

Abstract
The central nervous system is derived from the neural plate that undergoes a series of complex morphogenetic events resulting in formation of the neural tube in a process known as neurulation. The cellular behaviors driving neurulation in the cranial region involve forces generated by the neural tissue itself as well as the surrounding epithelium and mesenchyme. Of interest, the cranial mesenchyme underlying the neural plate undergoes stereotypical rearrangements hypothesized to drive elevation of the neural folds. As the neural folds rise, the hyaluronate-rich extracellular matrix greatly expands resulting in increased space between individual cranial mesenchyme cells. Based on inhibitor studies, expansion of the extracellular matrix has been implicated in driving neural fold elevation; however, since the surrounding neural and epidermal ectoderm were also affected by inhibitor exposure, these studies are inconclusive. Similarly, treatment of neurulating embryos with teratogenic doses of retinoic acid results in altered organization of the cranial mesenchyme but alterations in surrounding tissues are also observed. The strongest evidence for a critical role for the cranial mesenchyme in neural fold elevation comes from studies of genes expressed exclusively in the cranial mesenchyme that when mutated result in exencephaly associated with abnormal organization of the cranial mesenchyme. Twist is the best studied of these and is expressed in both the paraxial mesoderm and neural crest derived cranial mesenchyme. Here we review the evidence implicating the cranial mesenchyme in providing a driving force for neural fold elevation to evaluate whether there is sufficient data to support this hypothesis.
The central nervous system originates during embryogenesis with induction of the neural plate. The neural plate subsequently undergoes a complexly orchestrated series of morphogenetic movements resulting in formation of the neural tube (Figure 1). This process is highly susceptible to genetic and environmental perturbations resulting in failure of neural tube closure and neural tube defects (NTDs). NTDs represent one of the most common structural birth defects affecting anywhere from 1 in 50 to 0.6 in 1000 live births depending on the population in question (Gu et al., 2007; Parker et al., 2010). A failure of neural tube closure in the spinal or cranial regions of the embryo results in spina bifida or exencephaly, respectively. Spina bifida is associated with varied degrees of disability depending upon the level of the lesion and the involvement of the meninges, whereas exencephaly is fatal around birth. The causes of NTDs are multifactorial involving both environmental and genetic factors (Zohn, 2012; Zohn and Sarkar, 2008). One of the most well studied environmental factors influencing the occurrence of NTDs is folic acid supplementation during the periconception period (Zohn and Sarkar, 2010). The neural tube closes between the 3rd and 4th week of gestation in humans, which corresponds to 8.5 to 10.5 days post coitum in the mouse embryo and 9 to 12 in the rat. Neural tube closure is initiated at discrete closure points dubbed Closure 1, 2 and 3. Closure of the neural tube at these points is followed by zipping of the neural folds closed in both anterior and posterior directions (Copp et al., 2003). Closure 1 initiates at the hindbrain/spinal cord boundary and extends from there. In the cranial region, Closure 3 initiates at the anterior aspect of the forebrain and Closure 2 at the midbrain/forebrain boundary. The position of Closure 2 is variable between mouse strains and may also be variable in humans as it has been identified in some but not other specimens (Greene and Copp, 2009).
Figure 1. Morphogenesis of the cranial mesenchyme during neural fold elevation.

Embryos in whole mount are shown in panels on the left and in sections in panels on the right. The plane of section is illustrated by a red line. In the 3-somite staged mouse embryo, the biconvex neural folds have not yet begun to elevate and exhibit a columnar organization. NC-CM is induced in the dorsal neural tube and can be labeled by the Wnt1-cre driver (blue). Underlying the neural plate are PM-CM cells that can be labeled by the Mesp1-cre line (light grey) and are shown as dashes in panel to the right. Yellow shading in panels to the right indicates the even distribution of HA in the cranial mesenchyme underlying the neural folds. At 6-somite stages, the neuroepithelium transforms to a pseudostratified epithelium and rounded neural crest cells (dots in panel to the right) can be seen migrating from the dorsal neural folds within the subectodermal PM-CM to positions in the branchial arches and frontonasal mesenchyme (blue dashes in left panel). At the 8-somite stage, the neural folds exhibit a “V” shape as they continue to rise and HA concentrations (darker yellow shading) in the ECM increase evenly throughout the cranial mesenchyme. Neural crest cells are migrating and individual PM-CM cells underlying the neural plate are orientated parallel to the neural plate. By the 15-somite stage, NC-CM has migrated to their final destinations in the branchial arches and frontonasal mesenchyme. As the neural folds rise, the HA-rich ECM around the cranial mesenchyme cells in medial regions of the cranial mesenchyme expands resulting in reduced concentration of HA. The neural folds exhibit a “C” shape as the folds begin to converge in the dorsal midline. The PM-CM cells only orientate parallel to the neural tube in the most lateral regions. In medial regions the space between PM-CM cells has expanded greatly. Conceptual diagrams on left were made after Figures 1 and 2 in (Jiang et al., 2002; Yoshida et al., 2008), respectively. Conceptual diagrams on right were made after Figures 2, 4 and 5 in (Morris-Wiman and Brinkley, 1990a).
Morphogenesis of the neural tube has been an intensely studied area of research. As a consequence, a number of biochemical pathways and cell biological processes are implicated in driving neural tube closure (Colas and Schoenwolf, 2001; Copp and Greene, 2010; Copp et al., 2003). These include pathways that regulate changes in cellular shape, number, position and adhesion within in the neural tissue itself or in the surrounding epithelium and mesenchyme along with inductive signals from tissues adjacent to the neural tube. In the cranial region in particular, morphogenesis of the underlying cranial mesenchyme is postulated to be essential for elevation of the neural folds (Copp, 2005; Fleming et al., 1997; Morriss-Kay, 1981). However, unlike the other morphogenic events that drive neurulation, this topic has been considerably understudied with the advent of modern molecular biology and genetics. A number of experiments conducted in the 1970's and 1980's described in detail morphogenesis of the cranial mesenchyme during neurulation in the rat embryo. Yet, for the most part, the pathways that regulate cranial mesenchyme behavior during normal and abnormal neural tube closure have not been described at the molecular level. Here we review the evidence implicating the cranial mesenchyme in elevation of the cranial neural folds with the goal of evaluating whether sufficient evidence exists in support of the hypothesis that the cranial mesenchyme providing a driving force for neural fold elevation in the cranial region.
