

Abstract
Genetically encoded phosphoserine incorporation programmed by the UAG codon was achieved by addition of engineered elongation factor and an archaeal aminoacyl-tRNA synthetase to the normal Escherichia coli translation machinery (Park (2011) Science 333, 1151). However, protein yield suffers from expression of the orthogonal phosphoserine translation system and competition with release factor 1 (RF-1). In a strain lacking RF-1, phosphoserine phosphatase, and where 7 UAG codons residing in essential genes were converted to UAA, phosphoserine incorporation into GFP and WNK4 was significantly elevated, but with an accompanying loss in cellular fitness and viability.
Keywords: Synthetic Biology, Phosphoserine, Phosphoproteomics, Genetic Code, Genetic Code expansion, Genome engineering
1. Introduction
Phosphorylation at serine residues is the most abundant phosphorylation event in eukaryotic signaling pathways. Given that these signaling networks form the basis for regulating most physiological processes, there exists a continued scientific interest in resolving the nature of these phosphorylation events. Chemical modification of proteins has enabled the study of functionally relevant phosphorylation sites within proteins but the use of this method is limted [1]. Recently, a new method for the study of phosphorylation via translational insertion of O-phosphoserine (Sep) has been developed. This SEP-system employs UAG codon (amber) suppression, an orthogonal aminoacyl tRNA synthetase/tRNA pair (SepRS/tRNASep), and an engineered elongation factor (EF-Sep) [2].
The use of UAG as a sense codon for Sep insertion results in low protein yields and truncated protein products due to the primary function of UAG as a stop codon [2]. While natural suppressor tRNAs are capable of efficient amber codon read-through in the presence of release factors [3], orthogonal suppressor systems remain less efficient at this process. Reported protein yields vary from ∼micrograms with Sep-incorporation [2] to ∼milligrams for other systems [4], depending on the employed unnatural amino acid and associated suppressor system. Thus, to achieve high yield production of Sep-proteins with the relatively inefficient SEP-system, release factor competition must be minimized.
The recognition of stop codons by release factors 1 (RF-1, encoded by prfA) and 2 (RF-2, encoded by prfB) leads to peptide chain termination during translation. Amber and ochre (UAG & UAA) stop codons are recognized by RF-1 while ochre and opal (UAA & UGA) stop codons are recognized by RF-2. RF-1 has been thought to be essential [5,6] in Escherichia coli. However, prfA deletion has recently been successful as employed by two very different classes of approaches. Studies have reported that prfA deletion is possible if (I) the genomic background of E. coli is significantly reduced and specific, accommodating mutations are introduced within prfB [7,8] or (II) essential TAG-terminating genes are supplied in trans as TAA-terminating alleles in the presence of an amber suppressor tRNA [9-11]. Thus, a defined set of genetic manipulations in E. coli might afford an efficient system for incorporating phosphoserine at engineered UAG codons.
In this study, we present a strategy for enhanced translational insertion of Sep by the SEP-system. A derivative of the MG1655 E. coli strain harboring TAG-to-TAA genomic recoding at seven essential TAG-terminating genes (hda, lolA, lpxK, coaD, mreC, murF, and hemA) was constructed to permit the deletion of the prfA gene [9]. In this new genetic context, genetically encoded Sep insertion was significantly enhanced by converting the UAG stop codon to a sense codon for phosphoserine.
2. Materials and Methods
2.1. Generation of a partially recoded RF-1 knockout strain
The sequences of oligonucleotides used in this study are listed in the supplementary material (Table S1). The strains in this study were generated from a modified E. coli MG1655 strain (EcNR2: E. coli MG1655 ΔmutS:catΔ(ybhB-bioAB):[lcI857Δ(cro-ea59):tetR-bla]) [12]. The following modifications were made to EcNR2 and are described in greater detail in the supplementary material. The TAG stop codons terminating seven essential genes were recoded to TAA in EcNR2 with mutagenic oligonucleotides by multiplex automated genome engineering (MAGE) [12]. The gene encoding RF-1 (prfA) was replaced by the spectinomycin resistance gene (specR) to generate rEc7ΔprfA. The native tolC gene [13] was deleted to generate rEc7ΔprfAΔtolC and subsequently reintroduced within the bla locus to generate rEc7ΔprfAΔtolC.bla:tolC. A cassette encoding T7 polymerase was integrated within the bla locus, replacing tolC to create rEc7ΔprfAΔtolC.bla:T7. Phosphoserine phosphatase (serB) was disrupted with a premature stop codon [2]. The SEP-system was introduced to generate EcAR7.SEP [2] (Table S2). Alternatively, suppression of TAG codons with glutamine was enabled by transformation of EcAR7 with pGFIB-supE to generate EcAR7.supE.
