830 published cases. Arch Dermatol. 1987;123(2):241–250. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- 2.Ruenger TM, DiGiovanna JJ, Kraemer KH. Hereditary diseases of genome instability and DNA repair. In: Wolff K, Goldsmith LA, Katz SI, et al., editors. Fitzpatrick’s Dermatology in General Medicine. 7. New York, NY: McGraw Hill; 2008. pp. 1311–1325. [Google Scholar]
- 3.Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007;145(4):1388–1396. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rapin I, Lindenbaum Y, Dickson DW, Kraemer KH, Robbins JH. Cockayne syndrome and xeroderma pigmentosum. Neurology. 2000;55(10):1442–1449. doi: 10.1212/wnl.55.10.1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cleaver JE, Thompson LH, Richardson AS, States JC. A summary of mutations in the UV-sensitive disorders: xeroderma pigmentosum, Cockayne syndrome, and trichothiodystrophy. Hum Mutat. 1999;14(1):9–22. doi: 10.1002/(SICI)1098-1004(1999)14:1<9::AID-HUMU2>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 6.Kraemer KH, Ruenger TM. Genome instability, DNA repair and cancer. In: Wolff K, Goldsmith LA, Katz SI, et al., editors. Fitzpatrick’s Dermatology in General Medicine. 7. New York, NY: McGraw Hill; 2008. pp. 977–986. [Google Scholar]
- 7.Moriwaki S, Kraemer KH. Xeroderma pigmentosum–bridging a gap between clinic and laboratory. Photodermatol Photoimmunol Photomed. 2001;17(2):47–54. doi: 10.1034/j.1600-0781.2001.017002047.x. [DOI] [PubMed] [Google Scholar]
- 8.van Steeg H, Kraemer KH. Xeroderma pigmentosum and the role of UV-induced DNA damage in skin cancer. Mol Med Today. 1999;5(2):86–94. doi: 10.1016/s1357-4310(98)01394-x. [DOI] [PubMed] [Google Scholar]
- 9.Kleijer WJ, Laugel V, Berneburg M, et al. Incidence of DNA repair deficiency disorders in western Europe: xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophy. DNA Repair (Amst) 2008;7(5):744–750. doi: 10.1016/j.dnarep.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 10.Hirai Y, Kodama Y, Moriwaki S, et al. Heterozygous individuals bearing a founder mutation in the XPA DNA repair gene comprise nearly 1% of the Japanese population. Mutat Res. 2006;601(1–2):171–178. doi: 10.1016/j.mrfmmm.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 11.Nishigori C, Moriwaki S, Takebe H, Tanaka T, Imamura S. Gene alterations and clinical characteristics of xeroderma pigmentosum group A patients in Japan. Arch Dermatol. 1994;130(2):191–197. [PubMed] [Google Scholar]
- 12.Oh KS, Khan SG, Jaspers NG, et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum Mutat. 2006;27(11):1092–1103. doi: 10.1002/humu.20392. [DOI] [PubMed] [Google Scholar]
- 13.Satokata I, Tanaka K, Okada Y. Molecular basis of group A xeroderma pigmentosum: a missense mutation and two deletions located in a zinc finger consensus sequence of the XPAC gene. Hum Genet. 1992;88(6):603–607. doi: 10.1007/BF02265282. [DOI] [PubMed] [Google Scholar]
- 14.States JC, McDuffie ER, Myrand SP, McDowell M, Cleaver JE. Distribution of mutations in the human xeroderma pigmentosum group A gene and their relationships to the functional regions of the DNA damage recognition protein. Hum Mutat. 1998;12(2):103–113. doi: 10.1002/(SICI)1098-1004(1998)12:2<103::AID-HUMU5>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 15.Tamura D, DiGiovanna JJ, Kraemer KH. Xeroderma pigmentosum. In: Lebwohl MG, Heymann WR, Berth-Jones J, Coulson I, editors. Treatment of Skin Disease. 3. London, England: Elsevier; 2009. [Google Scholar]
- 16.Gozukara EM, Khan SG, Metin A, et al. A stop codon in xeroderma pigmentosum group C families in Turkey and Italy: molecular genetic evidence for a common ancestor. J Invest Dermatol. 2001;117(2):197–204. doi: 10.1046/j.1523-1747.2001.01424.x. [DOI] [PubMed] [Google Scholar]
