

Abstract
Background
Pineapple (Ananas comosus var. comosus), is an important tropical non-climacteric fruit with high commercial potential. Understanding the mechanism and processes underlying fruit ripening would enable scientists to enhance the improvement of quality traits such as, flavor, texture, appearance and fruit sweetness. Although, the pineapple is an important fruit, there is insufficient transcriptomic or genomic information that is available in public databases. Application of high throughput transcriptome sequencing to profile the pineapple fruit transcripts is therefore needed.
Methodology/Principal Findings
To facilitate this, we have performed transcriptome sequencing of ripe yellow pineapple fruit flesh using Illumina technology. About 4.7 millions Illumina paired-end reads were generated and assembled using the Velvet de novo assembler. The assembly produced 28,728 unique transcripts with a mean length of approximately 200 bp. Sequence similarity search against non-redundant NCBI database identified a total of 16,932 unique transcripts (58.93%) with significant hits. Out of these, 15,507 unique transcripts were assigned to gene ontology terms. Functional annotation against Kyoto Encyclopedia of Genes and Genomes pathway database identified 13,598 unique transcripts (47.33%) which were mapped to 126 pathways. The assembly revealed many transcripts that were previously unknown.
Conclusions
The unique transcripts derived from this work have rapidly increased of the number of the pineapple fruit mRNA transcripts as it is now available in public databases. This information can be further utilized in gene expression, genomics and other functional genomics studies in pineapple.
Introduction
The pineapple (Ananas comosus var. comosus), a member of the Bromeliaceae, is an economically important tropical fruit. Pineapple, along with three other dominant tropical fruits (mango, papaya and avocado), account for approximately 75% of global flesh tropical fruit production [1]. Despite its economic contribution, research on the genetics of the crop is scarce. This can be seen by the low number of pineapple genome sequences that are available in the public database. As of December 2011, the number of pineapple ESTs available in NCBI dbEST accounts to only about 6,000.
Pineapple is a non-climacteric fruit, as with grape, citrus, strawberry and pepper. In pineapple, there is no extra accumulation of starch during the ripening once the fruit is harvested or detached from the mother plant [2]. This is in contrast to climacteric fruits (such as tomato, papaya and banana), where a burst of respiration and a spike in ethylene causes the conversion of starch into sugar allowing the fruit to sweeten during fruit ripening. The unique ripening mechanism of non-climacteric fruit development is still unclear [3]–[4]. The underlying ripening pathways of the non-climacteric fruit can be deduced through the analysis of the transcripts being expressed.
Recent development in functional genomics has enabled the study of plant and organisms at the whole-transcriptomic level [5]. The emergence of the next generation sequencing (NGS) technology, allows for the rapid discovery of transcripts (including rare transcripts) and the quantification of gene expression. The applications of 454 (Roche), Solexa (Illumina) and/or Solid (Applied Biosystem) massively parallel sequencing, has been widely used to sequence any organism of choice at both the genomic or transcriptomic level. Utilization of short reads has been proven to effectively facilitate the de novo genome assembly [6], [7] and reference assembly of transcriptomic data [8]. It has also been shown that short reads have been effectively assembled and used in the discovery of genes and in the study of gene expression [9].
This paper reports on the millions of paired-end Solexa reads that were generated to reveal the transcriptomic profiles of the pineapple fruit. This dataset obtained through de novo assembly will be the first pineapple transcriptomic data generated from massively parallel sequencing. The information provides a good platform for further gene expression, genomics functional genomics and the non-climacteric ripening in pineapple.
Results and Discussion
Paired-end sequencing and de novo assembly
A total of about 4.7 (52.7%) million reads were obtained from mRNA-seq whole transcriptome sequencing of the pineapple fruit flesh. These were then assembled into 28,728 unique transcripts (UTs) using k-mer 47. The assembly produced 6.4 Mbp of reads with a N50 value of 223 bp. The length of the UTs generated from the de novo assembly ranged from 100 bp to 3.8 kb. The percentage of reads used in the generation of UTs obtained in this report was higher compared to those assembled (Solexa reads) in sweet potato root transcriptome [10]. On the other hand, in coral larval [11] and Artemisia annua [12] the percentage of reads used in the assembly were slightly higher compared to pineapple. However, this may be due to the different sequence length produced of the platforms i.e longer reads are produced from the 454/Roche compared to Illumina GA. In addition, the present of alternative splicing region in the transcripts also will hinder the assembly of long sequences [13]. Aside from this, the presence of repeats including both the simple and complex repeats was also likely factors contributing to be difficulty in assembly [14].
Estimating the number of genes and the level of transcripts coverage represented in an EST collection or assembly is important. To compute this, indirect evaluation of the pineapple transcriptome coverage was performed by comparing the number UTs generated from the assembly (28,728) and the mean length (n50 value = 223 bp) to the Arabidopsis thaliana genes.