Developmental origin of the cranial mesenchyme
The cranial mesenchyme is derived from the anterior paraxial mesoderm (PM-CM) and the neural crest (NC-CM; Figure 1). Experiments in rodent and chick embryos suggest that both the PM-CM and NC-CM are essential for neural fold elevation (Colas and Schoenwolf, 2001; Copp, 2005; Fleming et al., 1997; Morriss-Kay, 1981). These populations are induced at different times in development, localized in different positions in the embryo and will develop into different structures (Noden and Trainor, 2005; Yoshida et al., 2008). The PM-CM and NC-CM lineages can be differentially marked by the Mesp1-cre and Wnt1-cre transgenic mouse lines, respectively (Yoshida et al., 2008). In addition to providing fate-mapping information for these populations, these reagents will be important for dissecting the relative contribution of the PM-CM and NC-CM in neural fold elevation. The PM-CM originates from the primitive streak and migrates to the anterior region of the embryo to underlie the presumptive neural plate. The NC-CM is induced at the junction of the neural plate and epidermal ectoderm, undergoes an epithelial to mesenchyme transition and delaminates just prior to neural fold elevation in the rodent embryo. NC-CM cells migrate along stereotypic paths in the subectodermal PM-CM to the branchial arch, frontonasal and periocular mesenchyme. This is in contrast to the trunk neural crest that in addition to a subectodermal route, also migrates along a path adjacent to the spinal neural tube. The PM-CM will contribute to some of the bones of the skull vault and muscles of the face; whereas the NC-CM will contribute to other bones of the skull and face in addition to cranial nerves (Jiang et al., 2002; Noden and Trainor, 2005; Yoshida et al., 2008).
Morphogenesis of the neural tube – the “cranial mesenchymecentric” view
The behavior of the cranial mesenchyme during morphogenesis of the neural tube has been described in detail in the rat embryo (Morris-Wiman and Brinkley, 1990a; Morris-Wiman and Brinkley, 1990b; Morris-Wiman and Brinkley, 1990c; Morriss and Solursh, 1978a; Morriss and Solursh, 1978b). During neural fold elevation, both the number of cranial mesenchyme cells and space between cells increases (Morris-Wiman and Brinkley, 1990a; Morriss and Solursh, 1978a; Morriss and Solursh, 1978b). As described by Morris-Wiman and Brinkley (1990a) in the early somite stage rat embryo, two to three layers of evenly distributed PM-CM cells underlie the neural plate. At this stage, the neural and epidermal ectoderm are of the similar thickness. The neural groove forms in the center of the neural plate giving the neural folds a biconvex shape (Figure 1). By the 6-somite stage, the neural ectoderm thickens, changing from a columnar to pseudostratified epithelium. NC-CM emigration begins at this stage and rounded neural crest cells can be seen migrating from the dorsal neural tube within the subectodermally localized PM-CM. The PM-CM remains evenly distributed and the neural folds maintain their biconvex shape. From 7-10 somite stages, the neural folds begin to elevate, transforming from a biconvex to “V” shaped neural plate with the neural groove in the center. As the neural folds elevate, proliferation of the neuroepithelium results in lateral expansion of this tissue. Additionally, the PM-CM cells that underlie the neural plate begin to orientate parallel to the neural epithelium. By the 11-13 somite stages, dorsal lateral hinge points begin to form as the neuroepithelial cells in the hinge point region become wedge shaped. PM-CM cells become more widely spaced and in medial regions are no longer orientated parallel to the neuroepithelium. From 14-17 somite stages, the dorsal lateral hinge points become more pronounced and the neural folds take on a “C” shape. During this time, widely spaced PM-CM cells with random orientations sparsely populate the central and medial regions of the cranial mesenchyme. In lateral regions, PM-CM cells are more closely packed and are orientated parallel to the neural tube. By 18/19 somite stages the neural folds are fully elevated and fuse in the dorsal midline. The PM-CM has greatly expanded in the dorsal ventral direction with the distance between the notochord/gut and neuroepithelium increasing as the neural folds elevate. Expansion of the PM-CM cannot be explained by differential proliferation or apoptosis (Morris-Wiman and Brinkley, 1990b). In fact, proliferation is highest in the central versus lateral regions where the PM-CM expansion is greatest. Similarly, no appreciable cell death is detected in the central region. Instead differential expansion is likely due to cell translocation driven by expansion of the extracellular matrix (ECM) surrounding the PM-CM as discussed below.
ECM composition during neural fold elevation and morphogenesis of the cranial mesenchyme
During neurulation, the ECM and cells of the cranial mesenchyme form a porous meshwork made of intermingling ECM strands and stellate shaped cells that support the elevating neural folds (Morris-Wiman and Brinkley, 1990a; Morriss and Solursh, 1978a; Morriss and Solursh, 1978b). The ECM underlying the neural folds consists of fibronectin, laminin and is rich in glycosaminoglycans (Morriss and Solursh, 1978a; Morriss-Kay and Tuckett, 1989; Solursh and Morriss, 1977; Tuckett and Morriss-Kay, 1986). Hyaluronate (HA) is the predominant glycosaminoglycan with smaller amounts of chondroitin sulfate and heparin sulfate present (Solursh and Morriss, 1977). Changes in laminin and fibronectin concentrations in the ECM are not correlated with neural fold elevation and expansion of the cranial mesenchyme (Tuckett and Morriss-Kay, 1986); however, dynamic changes in HA concentrations are associated with these morphogenetic events (Morris-Wiman and Brinkley, 1990a; Morriss and Solursh, 1978a; Morriss and Solursh, 1978b). As described by Morris-Wiman and Brinkley (1990a) and illustrated in Figure 1, in the cranial mesenchyme of 3-5 somite stages HA is evenly distributed with slightly higher concentrations in the medial region. By 7/8 somite stages, when the neural folds begin to elevate, HA distribution increases underlying the neural folds with a decreasing medial to lateral gradient. By 11 somites, as the neural folds form a “V” shape, the concentration of HA increases further. At 12-13 somite stages, HA levels increase in medial and lateral regions but decrease in the intervening region as cells loose their orientation parallel to the neuroepithelium. In subsequent stages, relative HA concentrations are greater beneath the lateral neural folds. Regional differences in HA concentration during neural fold elevation are not due to differential synthesis, which is uniform across the cranial mesenchyme (Morris-Wiman and Brinkley, 1990c). Instead, the reduced concentration in medial and central regions is likely due to expansion of the HA rich ECM (Morris-Wiman and Brinkley, 1990c). This expansion would result in the decreased cell density and the decreased concentration of HA observed in this region.