- 17.Tanaka K, Satokata I, Ogita Z, Uchida T, Okada Y. Molecular cloning of a mouse DNA repair gene that complements the defect of group-A xeroderma pigmentosum. Proc Natl Acad Sci U S A. 1989;86(14):5512–5516. doi: 10.1073/pnas.86.14.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tanaka K, Miura N, Satokata I, et al. Analysis of a human DNA excision repair gene involved in group A xeroderma pigmentosum and containing a zinc-finger domain. Nature. 1990;348(6296):73–76. doi: 10.1038/348073a0. [DOI] [PubMed] [Google Scholar]
- 19.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. Washington, DC: ASM Press; 2006. [Google Scholar]
- 20.Nishigori C, Zghal M, Yagi T, Imamura S, Komoun MR, Takebe H. High prevalence of the point mutation in exon 6 of the xeroderma pigmentosum group A-complementing (XPAC) gene in xeroderma pigmentosum group A patients in Tunisia. Am J Hum Genet. 1993;53(5):1001–1006. [PMC free article] [PubMed] [Google Scholar]
- 21.Satokata I, Tanaka K, Miura N, et al. Characterization of a splicing mutation in group A xeroderma pigmentosum. Proc Natl Acad Sci U S A. 1990;87(24):9908–9912. doi: 10.1073/pnas.87.24.9908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Satokata I, Tanaka K, Miura N, et al. Three nonsense mutations responsible for group A xeroderma pigmentosum. Mutat Res. 1992;273(2):193–202. doi: 10.1016/0921-8777(92)90080-m. [DOI] [PubMed] [Google Scholar]
- 23.Satokata I, Uchiyama M, Tanaka K. Two novel splicing mutations in the XPA gene in patients with group A xeroderma pigmentosum. Hum Mol Genet. 1995;4 (10):1993–1994. doi: 10.1093/hmg/4.10.1993. [DOI] [PubMed] [Google Scholar]
- 24.Driggers WJ, Grishko VI, LeDoux SP, Wilson GL. Defective repair of oxidative damage in the mitochondrial DNA of a xeroderma pigmentosum group A cell line. Cancer Res. 1996;56(6):1262–1266. [PubMed] [Google Scholar]
- 25.Reardon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in xeroderma pigmentosum patients. Proc Natl Acad Sci U S A. 1997;94(17):9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brooks PJ. The case for 8,5′-cyclopurine-2′-deoxynucleosides as endogenous DNA lesions that cause neurodegeneration in xeroderma pigmentosum. Neuroscience. 2007;145(4):1407–1417. doi: 10.1016/j.neuroscience.2006.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuraoka I, Bender C, Romieu A, Cadet J, Wood RD, Lindahl T. Removal of oxygen free-radical-induced 5′, 8-purine cyclodeoxynucleosides from DNA by the nucleotide excision-repair pathway in human cells. Proc Natl Acad Sci U S A. 2000;97(8):3832–3837. doi: 10.1073/pnas.070471597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harper PS. Practical Genetic Counselling. 3. Oxford, England: Butterworth-Heinemann Ltd; 1988. pp. 26–29. [Google Scholar]
- 29.Arcos-Burgos M, Muenke M. Genetics of population isolates. Clin Genet. 2002;61(4):233–247. doi: 10.1034/j.1399-0004.2002.610401.x. [DOI] [PubMed] [Google Scholar]
- 30.Zeegers MP, van Poppel F, Vlietinck R, Spruijt L, Ostrer H. Founder mutations among the Dutch. Eur J Hum Genet. 2004;12(7):591–600. doi: 10.1038/sj.ejhg.5201151. [DOI] [PubMed] [Google Scholar]
- 31.Committee on Ethics, American College of Obstetricians and Gynecologists; Committee on Genetics, American College of Obstetricians and Gynecologists. ACOG Committee Opinion No. 410: Ethical issues in genetic testing. Obstet Gynecol. 2008;111(6):1495–1502. doi: 10.1097/AOG.0b013e31817d252f. [DOI] [PubMed] [Google Scholar]
- 32.Slor H, Batko S, Khan SG, et al. Clinical, cellular and molecular features of an Israeli xeroderma pigmentosum family with a frameshift mutation in the XPC gene: sun protection prolongs life. J Invest Dermatol. 2000;115(6):974–980. doi: 10.1046/j.1523-1747.2000.00190.x. [DOI] [PubMed] [Google Scholar]