Assuming a similar number of genes occur in pineapple as in A. thaliana, 25,000 genes with an average length of 2,000 bp [15]. This assembly was able to cover 0.13× transcripts of the pineapple transcriptome. The results was fairly good as the number of pineapple genes was solely evaluated from assembly of one fruit tissue and the UTs generated were assembled without a reference genome. Deep sequencing with various pineapple tissues and stages or reference assembly by using pineapple ESTs or nearest genome sequence will eventually fill up the gaps and give a more thorough transcripts representation in pineapple [16].
Characterization by Similarity Search
In order to make an assessment for the putative identities of the assembly, all the UTs were subjected to BLASTx similarity search. A similarity search against the non-redundant (nr) NCBI database showed that 16,932 (59%) UTs had significant matches with e-values of ≤10−6 (Table 1). Of these, 13,407 matched to known genes while 3,525 matched to unknown genes. A large portion of the unknown genes were hypothetical proteins. The remaining 11,796 (41%) UTs resulted in no significant hits. The identification of un-characterized sequences from cDNA libraries ranges considerably with most being reported to be between 35–50% [17]. In the characterization of apple EST sequences, approximately 40% of the unique transcripts from the assembly did not exhibit significant similarity [18] while in strawberry ESTs, 43.9% were without significant similarity [19]. This was similar with pineapple, where 40% of the UTs were above the cut off value (Table 1). The high percentage of the pineapple transcripts in this study with no hits to the non-redundant (nr) protein database indicate that there is enormous potential for the discovery of new genes in this plant besides providing the possibility of new identification of gene networks and also the fruit ripening pathways. The portion of the UTs that resulted in ‘no hits’ to the nr database is mainly due to the short UTs length although obtaining 30–40% of sequences more than the cut-off value is quite common in many studies, especially in studies involving large-scale sequencing [11], [20]–[21].
Table 1. Summary of the assembly and annotation of pineapple UTs.
| Number of sequences (%) | |
| 1. Assembly | |
| Total number of reads | 9,000,000 |
| Total number of reads used for assembly | 4,745,237 |
| Total unigenes generated | 28,728 |
| 2. Similarity Search | |
| Unigenes matching to known genes | 13, 407 (46.7%) |
| Unigenes matching to unknown genes | 3,525 (12.3%) |
| Unigenes matching to no hits | 11,796 (41%) |
| 3. Functional Annotation | |
| Unigenes without mapping | 1,425 |
| Unigenes with annotation | 14,510 |
| Number of GO terms assigned | 65,637 |
Of the UTs with matched similarities to the nr protein database, 452 (2.7%) had an e-value of e-100 or less and are considered highly significant. The moderately significant are those with e-values between e-20 to e-99. About 8,957 (52.9%) of the pineapple UTs were moderately similar to the return hits. Finally, the remaining 7,523 (44.4%) UTs were assigned weak homology i.e. e-values between e-7 to e-19. The overall e-value distribution pattern was similar with those characterized from oyster ESTs from oyster [21]. Organism distribution based on the BLASTx analysis of the UTs showed that the UTs hit a range of plant species. Among the various plants that have protein sequences in GenBank, the pineapple UTs had the highest number of hits to rice (Oryza sativa), by almost a quarter portion (26.2%). This was followed by Sorghum bicolor (17.52%), Vitis vinifera (16.84%), Zea mays (8.33%), Ricinus communis (7.94%) and Populus trichocarpa (7.59%). The hit to Ananas comosus was only 0.52% while, hits to non-plants organisms make up to a total of 0.25% (Figure 1).
Figure 1. Species distribution of pineapple UTs from the velvet de novo assembly.

Both the pineapple and rice are monocotyledons, which may explain for the high similarities that were observed. The high similarity of the pineapple UTs to the rice genes may provide for the possibility of using the rice's ESTs as a reference for future assembly using next generation sequencing. Collins and co-workers [22] showed that they managed to efficiently assemble the Pachycladon enysii, a polyploid plant by using a diploid plant as a reference genome. It is worth mentioning that the relatively small number of UTs matching to the pineapple protein sequences suggests that this assembly identified a considerable large group of genes previously unidentified in pineapple.
Functional characterization by GO annotation
GO database, is a platform allowing a standardized gene functional annotation of the properties of genes using controlled vocabulary annotations which is categorised into the three main ontologies i.e. Molecular Function, Biological Processes and Cellular Component [23]. By using the Blast2GO program, the UTs were annotated against the non-redundant (nr) Genbank database. Next, the nr annotated UTs were then mapped against the GO database to retrieve the GO terms. In total 65,637 GO terms were assigned to all 15,507 mapped UTs with an average of one UT assigned to four GO terms. Of these, the majority of the GO terms were assigned to Molecular function (25,033, 38.1%) followed by Biological process (20,672, 31.5%) and the least were categorized under the Cellular component (19,932, 30.4%).