These dynamic changes in HA concentration are very interesting with respect to expansion of the cranial mesenchyme. HA-rich matrix can become heavily hydrated causing swelling of the ECM and expansion of the spaces between cells (Spicer and Tien, 2004; Toole, 2004). In addition, HA interacts with the cell surface receptors Cd44 and RHAMM (Receptor for Hyaluronan-Mediated Motility) which mediate signal transduction by HA (Toole, 2004). Expansion of HA-rich ECM provides the driving force for morphogenesis of other structures including morphogenesis of the palate, endocardial cushion and chondrogenesis (Spicer and Tien, 2004). Thus, nonrandom distribution of HA and unequal expansion of the cranial mesenchyme could potentially drive neural fold elevation as proposed by Morris-Wiman and Brinkley (1990a). According to the model proposed in their paper, the polarization of cranial mesenchyme underlying the neuroepithelium is coincident with the lateral expansion of the neuroepithelium. This expansion disrupts cell interactions that restrain swelling of the cranial mesenchyme in central regions, allowing the HA-rich ECM to expand. Since at this stage HA is most highly concentrated in the central cranial mesenchyme, this region will expand the most. The direction of expansion is restricted by the neural groove and notochord medially and the surface ectoderm laterally. These restraints combined with differential expansion of the cranial mesenchyme in central regions may drive neural fold elevation. Subsequently, the uneven expansion of the HA-rich ECM results in decreased concentration of HA in the central region. As the neural groove and notochord become separated, dorsal-ventral cranial mesenchyme expansion is allowed resulting in tremendous expansion of the cranial mesenchyme in this direction.
Essential role of the expansion of the cranial mesenchyme in neural fold elevation
The correlative data described above implicates expansion of the cranial mesenchyme in neural fold elevation. This is further supported by inhibitor studies where embryos are cultured in the presence of ECM inhibitors during neurulation. For example, culture of rat embryos in Streptomyces hyaluronidase, an enzyme that preferentially degrades HA, resulted in delayed elevation of the neural folds (Morriss-Kay et al., 1986). Histological analyses of these embryos revealed increased density of cranial mesenchyme. However, cell proliferation and consequently cell number was also greatly affected in the cranial mesenchyme. Hyaluronidase also inhibits neurulation in chicken embryos (Schoenwolf and Fisher, 1983). Further evidence that HA is essential for neural fold elevation comes from experiments where rat embryos were cultured with the HA synthesis inhibitor DON (Morris-Wiman and Brinkley, 1990a; Morris-Wiman and Brinkley, 1990b; Morris-Wiman and Brinkley, 1990c). Similar to the hyaluronidase experiments, DON treatment resulted in increased density of the cranial mesenchyme; however, unlike with hyaluronidase, no change in mitosis was observed. In DON treated embryos the neural folds fail to transition from the biconvex to “V” shape and cranial mesenchyme cells fail to orientate parallel to the neuroepithelium. Treatment of embryos during neurulation with other ECM disrupting compounds including chondroitinase ABC, bD-xyloside and heparitinase inhibits neural tube closure and affects morphogenesis of the cranial mesenchyme (Morriss-Kay and Tuckett, 1989; Morriss-Kay and Crutch, 1982; Tuckett and Morriss-Kay, 1989). One important caveat of these experiments is that these inhibitors have access to all tissues and in most of these experiments the morphology of neural and surface ectoderm was also affected. Thus whether morphogenesis of the cranial mesenchyme alone is responsible for neural fold elevation still remains to be definitively demonstrated.
Excess retinoic acid also affects cranial mesenchyme organization and neural fold elevation
The teratogenic effect of retinoic acid on closure of the neural tube has been documented as early as the 1950s (Cohlan, 1953). In the proceeding decades, the observation that failure of neural fold elevation in retinoic acid exposed embryos is associated with reduced cranial mesenchyme and increased extracellular space was made (see (Marin-Padilla, 1966; Morriss, 1972; Morriss and Steele, 1974) for examples). These studies noted that both PM-CM and NC-CM populations are affected by excess retinoic acid exposure during neurulation. Migration of PM-CM cells to positions underlying the cranial neural tube following gastrulation is reduced in retinoic acid exposed embryos (Morriss, 1972; Morriss and Steele, 1974; Morriss and Steele, 1977). Similarly, migration of NC-CM is affected with many NC-CM cells failing to undergo the epithelial to mesenchyme transition and delaminate from the dorsal neural tube (Geelen, 1979; Moro Balbas et al., 1993; Pratt et al., 1987; Shankar et al., 1994; Thorogood et al., 1982; Webster et al., 1986).
Since these initial observations, the molecular mechanism of retinoic acid action during embryogenesis has been elucidated. In a simplistic model during embryogenesis, the bioavailability of retinoic acid is tightly controlled by the precise temporal and spatial regulated expression of the retinoic acid activating and deactivating enzymes retinaldehyde dehydrogenase (Raldh) and cytochrome P450 (Cyp26s), respectively (Rhinn and Dolle, 2012). At the start of neurulation, the PM-CM underlying the anterior portion of the cranial neural tube expresses high levels of Cyp26c1 and little Raldh2, resulting in the PM-CM being a relatively retinoic acid free region of the embryo (Bothe et al., 2011). Thus, exposure of these cells to retinoic acid results in inappropriate expression of retinoic acid-responsive genes. Retinoic acid binds to retinoic acid receptors (RARa, b, g and RXRa, b, g) which heterodimerize and bind to retinoic acid responsive elements (RAREs; (Rhinn and Dolle, 2012). Retinoic acid both positively and negatively regulates the expression of a number of genes including enzymes that synthesize ECM components and homeobox genes (Ackermans et al., 2011; Rhinn and Dolle, 2012).