Growth curves were obtained using a Biotek Synergy HT Plate reader. All strains were grown at 34°C in 150 μL of LB medium supplemented with 2 mM Sep, 50 μL IPTG and antibiotics for plasmid maintenance where indicated. Growth media was inoculated with pre-cultures to an initial A600 of ∼0.1 and A600 was measured at ten-minute intervals for 27 hours. All data were obtained in triplicate and averaged to construct a representative growth curve for each strain. Doubling times were calculated in triplicate and averaged to obtain representative values for each strain.
2.2 Plasmids and strains for incorporation of Sep
Plasmids and methods for Sep incorporation have been described previously [2]. Sep incorporation into recombinant reporter proteins green fluorescent protein (GFP) and mouse serine/threonine protein kinase (WNK4) was monitored in the newly created strain EcAR7, as well as the previously published strains of E. coli BL21 (Table S2) [2].
C-terminally His-tagged GFP under the control the PLtetO promoter was subcloned from pZE21G [14] and blunt-end cloned into pCR®-Blunt II-TOPO (Invitrogen). The kanamycin resistance cassette was deleted from the resulting construct by digestion with BsaI and RsrII followed by blunting of the sticky ends and religation. Codons encoding residues E17, Y66, Q94, E132, E142, Q157, S202, S205, E213 and E222 were mutated to TAG using the QuikChange site-directed mutagenesis kit (Agilent) to yield single or double TAG sites within the protein.
The C-terminal regulatory domain (amino acids 1000-1222) of mouse WNK4 was used to test translational phosphoserine incorporation in a mammalian protein. The vector PCR T7/NT-TOPO was modified by replacing the multicloning site with PLtetO-GFP from pZE21G [14]. An extra copy of the Tet-repressor was also introduced [14,15]. The PLtetO-GFP KpnI and HindIII were used to replace GFP with the WNK4 construct. The corresponding serine codons, S172 and S199, were reassigned to TAG by QuikChange to create coding sequences for mutants containing one or two TAG codons, respectively. Detailed methods can be found in supplementary materials.
3. Results
3.1 Phosphoserine incorporation imposes a severe growth phenotype in wild type and RF-1 deletion strains
In this study, we sought to identify important factors that enable enhanced production of site-specific phosphorylated proteins in engineered strains of E. coli. We hypothesized that several classes of modifications would collectively increase the efficiency of site-specific phosphoserine incorporation in the EcNR2 strain of E. coli [12,16]. First, we performed targeted TAG codon reassignment to the synonymous TAA codon across seven annotated essential genes [9,17]. EcNR2 was used as the parent strain because it allows the use of MAGE for targeted, seamless and efficient genetic modifications directly at each genomic locus (Fig. 1 A and B) [12,16]. Seven codon reassignments were performed at their natural positions of seven essential ORFs (coaD, hda, hemA, mreC, murF, lolA, and lpxK) via MAGE and verified by MASC-PCR as described previously (Fig. 1B) [16]. These seven TAG-to-TAA codon reassignments were introduced to re-direct chain termination of these stop codons from RF-1 to RF-2. Consistent with prior work, we sought to determine if these seven reassigned codons enabled us to delete the prfA gene, which encodes for the release factor 1 protein (RF-1) [9]. Specifically, these modifications allowed for the replacement of prfA by a spectinomycin resistance gene (specR). Successful replacement was demonstrated by PCR with primers binding within and adjacent to the prfA site (Fig. 1C and D). Prior studies have shown that the RF-1 deletion either requires reassignment of at least seven TAG codons to TAA stop codons and [10,11,18] modifications to RF-2 [7,8]. Since mutations to RF-2 could have unexpected secondary effects, such as broadening its ability to include TAG termination, we decided to leave RF-2 unaltered and focused on the recoding of TAG codons to TAA codons in the genome to delete RF-1.
Fig. 1. Construction of EcAR7.

(A) Genomic locations of seven essential genes where the stop codons were recoded from TAG to TAA (green). Additional genomic modifications are shown in red. (B) Verification of TAG-to-TAA recoding at seven essential genes, using multiplex allele specific PCR. (C) Illustration of the genomic context of prfA and its replacement with a gene that confers resistance to spectinomycin. Primers that were used to scrutinize prfA for successful integration of specR are indicated with arrows and color-coded for reference in (D). (D) Replacement of prfA with specR. Three unique primer pairs were used to verify successful specR integration at the prfA locus by PCR. All reactions were performed using a forward primer that anneals upstream of prfA (within hemA), and are distinguished by the use of a unique reverse primer depicted above the image and color-coded for reference in (C).