Under the Cellular component ontology, proteins involved in cell and organelle development was dominant while in Molecular function ontology, proteins for binding and catalytic activity were generally encoded by the pineapple UTs. Biological process ontology distribution on the other hand, contains mainly proteins involved in cellular processes and metabolic processes (Figure 2). This pattern of distribution is typically seen in the transcriptome of samples undergoing development process [24]. During the development process of pineapple until it fully ripen on plant, biological processes involves cell development and cellular changes. Other proteins involved in the biological process ontology observed were those responsible for stress response, biological regulation, reproduction, developmental processes and multicellular organismal process. All these categories indicate that the pineapple fruit undergoes multiple processes of development and stresses.
Figure 2. Gene Ontology classification of the pineapple UTs distribution in Cellular component, Molecular function and Biological process ontology.

Functional classification by KEGG
In order to identify the active biological pathways in pineapple, the assembled UTs were used to obtain the Enzyme Commission (EC) when annotated against the KEGG database. A total of 5579 UTs were assigned to 7712 EC. The ECs were subsequently grouped into 126 biochemical pathways, of which 122 pathways were found to be involved in fruit metabolism. The presence of abundant metabolic pathways was also seen in the transcriptome of Citrus sinensis during fruit development [25]. Also, in the proteomic analysis of grape skin, the changes in the proteome among different stages of ripening mainly involve metabolic pathways [26]. This suggests that there are a high number of metabolic activities occurring during the fruit development of pineapple.
Enzymes involved in pineapple metabolism were further classified into 11 sub-categories. There are considerably higher portions of enzymes participate in the metabolism of lipid (15 pathways), carbohydrate (15 pathways), and xenobiotics biodegradation and metabolism (156 pathways) (Figure 3). This indicates that there are enormous cell walls activities, starch and sucrose synthesis and many other health-related compounds being synthesis during the ripe stage pineapple fruit. This is particularly similar with the analysis of Expressed Sequence Tags in apple [27] and kiwi fruit [28]. In transcriptome analysis of the mutant sweet orange, many genes were found to be involved in carbohydrates metabolism and were differentially expressed during fruit development [25]. The details of all pathways together with total UTs annotated to the pathways and the number of EC assigned to the pathways for of the sub-categories is provided in Additional file 1.
Figure 3. Classification of the sub-categories involved in the pineapple metabolism.

Analysis of genes encoding important traits in pineapple using the assembled UTs
Fragrance Biosynthesis
In the synthesis of fragrance in fruits, the volatiles compounds contributing aroma volatiles include the terpenes, alcohols, aldehydes, esters and aromatic acid [29]. Terpenoid and esteric compounds were predominantly found to contribute in the aroma biosynthesis of plants and fruits [30]. The terpene biosynthesis pathways synthesize compounds that include the monoterpenes, sesquiterpenes, diterpenes, triterpenes, carotenoids, ubiquinone and other terpenoid-quinones. The key enzyme that starts the terpene biosynthesis is the mevalonate kinase (Figure 4). The mevalonate kinase with the EC number of 2.7.1.36 were encoded by 4 UTs (EC:2.7.1.36; 4 UTs) react to convert mevalonate to 5-phosphomevalonate. Next, phosphomevalonate kinase (EC:2.7.4.2; 6 UTs) convert the compound to 5-diphosphomevalonate. Melavonate diphosphate decarboxylase (EC:4.1.1.33) which are responsible for the production of isopentenyl diphosphate were not detected in the annotated UTs.
Figure 4. The terpene biosynthesis pathway.
The pineapple transcripts encoding enzymes (highlighted) involved in the pathway was identified by BLASTx (e-06 cutoff). Numbers in bracket are the total number of UTs encode for the enzyme.
However, isopentenyl diphosphate can be synthesized via the MEP/DOXP pathway where pyruvate is converted through a number of enzymatic steps. The enzymes involved and found to be encoded by the annotated UTs are 1-deoxy-D-xylulose-5-phosphate synthase (EC:2.2.1.7; 3 UTs), 1-deoxy-D-xylulose-5-phosphate reductoisomerase (EC:1.1.1.267; 3 UTs), 4-(cytidine 5′-diphospho)-2-C-methyl-D-erythritol kinase (EC:2.7.1.148; 3 UTs), 2-C-methyl-D-erythritol 2,4-cyclodiphosphate synthase (EC:4.6.1.12; 2 UTs), 4-hydroxy-3-methylbut-2-enyl diphosphate reductase (EC:1.17.1.2; 4 UTs) and isopentenyl-diphosphate Delta-isomerase (EC:5.3.3.2; 4 UTs) (Figure 4).