The regulation of ECM composition by retinoic acid is very interesting in light of the fact that the ECM plays an essential role in neural fold elevation. The composition of the ECM changes in the cranial mesenchyme following retinoic acid exposure (see (Moro Balbas et al., 1993; Shankar et al., 1994) for examples). This could be due to direct induction of HA synthesis enzymes in response to retinoic acid exposure. While not yet demonstrated in the cranial mesenchyme, retinoic acid has been shown to increase HA levels in a number of cell types (Akiyama et al., 1994; King, 1984; Margelin et al., 1996; Tammi and Tammi, 1986). Upregulation of HA may occur by increased expression of the HA synthesis enzyme Has2 which contains RAREs in its promoter (Saavalainen et al., 2005). In addition to Has2, retinoic acid increases expression of the HA receptor Cd44 (Pasonen-Seppanen et al., 2008; Rousche and Knudson, 2002). Thus retinoic acid exposure can potentially alter the ECM composition of the cranial mesenchyme and the expression of ECM receptors, which could alter NC-CM migration and morphogenesis of the PM-CM contributing to failure of neural fold elevation. Whether this sequence of events occurs in retinoic acid exposed cranial mesenchyme during neurulation and if these changes result in failure of neural tube closure remains to be determined.
Exencephaly in Twist mutant mice is associated with reduced cranial mesenchyme
Abnormal cranial mesenchyme morphogenesis has been suggested to underlie failure of cranial neurulation in a number of mouse mutants including Ski, Cart1/Alx1, Alx3, Inka1 and Twist (Berk et al., 1997; Chen and Behringer, 1995; Lakhwani et al., 2010; Reid et al., 2010; Zhao et al., 1996). Of these, the Twist mutant mouse model has been best studied. Embryos with homozygous mutation in the basic helix-loop-helix transcription factor Twist exhibit exencephaly associated with defects in development of the cranial mesenchyme (Chen and Behringer, 1995). Though not examined during neural fold elevation, immediately following neural tube closure, Twist mutant cranial mesenchyme cells do not exhibit the typical stellate morphology. Rather mutant cells underlying the forebrain (NC-CM) and midbrain (PM-CM) are rounded with altered cellular contacts and expanded extracellular space (Chen and Behringer, 1995). Twist is expressed in both the PM-CM and NC-CM lineages and expression is not detected in other tissues involved in cranial neurulation such as the neural and surface ectoderm (Fuchtbauer, 1995; Stoetzel et al., 1995). Thus the observation that in Twist mutants the neural folds fail to elevate provides the best evidence in support of the hypothesis that morphogenesis of the cranial mesenchyme drives neural fold elevation. While the role of Twist in cranial mesenchyme morphogenesis is clear, it remains unknown if the PM-CM or NC-CM or both is essential for cranial neural fold elevation.
The role of Twist in development of both the PM-CM and NC-CM has been extensively studied. The neural crest appears to form properly in Twist mutants and they migrate to the facial primordia albeit with reduced numbers (Soo et al., 2002). Furthermore, in Twist mutants the neural crest cells migrate to positions deeper in the PM-CM (Soo et al., 2002). Interestingly, cell transplantation experiments indicate that Twist is required both cell autonomously in the NC-CM and nonautonomously in the PM-CM for guidance of neural crest (Soo et al., 2002). In addition, Twist is required for the expression of a number of genes including Alx1/Cart1 and Alx3 in the NC-CM (Soo et al., 2002). Interestingly, the expression of Alx1 and Alx3 is restricted to the NC-CM during neural fold elevation (Beverdam and Meijlink, 2001) and null mutations in these result in exencephaly with reduced NC-CM density around the forebrain primordium and elevated apopotosis, very similar to that observed in Twist mutants (Lakhwani et al., 2010; Zhao et al., 1996). In particular, the neural crest undergoes increased apoptosis in Alx1 and Alx3 mutants (Lakhwani et al., 2010; Zhao et al., 1996). Thus, neural tube defects in Twist mutants could potentially be explained by a cell autonomous role of Twist in the neural crest affecting expression of Alx1 and Alx3 in the NC-CM. Surprisingly, however, conditional inactivation of Twist in the neural crest lineage using the Wnt1-Cre driver does not result in neural tube closure defects (Bildsoe et al., 2009). This result argues for an essential role for Twist in the PM-CM as suggested by the cell transplantation experiments (Soo et al., 2002). Importantly, in the Wnt1-Cre;Twistflox conditionally deleted embryos, Twist expression is absent in the NC-CM assessed at later stages of development demonstrating significant recombination of the floxed allele in the neural crest lineage (Bildsoe et al., 2009). However, since expression of Twist protein was not examined during neural fold elevation, it is possible that perdurance of the Twist transcript or protein in the NC-CM lineage allows for normal function during this time. Determination of Twist protein expression during neural fold elevation in these conditional knockout embryos would resolve this issue. The reciprocal experiment where Twist is conditionally deleted in the PM-CM using the Mesp1-cre line would further address the important domain of Twist expression required for neural tube closure. Finally, whether the composition of the ECM in the cranial mesenchyme of Twist mutants during neural fold elevation is altered or defects are primarily due to enhance apoptosis remains to be determined.
Failure of cranial mesenchyme expansion is associated with exencephaly in Hectd1 mutant mice
Our laboratory has been investigating abnormal cranial mesenchyme morphogenesis as a potential mechanism underlying cranial neural tube defects in the openmind (opm) mutant mouse line. This mouse line was created in an ENU mutagenesis screen to identify genes required for neurulation (Kasarskis et al., 1998; Zohn et al., 2005). Positional cloning revealed that the opm mutation is in the uncharacterized HECT (Homologous to the E6-AP Carboxyl Terminus) domain ubiquitin ligase Hectd1 (HECT domain containing 1; (Zohn et al., 2007). Hectd1 is widely expressed in the neural and surface epithelium and the PM-CM and NC-CM during neurulation (Zohn et al., 2007). The neural folds fail to elevate in Hectd1opm mutants and the folds maintain the biconvex morphology characteristic of early somite stage embryos (Figure 2 and (Sarkar and Zohn, 2012; Zohn et al., 2007). Histological analyses of the cranial mesenchyme during the time when the neural folds should have elevated reveals that the cranial mesenchyme cells underlying the neural tube fail to undergo the characteristic expansion (Sarkar and Zohn, 2012; Zohn et al., 2007). These differences in cell density are not likely due to altered apoptosis which remains essentially the same in the cranial mesenchyme of wildtype and Hectd1opm mutant embryos (Zohn et al., 2007). Furthermore, proliferation as measured by phosphorylated histone H3 immunostaining was not changed between wildtype and mutants (Zohn et al., 2007). In addition to the failure to expand the space between cranial mesenchyme cells in Hectd1opm mutants, the organization of the cranial mesenchyme is altered. Specifically, the cells underlying the neural epithelium fail to orientate parallel to the neural epithelium (Figure 1), a cell behavior correlated with neural fold elevation (Morris-Wiman and Brinkley, 1990a; Sarkar and Zohn, 2012).