Next, we introduced a series of genetic and translational modifications (supplementary figures S1-S4) to permit site-specific incorporation of phosphoserine at in-frame TAG codons [2] (Fig. 1A). These modifications included introduction of a nonsense mutation in the phosphoserine phosphatase gene (serB) by a mutagenic oligonucleotide. All genomic alterations were verified by PCR amplification at the genetic locus for each modification and are depicted in Figure 1A. The resulting strain was termed EcAR7 and was transformed with either a plasmid containing a natural suppressor tRNA supE [18] or the phosphoserine incorporation system (SEP-system) [2] to yield EcAR7.supE or EcAR7.SEP, respectively.
To investigate the cellular fitness of the recoded E. coli strains, we obtained growth curves for all of the strains and calculated their respective doubling times (Table 1, Fig. 2). While recoding of seven TAG stop codons to TAA had no effect on the growth phenotype (79 min (EcNR2) and 82 min (rEc7) doubling time), the RF-1 knockout (rEc7.δprfA) strain showed a 1.6 fold increase in doubling time to 134 minutes. All further modifications did not have an impact on the growth phenotype (Fig. 2; Table 1).
Table 1.
Doubling times and maximum optical density of various strains grown in LB supplemented with 2 mM phosphoserine, 50 μM IPTG and required antibiotics. Doubling times and standard deviations were obtained from at least three independent growth curves.
| Strain | Doubling Time (minutes) | Maximal A600 |
|---|---|---|
| MG1655 | 71.06 ± 0.74 | 1.90 ± 0.06 |
| EcNR2 | 79.37 ± 0.80 | 1.72 ± 0.08 |
| rEc7 | 82.29 ± 2.53 | 1.92 ± 0.06 |
| rEc7.δprfA | 134.44 ± 2.59 | 0.77 ± 0.03 |
|
| ||
| EcAR7 | 136.75 ± 2.06 | 0.56 ± 0.02 |
| EcAR7.SEP | 379.41 ± 3.43 | 0.30 ± 0.01 |
| EcAR7.supE | 132.38 ± 0.52 | 1.16 ± 0.04 |
|
| ||
| BL21.WT | 81.81 ± 1.38 | 1.81 ± 0.03 |
| BL21.SEP | 114.11 ± 2.85 | 1.34 ± 0.04 |
| BL21.L11C.SEP | 104.66 ± 5.34 | 1.66 ± 0.03 |
Fig. 2. Cellular fitness.

Growth curves were obtained in triplicate in LB-Medium containing 2 mM phosphoserine and 50 μM IPTG. A representative growth curve is depicted.
We next investigated whether the growth defect imposed by RF-1 deletion can be restored by introducing the suppressor tRNA supE, as described previously [11]. We used tRNA-scanSE [19] to verify that EcAR7 does not encode an annotated natural amber suppressor tRNA [20]. When the suppressor tRNA supE was supplied in trans, EcAR7.supE displayed a similar doubling time but demonstrated enhanced viability by reaching a higher final OD (Table 1, Fig. 2). These results indicate that a suppression system for glutamine can partially rescue the RF-1 induced phenotype. These results suggest that supE permits glutamine incorporation at a subset of natural TAG stop codons that relieves ribosomal stalling and results in the production of elongated proteins, which we posit maintain function.
Next, we investigated fitness and viability of EcAR7.SEP and two BL21-derived strains (BL21.SEP and BL21.L11C.SEP) upon introduction of the SEP-system. Overexpression of the L11C mutant ribosomal subunit has been shown to decrease RF-1 activity [21]. A striking reduction in fitness and cell viability were observed (Table 1, Fig. 2). The doubling time for EcAR7.SEP was reduced 2.8 fold to 379 minutes. The maximum optical density reached under these conditions was A600=0.3, which is a 2.6 fold decrease compared to EcAR7. Thus, while the pleiotropic effects associated with RF-1 deletion can be compensated by suppression of TAG with glutamine, Sep incorporation places an additional burden on the cell. We observed a similar phenomenon for BL21.SEP and BL21.L11C.SEP, where doubling times increased upon induction by 1.33 and 1.4 fold respectively, and final cell densities decreased (Table 1).