Next, conversion of isopentenyl diphosphate or/and dimethylallyl diphosphate to geranyl diphosphate (geranyl-PP) was carried out by dimethylallyltranstransferase (EC:2.5.1.1; 1 UT) for the synthesis of monoterpenes. The enzyme, Polyisoprene synthase, which is responsible for both the farnesyl diphosphate (farnesyl-PP) and geranyl geranyl diphosphate (geranyl geranyl-PP) synthesis of sesquiterpenes and diterpenes respectively, was not identified in the pineapple fruit assembly. However, the enzymes for triterpenes and carotenoids synthesis were found to be present in the fruit transcriptome. Two sequences encoding enzyme squalene monooxygenase (EC: 1.14.99.7; 2 UTs), which is involved in the conversion of squalene to sterol for the synthesis of triterpenes were identified. Another two sequences encoding phytoene synthase (EC:2.5.1.32; 2 UTs) which is involved in carotenoid synthesis was also discovered.
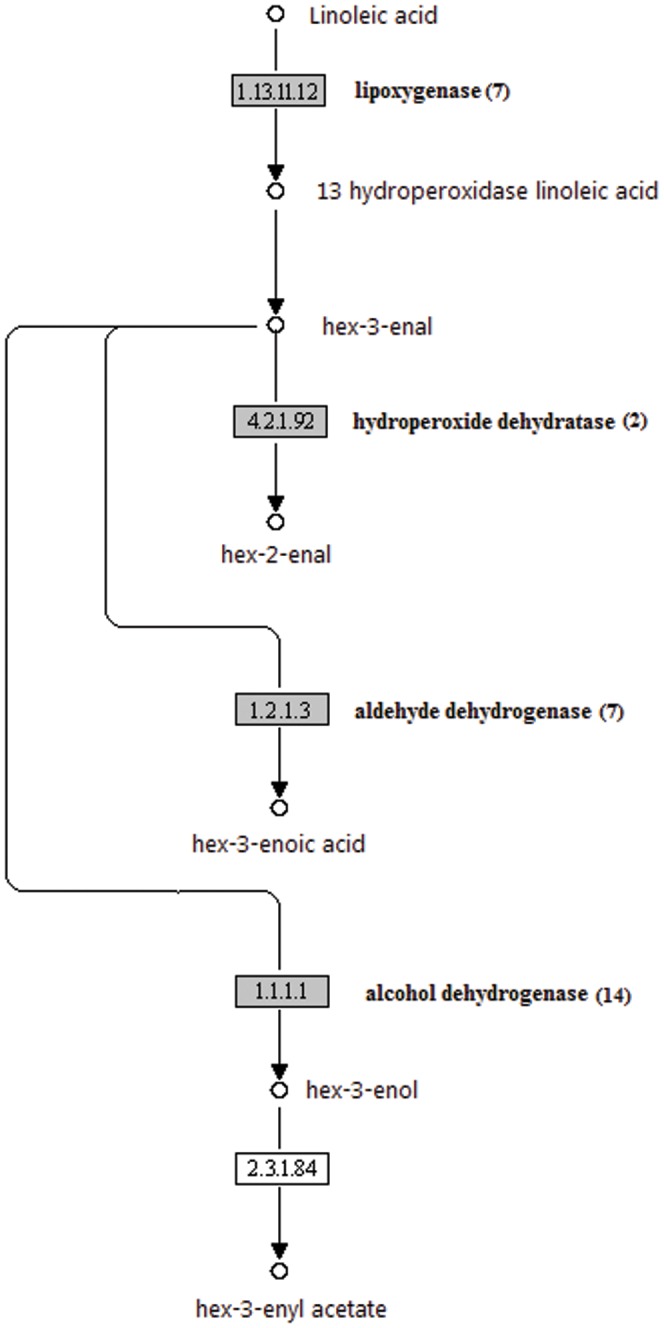
Ester compounds have been reported to be commonly synthesized in fruits,such as Actinidia [28], Malus domestica [27] and pineapple [31]. Four out of five enzyme involved in the straight chain ester biosynthesis were identified in pineapple. The first step in the synthesis involves the activity of the lipoxygenase enzyme (EC: 1.13.11.12; 7 UTs). It converts linoleic acid to 13 hydroperoxidase linoleic acid. From 13 hydroperoxidase linoleic acid, hydroperoxide lyase enzyme converts the compound to hex3-enal, which is then converted to hex-2-enal by hydroperoxide dehydratase (EC: 4.2.1.92; 2 UTs). The aldehydes, can either be converted to its acids by aldehyde dehydrogenases (EC: 1.2.1.3; 7 UTs) or to alcohols by alcohol dehydrogenese (EC: 1.1.1.1; 14 UTs). The alcohol, hex-3-enol, is later converted to ester by alcohol acyl transferases (Figure 5).