Figure 2. Failure of neural fold elevation in Hectd1opm mutant embryos is correlated with disorganized cranial mesenchyme.
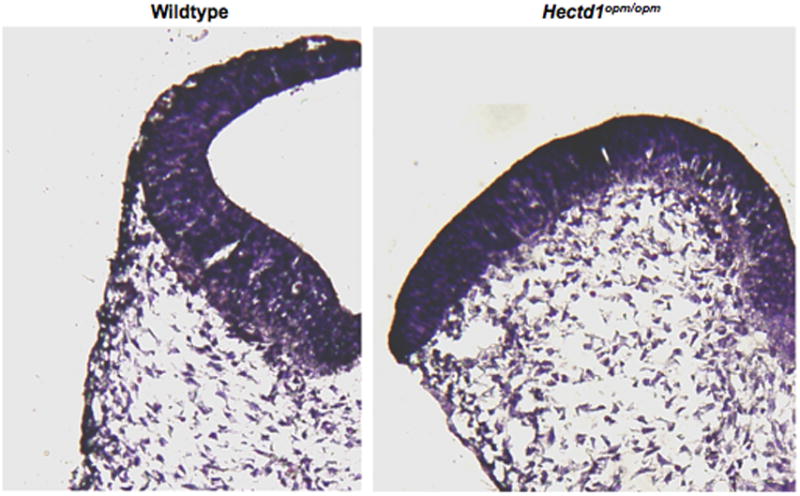
The neural plate in E9.5 Hectd1opm mutant embryos does not transform from the biconvex to “V” then “C” shape demonstrating failure of neural fold elevation. The PM-CM in Hectd1opm mutant embryos fails to undergo the characteristic expansion seen in wildtype embryos and the PM-CM cells underlying the neural plate do not orientate parallel to the neuroepithelium.
We have further characterized the behavior of Hectd1opm mutant cranial mesenchyme cells in an artificial explant assay where cranial mesenchyme explants dissected at E8.5 (during neural fold elevation) are plated on an ECM-coated plate (Sarkar and Zohn, 2012). Cells will exit and migrate from the explant. Importantly, more Hectd1opm mutant cells will exit the explant than from wildtype explants and these mutant cells will migrate further. This experiment indicates that the behavior of Hectd1opm mutant cranial mesenchyme cells is different than wildtype cells. Along with our histological data, this finding provides a potential cellular basis for the abnormal organization of the cranial mesenchyme and possibly failure of cranial neurulation in Hectd1opm mutants.
Hectd1 encodes an E3 ubiquitin ligase and our data indicate that the ubiquitin ligase activity of Hectd1 is essential for its biological function (Sarkar and Zohn, 2012; Zohn et al., 2007). Ubiquitin ligases catalyze the addition of ubiquitin to target proteins (Mukhopadhyay and Riezman, 2007; Pickart, 2001). Ubiquitination of proteins results in a myriad of consequences depending on whether ubiquitin is added singly (monoubiquitination) or in chains (polyubiquitination). To create polyubiquitin chains, additional ubiquitins are added to one of the multiple Lysine residues on the ubiquitin protein. If ubiquitin chains are made utilizing Lysine-48 linkages, the substrate protein is typically targeted to the proteasome for degradation; however, if Lysine-63 linkages are used or the substrate is monoubiquitinated then the modification typically results in altered localization or activity of the substrate (Mukhopadhyay and Riezman, 2007). Our data indicate that Hectd1 primarily catalyzes the addition of Lysine-63 linked polyubiquitin chains to its substrates (Sarkar and Zohn, 2012). While other ubiquitin ligases including Mdm4, Mib2 and Smurf1;Smurf2 double knockouts have been shown to be regulate neurulation, none of these have been implicated in cranial mesenchyme morphogenesis (Migliorini et al., 2002; Narimatsu et al., 2009; Wu et al., 2007).
To understand the pathways regulated by Hectd1 during neurulation we performed proteomic screens to identify the substrates of Hectd1 (Sarkar and Zohn, 2012). One interesting substrate identified from this approach is Heat shock protein 90 (Hsp90). Heat shock proteins are cellular chaperones that interact with client proteins, promoting their active confirmations (Taipale et al., 2010). Hsp90 has a number of clients that regulate a multitude of processes including cell migration (Tsutsumi and Neckers, 2007). In addition, Hsp90 is secreted from the cell where it can act upon clients on the cell surface and in the ECM to stimulate migration (Tsutsumi and Neckers, 2007). Our data indicate that the addition of Lysine-63 linked polyubiquitin chains onto Hsp90 does not target it to the proteasome, but rather traffics it away from the secretory pathway (Sarkar and Zohn, 2012). Thus in the absence of Hectd1 activity in Hectd1opm mutants, Hsp90 secretion is increased. Utilizing the cranial mesenchyme explant assay described above, we demonstrated that the increased migration of cranial mesenchyme cells from mutant explants is due to the increased Hsp90 secreted from these cells. These results provide a potential molecular mechanism for the abnormal behavior of the cranial mesenchyme during neural fold elevation. Interestingly, in tumor cells, Hsp90 plays an essential role in interaction of HA with its receptor Cd44 and activation of extracellular matrix metalloproteinases (MMPs) to promote invasion (Ghatak et al., 2005; Kim et al., 2008; Lagarrigue et al., 2010). Thus it is possible that the enhanced extracellular Hsp90 in Hectd1opm mutant cranial mesenchyme may be acting on the HA-rich ECM to promote disorganized migration.