Figure 5. The straight chain ester biosynthesis from fatty acids.

The pineapple transcripts encoding enzymes (highlighted) involved in the pathway was identified by BLASTx (e-06 cutoff). Numbers in bracket are the total number of UTs encode for the enzyme.
Branched chain esters are synthesized via isoleucine, valine and leucine [27]. The chain of enzymes involved in converting threonine and/or pyruvate to valine, leucine and isoleucine were identified in the pineapple UTs (Figure 6). They were threonine ammonia-lyase (EC: 4.3.1.19; 3 UTs), acetolactate synthase (EC: 2.2.1.6; 5 UTs), ketol-acid reductoisomerase (EC: 1.1.1.86; 4 UTs), dihydroxy-acid dehydratase (EC: 4.2.1.9; 3 UTs), branched-chain-amino-acid transaminase (EC: 2.6.1.42; 9 UTs), 2-isopropylmalate synthase (EC: 2.3.3.13; 3 UTs), 3-isopropylmalate dehydratase (EC: 4.2.1.33; 4 UTs), 3-isopropylmalate dehydrogenase (EC: 1.1.1.85; 11 UTs) and branched-chain-amino-acid transaminase (EC: 2.6.1.42; 9 UTs). The three acid syntheses are then converted to its aldehydes and alcohols by decarboxylase enzyme (EC: 4.1.1.1; 10 UTs) and alcohol dehydrogenase (EC: 1.1.1.1; 14 UTs), respectively. Alcohol acyl transferase will then convert the alcohols to ester.
Figure 6. The branched chain ester biosynthesis.

The pineapple transcripts encoding enzymes (highlighted) involved in the pathway was identified by BLASTx (e-06 cutoff). Numbers in brackets are the total number of UTs encode for the enzyme.
Flavor Biosynthesis
Flavor and aroma are closely related traits which give the best eating quality of the pineapple. Fruit acidity measured by the synthesis of the organic acid and the sweetness (sucrose metabolism) are the major factors of concern in the biosynthesis of flavor compounds. The organic acid synthesis (malic acid and citric acid) accumulations in the fruit involves the citrate acid cycle (Figure 7). The enzymes, citrate synthase (EC:2.3.3.1; 4 UTs), aconitate hydratase (EC:4.2.1.3; 20 UTs), phosphoenolpyruvate carboxylase (PEPC) (EC:4.1.1.31; 21 UTs), malate dehydrogenase (MDH) (EC:1.1.1.37; 24 UTs) and malic enzyme (EC:1.1.1.40; 5 UTs) were found to be encoded by the assembled UTs. Three of these enzymes, citrate synthase, aconitate hydratase and malate dehydrogenase were found be involved in the citrate acid cycle.
Figure 7. The citrate acid cycle.

The pineapple transcripts encoding enzymes (highlighted) involved in the pathway was identified by BLASTx (e-06 cutoff). Numbers in brackets are the total number of UTs encode for the enzyme.
A great number of enzymes involved in the starch and sucrose metabolism have been identified encoded by the pineapple UTs. The sucrose metabolism route involves four main enzymes. Sucrose phosphate synthase (SPS) (EC:2.4.1.14; 11 UTs) synthesizes sucrose-6-phosphate from UDP-glucose and fructose-6-phosphate. Next, sucrose-6-phosphate undergo hydrolysis to form sucrose by the action of sucrose-phosphate phosphatase (EC:3.1.3.24; 1 UT). The sucrose are then cleaved to sugar nucleotides either by sucrose synthase (EC:2.4.1.13; 18 UTs) and/or invertase (EC:3.2.1.26; 19 UTs) (Figure 8).
Figure 8. The sucrose metabolism.

The pineapple transcripts encoding enzymes (highlighted) involved in the pathway was identified by BLASTx (e-06 cut off). Numbers in brackets are the total number of UTs encode for the enzyme.
Texture/Structural Biosynthesis
Lignin is a type of polymer derived from the combination of aromatic monomers subunits [32]. Lignin was the second most abundant molecule after cellulose that provides plant with structural rigidity. Aside from the structural function, lignin also acts as an important component of fiber in the fruit and is beneficial for human health [33]. The initial step in its biosynthesis involves the deamination of phenylalanine to cinnamic acid. Phenylalanine are then converted to p-coumaric acid by trans-cinnamate 4-monooxygenase (EC:1.14.13.11; 2 UTs). p-coumaric acid are then converted to caffeic acid by coumarate 3-hydroxylase. From caffeic acid, caffeate O-methyltransferase (C-OMT) (EC: 2.1.1.68; 3 UT) then converts this compound to ferulic acid. Conversion from ferulic acid to 5-hydroxyferulic acid and then to sinapic acid was carried out by ferulate 5-hydroxlase (FSH) and C-OMT, respectively (Figure 9).
Figure 9. The lignin biosynthesis.

The pineapple transcripts encoding enzymes (highlighted) involved in the pathway was identified by BLASTx (e-06 cutoff). Numbers in brackets are the total number of UTs encode for the enzyme.
All the acid synthesized as mentioned above are then ligated with CoA to form it respective CoA acid derivatives by 4-coumarate-CoA ligase (EC:6.2.1.12; 5 UTs). These acids are then reduced to its corresponding aldehydes by cinnamoyl-CoA reductase (EC:1.2.1.44; 13 UTs). Next, the aldehydes are then reduced to its alcoholic compound by cinnamyl alcohol dehydrogenase (CAD) (EC: 1.1.1.195; 4 UTs). The final step of the lignin biosynthesis is the polymerization the monolignols by peroxidase (EC:1.11.1.7; 28 UTs). Other intermediate compound conversion enzyme found in pineapple was caffeoyl-CoA O-methyltransferase (EC: 2.1.1.104; 3 UTs) which convert coffeoyl-CoA to feruloyl-CoA and 5-hydroxy-feruloyl-CoA to sinapoyl-CoA.
Health-Related Compound Biosynthesis
The general vitamins that were found in many types of vegetables and fruits include thiamin (vitamin B1), riboflavin (vitamin B2), nicotinamide (vitamin B3), biotin (vitamin B7), folate (vitamin B9) and ascorbate (vitamin C) [34]–[35]. The pineapple UTs encoded some of the enzymes in the synthesis of the above health-related compounds. Only a small portion of the enzymes involved in the synthesis of thiamin, nicotinamide, biotin and ascorbate was identified. A more complete enzymatic activity was observed in the synthesis of riboflavin and folate. Many researchers have focus to increase folate in staple fruit as the intake is essential in normal diet [36]. Despite its importance, human and animals are not able to synthesis folate. This drives the identification of high folate content food and the biofortication of folate (enhancement of folate in plants). Biofortication of folate in tomato was has been conducted by crossing of transgenic tomato with PABA- (p-aminobenzoate) and pteridine-overproduction traits which managed to increase the folate content to more than 25-folds [37]. The same method may possibly be applied in pineapple, whereby the increase in the nutritional content of pineapple fruit may eventually increase its economic value.
Materials and Methods
Plant Material and mRNA Sequencing
Pineapple was collected from an experimental plot at a pineapple farm managed by a local farmer located at Kampung Babagon, Sabah. The pineapple sample was kindly provided and permitted by the farm owner. The sample was excised, packed and stored at −80°C. The total RNA of the ripe pineapple fruit tissue was extracted using method described by [38] with minor modifications. The RNA isolated was checked for yield (≥500 ng/µl) and quality by measuring both the A260/280 and A260/230 ratios (with a ratio of more than 1.6) using a NanoVue spectophotometer (GE Healthcare, USA). Total RNA volume of 30 µl (with the required yield and purity) was sent on May 2009 for paired-end mRNA sequencing with Genome Analyzer IIx, using the service provided by Illumina FastTrack, USA. A 75 bp paired-end sequencing protocol with insert sizes of 200 bp was employed.
De novo Assembly by Velvet Software
Velvet de novo software was used to assemble the 75 bp paired-end reads. The software algorithm involved the construction of de Bruijn graph for the sequence assembly [39]. Therefore, different k-mers (or hash number) were initially used to assess the best assembly of the paired-end reads. It was found that the assembly with k-mer of 47 provided the best output for the transcriptomic assembly. The unique transcripts (UTs) obtained from assembly using k-mers of 47 were then used for subsequent downstream analysis. The assembled UTs with sequence length longer than 200 bp have been deposited in the Transcriptome Shotgun Assembly (TSA) at NCBI under the accession number: JI400295–JI412109.
Functional Characterization and Gene Ontology (GO) Annotation
All the UTs were then used for blast search against GenBank's non-redundant database. Blast matches were considered significant with E-value scores ≤10−6 using the BLASTx tool (http://blast.ncbi.nlm.nih.gov/Blast.cgi) [40]. Gene Ontology (GO) annotations were performed to retrieve molecular function, biological process and cellular component terms using Blast2GO (http://www.blast2go.org/) [23]. The sequences were loaded into the Blast2GO program, and BLASTx with a minimum E-value of 10−6 was performed by the program prior to mapping. The mapping step allowed the annotation of the sequences to the GO database for GO terms.