Summary and Conclusions
The cranial mesenchyme undergoes stereotypical morphogenesis during neural fold elevation. This morphogenesis is associated with alterations in the HA-rich ECM. Based on experiments where embryos were cultured with HA and other ECM disrupting agents, interactions of the cells of the cranial mesenchyme and the HA-rich ECM have been implicated in driving cranial mesenchyme morphogenesis and neural fold elevation. However, these experiments are flawed as all tissues were exposed to the inhibitors and their morphogenesis was similarly affected. Here we reviewed further evidence that the cranial mesenchyme is important for neural fold elevation. In retinoic acid treated embryos and some genetic mutants, defects in cranial mesenchyme morphogenesis are associated with failure of neural tube closure. Exencephaly in the Twist mutant mouse illustrates that it is indeed the cranial mesenchyme that is essential for neural fold elevation since Twist is not expressed in the neural or surface ectoderm. Future studies, will determine whether morphogenesis of the PM-CM, the NC-CM or both are important for driving neural fold elevation. Interestingly, both retinoic acid and Hectd1 may influence interactions between cranial mesenchyme cells and the HA matrix. Additional experiments will determine whether these interactions are affected in the cranial mesenchyme resulting in failure of neural fold elevation in these mouse models. Finally, the careful analyses of cranial mesenchyme behaviors in additional mouse mutants will further elucidate the molecular pathways that control cranial mesenchyme morphogenesis. Importantly, these will provide added evidence as to whether morphogenesis of the CM in fact provides a driving force for neural fold elevation in the cranial region.
Acknowledgments
This work was funded by grant NICHD-R01-HD058629 (from the National Institute of Child Health and Human Development) to I.E. Zohn.
Footnotes
Presented at the 7th International Conference on Neural Tube Defects, Austin TX, November 2011.
References
- Ackermans MM, et al. Vitamin A and clefting: putative biological mechanisms. Nutr Rev. 2011;69:613–24. doi: 10.1111/j.1753-4887.2011.00425.x. [DOI] [PubMed] [Google Scholar]
- Akiyama H, et al. Analytical studies on hyaluronic acid synthesis by normal human epidermal keratinocytes cultured in a serum-free medium. Biol Pharm Bull. 1994;17:361–4. doi: 10.1248/bpb.17.361. [DOI] [PubMed] [Google Scholar]
- Berk M, et al. Mice lacking the ski proto-oncogene have defects in neurulation, craniofacial, patterning, and skeletal muscle development. Genes Dev. 1997;11:2029–39. doi: 10.1101/gad.11.16.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beverdam A, Meijlink F. Expression patterns of group-I aristaless-related genes during craniofacial and limb development. Mech Dev. 2001;107:163–7. doi: 10.1016/s0925-4773(01)00450-6. [DOI] [PubMed] [Google Scholar]
- Bildsoe H, et al. Requirement for Twist1 in frontonasal and skull vault development in the mouse embryo. Dev Biol. 2009;331:176–88. doi: 10.1016/j.ydbio.2009.04.034. [DOI] [PubMed] [Google Scholar]
- Bothe I, et al. Dynamic control of head mesoderm patterning. Development. 2011;138:2807–21. doi: 10.1242/dev.062737. [DOI] [PubMed] [Google Scholar]
- Chen ZF, Behringer RR. Twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686–99. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Cohlan SQ. Excessive intake of vitamin A as a cause of congenital anomalies in the rat. Science. 1953;117:535–6. doi: 10.1126/science.117.3046.535. [DOI] [PubMed] [Google Scholar]
- Colas JF, Schoenwolf GC. Towards a cellular and molecular understanding of neurulation. Dev Dyn. 2001;221:117–45. doi: 10.1002/dvdy.1144. [DOI] [PubMed] [Google Scholar]
- Copp AJ. Neurulation in the cranial region--normal and abnormal. J Anat. 2005;207:623–35. doi: 10.1111/j.1469-7580.2005.00476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, Greene ND. Genetics and development of neural tube defects. J Pathol. 2010;220:217–30. doi: 10.1002/path.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copp AJ, et al. The genetic basis of mammalian neurulation. Nat Rev Genet. 2003;4:784–93. doi: 10.1038/nrg1181. [DOI] [PubMed] [Google Scholar]
- Fleming A, et al. Mechanisms of normal and abnormal neurulation: evidence from embryo culture studies. Int J Dev Biol. 1997;41:199–212. [PubMed] [Google Scholar]
- Fuchtbauer EM. Expression of M-twist during postimplantation development of the mouse. Dev Dyn. 1995;204:316–22. doi: 10.1002/aja.1002040309. [DOI] [PubMed] [Google Scholar]
- Geelen JA. Hypervitaminosis A induced teratogenesis. CRC Crit Rev Toxicol. 1979;6:351–75. doi: 10.3109/10408447909043651. [DOI] [PubMed] [Google Scholar]
- Ghatak S, et al. Hyaluronan constitutively regulates ErbB2 phosphorylation and signaling complex formation in carcinoma cells. J Biol Chem. 2005;280:8875–83. doi: 10.1074/jbc.M410882200. [DOI] [PubMed] [Google Scholar]
- Greene ND, Copp AJ. Development of the vertebrate central nervous system: formation of the neural tube. Prenat Diagn. 2009;29:303–11. doi: 10.1002/pd.2206. [DOI] [PubMed] [Google Scholar]
- Gu X, et al. High prevalence of NTDs in Shanxi Province: a combined epidemiological approach. Birth Defects Res A Clin Mol Teratol. 2007;79:702–7. doi: 10.1002/bdra.20397. [DOI] [PubMed] [Google Scholar]
- Jiang X, et al. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–16. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Kasarskis A, et al. A phenotype-based screen for embryonic lethal mutations in the mouse. Proc Natl Acad Sci U S A. 1998;95:7485–90. doi: 10.1073/pnas.95.13.7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MS, et al. 17-Allylamino-17-demethoxygeldanamycin down-regulates hyaluronic acid-induced glioma invasion by blocking matrix metalloproteinase-9 secretion. Mol Cancer Res. 2008;6:1657–65. doi: 10.1158/1541-7786.MCR-08-0034. [DOI] [PubMed] [Google Scholar]
- King IA. Increased epidermal hyaluronic acid synthesis caused by four retinoids. Br J Dermatol. 1984;110:607–8. doi: 10.1111/j.1365-2133.1984.tb04685.x. [DOI] [PubMed] [Google Scholar]
- Lagarrigue F, et al. Matrix metalloproteinase-9 is upregulated in nucleophosmin-anaplastic lymphoma kinase-positive anaplastic lymphomas and activated at the cell surface by the chaperone heat shock protein 90 to promote cell invasion. Cancer Res. 2010;70:6978–87. doi: 10.1158/0008-5472.CAN-10-0861. [DOI] [PubMed] [Google Scholar]
- Lakhwani S, et al. Alx3-deficient mice exhibit folic acid-resistant craniofacial midline and neural tube closure defects. Dev Biol. 2010;344:869–80. doi: 10.1016/j.ydbio.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Margelin D, et al. Hyaluronic acid and dermatan sulfate are selectively stimulated by retinoic acid in irradiated and nonirradiated hairless mouse skin. J Invest Dermatol. 1996;106:505–9. doi: 10.1111/1523-1747.ep12343819. [DOI] [PubMed] [Google Scholar]
- Marin-Padilla M. Mesodermal alterations induced by hypervitaminosis A. J Embryol Exp Morphol. 1966;15:261–9. [PubMed] [Google Scholar]
- Migliorini D, et al. Mdm4 (Mdmx) regulates p53-induced growth arrest and neuronal cell death during early embryonic mouse development. Mol Cell Biol. 2002;22:5527–38. doi: 10.1128/MCB.22.15.5527-5538.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moro Balbas JA, et al. Retinoic acid induces changes in the rhombencephalic neural crest cells migration and extracellular matrix composition in chick embryos. Teratology. 1993;48:197–206. doi: 10.1002/tera.1420480303. [DOI] [PubMed] [Google Scholar]
- Morris-Wiman J, Brinkley LL. Changes in mesenchymal cell and hyaluronate distribution correlate with in vivo elevation of the mouse mesencephalic neural folds. Anat Rec. 1990a;226:383–95. doi: 10.1002/ar.1092260316. [DOI] [PubMed] [Google Scholar]
- Morris-Wiman J, Brinkley LL. The role of the mesenchyme in mouse neural fold elevation. I. Patterns of mesenchymal cell distribution and proliferation in embryos developing in vitro. Am J Anat. 1990b;188:121–32. doi: 10.1002/aja.1001880203. [DOI] [PubMed] [Google Scholar]
- Morris-Wiman J, Brinkley LL. The role of the mesenchyme in mouse neural fold elevation. II. Patterns of hyaluronate synthesis and distribution in embryos developing in vitro. Am J Anat. 1990c;188:133–47. doi: 10.1002/aja.1001880204. [DOI] [PubMed] [Google Scholar]
- Morriss GM. Morphogenesis of the malformations induced in rat embryos by maternal hypervitaminosis. A J Anat. 1972;113:241–50. [PMC free article] [PubMed] [Google Scholar]
- Morriss GM, Solursh M. Regional differences in mesenchymal cell morphology and glycosaminoglycans in early neural-fold stage rat embryos. J Embryol Exp Morphol. 1978a;46:37–52. [PubMed] [Google Scholar]
- Morriss GM, Solursh M. The role of primary mesenchyme in normal and abnormal morphogenesis of mammalian neural folds. Zoon. 1978b;6:33–38. [Google Scholar]
- Morriss GM, Steele CE. The effect of excess vitamin A on the development of rat embryos in culture. J Embryol Exp Morphol. 1974;32:505–14. [PubMed] [Google Scholar]
- Morriss GM, Steele CE. Comparison of the effects of retinol and retinoic acid on postimplantation rat embryos in vitro. Teratology. 1977;15:109–19. doi: 10.1002/tera.1420150115. [DOI] [PubMed] [Google Scholar]
- Morriss-Kay G, Tuckett F. Immunohistochemical localisation of chondroitin sulphate proteoglycans and the effects of chondroitinase ABC in 9- to 11-day rat embryos. Development. 1989;106:787–98. doi: 10.1242/dev.106.4.787. [DOI] [PubMed] [Google Scholar]
- Morriss-Kay GM. Growth and development of pattern in the cranial neural epithelium of rat embryos during neurulation. J Embryol Exp Morphol. 1981;(65):225–41. [PubMed] [Google Scholar]
- Morriss-Kay GM, Crutch B. Culture of rat embryos with beta-D-xyloside: evidence of a role for proteoglycans in neurulation. J Anat. 1982;134:491–506. [PMC free article] [PubMed] [Google Scholar]
- Morriss-Kay GM, et al. The effects of Streptomyces hyaluronidase on tissue organization and cell cycle time in rat embryos. J Embryol Exp Morphol. 1986;98:59–70. [PubMed] [Google Scholar]
- Mukhopadhyay D, Riezman H. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science. 2007;315:201–5. doi: 10.1126/science.1127085. [DOI] [PubMed] [Google Scholar]
- Narimatsu M, et al. Regulation of planar cell polarity by Smurf ubiquitin ligases. Cell. 2009;137:295–307. doi: 10.1016/j.cell.2009.02.025. [DOI] [PubMed] [Google Scholar]
- Noden DM, Trainor PA. Relations and interactions between cranial mesoderm and neural crest populations. J Anat. 2005;207:575–601. doi: 10.1111/j.1469-7580.2005.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker SE, et al. Updated National Birth Prevalence estimates for selected birth defects in the United States, 2004-2006. Birth Defects Res A Clin Mol Teratol. 2010;88:1008–16. doi: 10.1002/bdra.20735. [DOI] [PubMed] [Google Scholar]
- Pasonen-Seppanen SM, et al. All-trans retinoic acid-induced hyaluronan production and hyperplasia are partly mediated by EGFR signaling in epidermal keratinocytes. J Invest Dermatol. 2008;128:797–807. doi: 10.1038/sj.jid.5701098. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–33. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Pratt RM, et al. Retinoic acid inhibits migration of cranial neural crest cells in the cultured mouse embryo. J Craniofac Genet Dev Biol. 1987;7:205–17. [PubMed] [Google Scholar]