Functional Classification by KEGG
Annotation against the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/) were performed to obtain the link of enzymes integrating together genomic, chemical and network information [41] using the Blast2GO program. All the mapped sequences were annotated to the KEGG database to obtain the enzyme commission (EC) number. The EC were then mapped to the KEGG Pathway to obtain the KEGG Pathway-Maps.
Supporting Information
List of metabolism pathways in ripe pineapple fruit UTs mapped against KEGG database.
(DOC)
Acknowledgments
The authors would like to thank the Ministry of Science, Technology and Innovation Malaysia for the funding, Malaysia Genome Institute and Science Vision Sdn. Bhd. for providing the facilities and support in the data analysis.
Funding Statement
This study was supported by the ScienceFund grant (02-01-10-SF0026) from the Ministry of Science, Technology and Innovation, Malaysia. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Zhou Y, Dahler JM, Underhill JR, Wills RBH (2003) Enzyme associated with blackheart development in pineapple fruit. Food Chemistry 80: 565–572. [Google Scholar]
- 2. Moyle R, Fairbairn DJ, Ripi J, Crowe M, Botella JR (2005) Developing pineapple fruit has a small transcriptome dominated by metallothionein. J Exp Bot 409: 101–112. [DOI] [PubMed] [Google Scholar]
- 3. Giovannoni JJ (2004) Genetic regulation of fruit development and ripening. The Plant Cell 16: S170–S180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Giovannoni JJ (2007) Fruit ripening mutants yield insights into ripening control. Curr Opin Plant Biol 10: 283–289. [DOI] [PubMed] [Google Scholar]
- 5. Wang X, Sun Q, McGrath SD, Mardis ER, Soloway PD, et al. (2008) Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. Plos ONE 3: e3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Shi C-Y, Yang H, Wei C-L, Yu O, Zhang Z-Z, et al. (2011) Deep sequencing of the Camelia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea specific compounds. BMC Genomics 12: 131–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang X-W, Luan J-B, Li J-M, Bao Y-Y, Zhang C-X, et al. (2010) De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genomics 11: 400–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hegedus Z, Zarrzewska A, Agoston VC, Ordas A, Racz P, et al. (2009) Deep sequencing of the zebrafish transcriptome response to mycobacterium infection. Mol Immunol 46: 2918–2930. [DOI] [PubMed] [Google Scholar]
- 9. Rosenkranz R, Borodina T, Lehrach H, Himmelbauer H (2008) Characterizing the mouse ES cell transcriptome with Illumina sequencing. Genomics 92: 187–194. [DOI] [PubMed] [Google Scholar]
- 10. Wang Z, Fang B, Chan J, Luo Z, Huang L, et al. (2010b) De novo assembly and characterization of root transcriptome using Illumina paired-end sequencing and development of cSSR markers in sweetpotato (Ipomoea batatas). BMC Genomics 11: 726–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Meyer E, Aglyamova GV, Wang S, Buchanan-Carter J, Abrego D, et al. (2009) Sequencing and de novo analysis of a coral larval transcriptome using 454 GSFlx. BMC Genomics 10: 219–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang W, Wang Y, Zhang Q, Qi Y, Guo D (2009) Global characterization of Artemisia annua glandular trichome transcriptome using 454 pyrosequencing. BMC Genomics 10: 465–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vera JC, Wheat CW, Fescemyer HW, Frilander MJ, Crawford DL, et al. (2008) Rapid transcriptome characterization for a non-model organism using 454 pyrosequencing. Mol Ecol 17: 1636–1647. [DOI] [PubMed] [Google Scholar]
- 14. Pop M, Salzberg SL (2008) Bioinformatics challenges of new sequencing technology. Trends in Genetics 24: 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bevan M, Walsh S (2005) The Arabidopsis genome: A foundation for plant research. Genome Research 15: 1632–1642. [DOI] [PubMed] [Google Scholar]
- 16. Parchman TL, Geist KS, Grahnen JA, Benkman CW, Buerkle CA (2010) Transcriptome sequencing in an ecologically important tree species: assembly, annotation, and marker discovery. BMC Genomics 11: 180–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Costa GGL, Cardoso KC, Del Bem LEV, Lima AC, Cunha MAS, et al. (2010) Transcriptome analysis of the oil-rich seed of the bioenergy crop Jatropha curcas L. BMC Genomics 11: 462–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Park S, Sugimoto N, Larson MD, Beaudry R, van Nocker S (2006) Identification of genes with potential roles in apple fruit development and biochemistry through large-scale statistical analysis of expressed sequence tags. Genome