- Reid BS, et al. Generation and characterization of a novel neural crest marker allele, Inka1-LacZ, reveals a role for Inka1 in mouse neural tube closure. Dev Dyn. 2010;239:1188–96. doi: 10.1002/dvdy.22248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M, Dolle P. Retinoic acid signalling during development. Development. 2012;139:843–58. doi: 10.1242/dev.065938. [DOI] [PubMed] [Google Scholar]
- Rousche KT, Knudson CB. Temporal expression of CD44 during embryonic chick limb development and modulation of its expression with retinoic acid. Matrix Biol. 2002;21:53–62. doi: 10.1016/s0945-053x(01)00189-5. [DOI] [PubMed] [Google Scholar]
- Saavalainen K, et al. The human hyaluronan synthase 2 gene is a primary retinoic acid and epidermal growth factor responding gene. J Biol Chem. 2005;280:14636–44. doi: 10.1074/jbc.M500206200. [DOI] [PubMed] [Google Scholar]
- Sarkar AA, Zohn IE. Hectd1 regulates intracellular localization and secretion of Hsp90 to control cellular behavior of the cranial mesenchyme. J Cell Biol. 2012;196:789–800. doi: 10.1083/jcb.201105101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenwolf GC, Fisher M. Analysis of the effects of Streptomyces hyaluronidase on formation of the neural tube. J Embryol Exp Morphol. 1983;73:1–15. [PubMed] [Google Scholar]
- Shankar KR, et al. Effect of in ovo retinoic acid exposure on forebrain neural crest: in vitro analysis reveals up-regulation of N-CAM and loss of mesenchymal phenotype. Dev Dyn. 1994;200:89–102. doi: 10.1002/aja.1002000202. [DOI] [PubMed] [Google Scholar]
- Solursh M, Morriss GM. Glycosaminoglycan synthesis in rat embryos during the formation of the primary mesenchyme and neural folds. Dev Biol. 1977;57:75–86. doi: 10.1016/0012-1606(77)90355-4. [DOI] [PubMed] [Google Scholar]
- Soo K, et al. Twist function is required for the morphogenesis of the cephalic neural tube and the differentiation of the cranial neural crest cells in the mouse embryo. Dev Biol. 2002;247:251–70. doi: 10.1006/dbio.2002.0699. [DOI] [PubMed] [Google Scholar]
- Spicer AP, Tien JY. Hyaluronan and morphogenesis. Birth Defects Res C Embryo Today. 2004;72:89–108. doi: 10.1002/bdrc.20006. [DOI] [PubMed] [Google Scholar]
- Stoetzel C, et al. Dorso-ventral and rostro-caudal sequential expression of M-twist in the postimplantation murine embryo. Mech Dev. 1995;51:251–63. doi: 10.1016/0925-4773(95)00369-x. [DOI] [PubMed] [Google Scholar]
- Taipale M, et al. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol. 2010;11:515–28. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- Tammi R, Tammi M. Influence of retinoic acid on the ultrastructure and hyaluronic acid synthesis of adult human epidermis in whole skin organ culture. J Cell Physiol. 1986;126:389–98. doi: 10.1002/jcp.1041260309. [DOI] [PubMed] [Google Scholar]
- Thorogood P, et al. Effects of vitamin A on the behaviour of migratory neural crest cells in vitro. J Cell Sci. 1982;57:331–50. doi: 10.1242/jcs.57.1.331. [DOI] [PubMed] [Google Scholar]
- Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–39. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- Tsutsumi S, Neckers L. Extracellular heat shock protein 90: a role for a molecular chaperone in cell motility and cancer metastasis. Cancer Sci. 2007;98:1536–9. doi: 10.1111/j.1349-7006.2007.00561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuckett F, Morriss-Kay GM. The distribution of fibronectin, laminin and entactin in the neurulating rat embryo studied by indirect immunofluorescence. J Embryol Exp Morphol. 1986;94:95–112. [PubMed] [Google Scholar]
- Tuckett F, Morriss-Kay GM. Heparitinase treatment of rat embryos during cranial neurulation. Anat Embryol (Berl) 1989;180:393–400. doi: 10.1007/BF00311170. [DOI] [PubMed] [Google Scholar]
- Webster WS, et al. Isotretinoin embryopathy and the cranial neural crest: an in vivo and in vitro study. J Craniofac Genet Dev Biol. 1986;6:211–22. [PubMed] [Google Scholar]
- Wu JI, et al. Targeted disruption of Mib2 causes exencephaly with a variable penetrance. Genesis. 2007;45:722–7. doi: 10.1002/dvg.20349. [DOI] [PubMed] [Google Scholar]
- Yoshida T, et al. Cell lineage in mammalian craniofacial mesenchyme. Mech Dev. 2008;125:797–808. doi: 10.1016/j.mod.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Zhao Q, et al. Prenatal folic acid treatment suppresses acrania and meroanencephaly in mice mutant for the Cart1 homeobox gene. Nat Genet. 1996;13:275–83. doi: 10.1038/ng0796-275. [DOI] [PubMed] [Google Scholar]
- Zohn IE. Mouse as a model for multifactorial inheritance of neural tube defects. Birth Defects Res C Embryo Today. 2012;96:193–205. doi: 10.1002/bdrc.21011. [DOI] [PubMed] [Google Scholar]
- Zohn IE, et al. Using genomewide mutagenesis screens to identify the genes required for neural tube closure in the mouse. Birth Defects Res A Clin Mol Teratol. 2005;73:583–90. doi: 10.1002/bdra.20164. [DOI] [PubMed] [Google Scholar]
- Zohn IE, et al. The Hectd1 ubiquitin ligase is required for development of the head mesenchyme and neural tube closure. Dev Biol. 2007;306:208–21. doi: 10.1016/j.ydbio.2007.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohn IE, Sarkar AA. Modeling neural tube defects in the mouse. Curr Top Dev Biol. 2008;84:1–35. doi: 10.1016/S0070-2153(08)00601-7. [DOI] [PubMed] [Google Scholar]
- Zohn IE, Sarkar AA. The visceral yolk sac endoderm provides for absorption of nutrients to the embryo during neurulation. Birth Defects Res A Clin Mol Teratol. 2010;88:593–600. doi: 10.1002/bdra.20705. [DOI] [PubMed] [Google Scholar]