Analysis 141: 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bombarely A, Merchante C, Csukasi F, Cruz-Rus E, Caballero JL, et al. (2010) Generation and anlysis of ESTs from strawberry (Fragaria xananassa) fruits and evaluation of their utility in genetic and molecular studies. BMC Genomics 11: 503–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Beldade P, Rudd S, Gruber JD, Long AD (2006) A wing expressed sequence tags resource for Bicyclus anynana butterflies, an evo-devo model. BMC Genomics 7: 130–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Quilang J, Wang S, Li P, Abernathy J, Peatman E, et al. (2007) Generation and analysis of ESTs from the eastern oyster, Crassostrea virginca Gmelin and identification of microsatellites and SNP markers. BMC Genomics 8: 157–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Collins LL, Biggs PJ, Voelckel C, Joly S (2008) An approcah to transcriptome analysis of non-model organisms using short-read sequences. Genome Informatics 12: 3–14. [PubMed] [Google Scholar]
- 23. Conesa A, Gotz S, Garcia-Gornez JM, Terol J, Talon M, et al. (2005) Blast2GO: a universal tool for annotation, visualization and analysis of functional genomics research. Bioinformatics 21: 3674–3676. [DOI] [PubMed] [Google Scholar]
- 24. Lokanathan Y, Mohd-Adnan A, Wan K-L, Nathan S (2010) Transcriptome analysis of the Cryptocaryon irritans tomont stage identifies potential genes for the detection and control of cryptocaryonois. BMC Genomics 11: 76–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Q, Zhu A, Chai L, Zhou W, Yu K, et al. (2009) Transcriptome analysis of a spontaneous mutant in sweet orange [Citrus sinensis (L.) Osbeck] during fruit development. J Exp Bot 60: 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Negri AS, Prinsi B, Rossoni M, Failla O, Scienza A, et al. (2008) Proteome changes in the skin of the grape cultivar Barbera among different stages of ripening. BMC Genomics 9: 378–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Newcomb RD, Crowhurst RN, Gleave AP, Rikkerink EHA, Allan AC, et al. (2006) Analyses of expressed sequence tags from apple. Plant Physiol 141: 147–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Crowhurst RN, Gleave AP, MacRae EA, Ampomah-Dwamena C, Atkinson RG, et al. (2008) Analysis of expressed sequence tags from Actinidia: applications of a cross species EST database for gene discovery in the areas of flavor, health, color and ripening. BMC Genomics 9: 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Dudareva N, Pichersky E, Gershenzon J (2004) Biochemistry of plant volatiles. Plant Physiol 135: 1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dudareva N, Pichersky E (2008) Metabolic engineering of plant volatiles. Curr Opin Biotechnol 19: 1–9. [DOI] [PubMed] [Google Scholar]
- 31. Elss S, Preston C, Hertzig F, Richling E, Schreier P (2005) Aroma prolies of pineapple fruit (Ananas comosus [L.] Merr.) and pineapple products. LWT-Food Science and Technology 38: 263–274. [Google Scholar]
- 32. Whetten R, Sederoff R (1995) Lignin Biosynthesis. The Plant Cell 7: 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aggarwal BB, Shishodia S (2006) Molecular targets of dietary agents for prevention and therapy of cancer. Biochemical Pharmacology 71: 1397–1421. [DOI] [PubMed] [Google Scholar]
- 34. Smith AG, Croft MT, Moulin M, Webb ME (2007) Plants need their vitamins too. Curr Opin Plant Biol 10: 266–275. [DOI] [PubMed] [Google Scholar]
- 35. Vahteristo L, Lehikoinen K, Ollilainen V, Varo P (1997) Application of an HPLC assay for the determination of folate derivatives in some vegetables, fruits and berries consumed in Finland. Food Chemistry 59: 589–597. [Google Scholar]
- 36. Stover PJ (2004) Physiology of folate and vitamin B12 in health and disease. Nutrition Reviews 62: S3–S12. [DOI] [PubMed] [Google Scholar]
- 37. Díaz de la Garza RI, Gregory JF III, Hanson AD (2007) Folate biofortication of tomato fruit. PNAS 104: 4218–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li JH, Tang CH, Song CY, Chen MJ, Feng ZY, et al. (2006) A simple, rapid and effective method for total RNA extraction from Lentinula edodes . Biotechnol Lett 26: 1193–1197. [DOI] [PubMed] [Google Scholar]
- 39. Zerbino DR, Birney E (2008) Velvet: Algorithms for de novo short read assembly using de Bruijin graphs. Genome Research 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 41. Minoru K, Susumu G (2000) KEGG:Kyoto Encyclopedia of Genes and Genomes. Nucl Acids Res 28: 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of metabolism pathways in ripe pineapple fruit UTs mapped against KEGG database.
(DOC)