3.2 Phosphoserine is incorporated into the proteome in response to native UAG codons
While the incorporation of unnatural amino acids into recombinant proteins has been widely studied [22], the global effect of an amber suppressor system on the host proteome has only been considered recently [7,11,16]. Here, we observe that EcAR7.SEP has a significant growth defect upon induction of the Sep expression system. We hypothesize that widespread Sep incorporation at endogenous TAG codons partially explains the reduced phenotype. Total proteins from IPTG- induced EcAR7.SEP strains were isolated, digested with trypsin, subjected to large scale phosphopeptide enrichment, and analyzed with LC-MS to directly measure Sep incorporation in the natural proteome. Analysis of both phosphopeptide and non-phosphopeptide fractions of the EcAR7.SEP proteome identified approximately 1,100 E. coli proteins with 82 TAG [16] containing open reading frames identified. We investigated the genomic context of several of the most abundant TAG containing ORFs in our proteome data and identified a region of the luxS gene (encoding S-ribosylhomocysteine lyase) that could produce a detectable tryptic phosphopeptide upon TAG suppression with Sep (Fig. 3A). We predicted that the LuxS protein would be extended by a total of eight amino acids followed by natural termination at a second downstream TAA stop codon (Fig. 3A). The peptide LGELHISPSVNYLHN was indeed observed and phosphoserine insertion at the amber codon was unambiguously identified in the enriched phosphopeptide fractions of the E. coli proteome (Fig. 3B). We then repeated this experiment and used label free quantitation to examine the abundance of the Sep-extended LuxS peptide in strains BL21, BL21.SEP, BL21.L11C.SEP, and EcAR7.SEP. As expected, no Sep-extended LuxS peptide was detected in BL21. WT cells without the SEP-system (Fig. 3C, blue). However, the peptide was readily detected in both BL21.SEP (red) and BL21.L11C.SEP (pink) with the total peptide yields slightly increased by the overexpression of the ribosomal subunit L11C. L11C overexpression was previously shown to quantitatively enhance recombinant Sep protein production [2]. We also observed a striking increase of Sep-mediated luxS suppression in the EcAR7.SEP strain (Fig. 3C, black). These results provide direct evidence of Sep insertion at a native TAG site in an unmodified E. coli locus and suggest that other sites are open to Sep suppression and global proteome extension. Furthermore, the dramatic increase in Sep-extended LuxS protein production showed that our engineering strategy had indeed boosted the efficiency of genetically encoded Sep insertion at amber codons.
Fig. 3. Natural UAG codon suppression in luxS monitored by mass spectrometry.
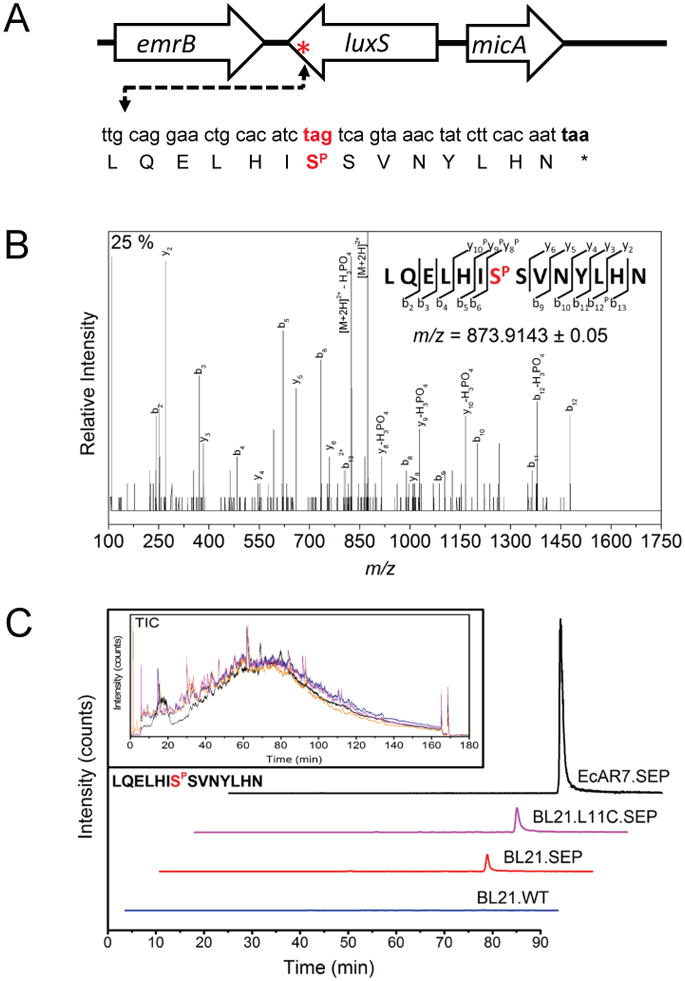
A) The genetic context of the luxS gene is shown. Suppression of the natural amber stop codon (tag=red) with phosphoserine (SP) is predicted to extend protein synthesis to the next in-frame non-amber stop codon (taa). B) Annotated tandem MS spectra and sequence coverage (y and b ions) for the predicted phosphopeptide LQELHISPSVNYLHN from LuxS detected in EcAR7.SEP. Fragment ions ([M+2H]2+ precursor = 873.914) showing neutral losses consistent with phosphorylation are indicated with a P in the sequence coverage map. C) Extracted ion chromatograms of the same peptide across multiple E. coli strains demonstrating improved suppression in the recoded strain. Total ion chromatograms of each experiment demonstrate equal loading (inset).
3.3 Phosphoserine incorporation is enhanced in a RF-1 deletion strain
We investigated the efficiency of Sep incorporation in the EcAR7.SEP strain in detail by examining the efficiency of UAG read-through directly compared to our previously reported BL21 strains [2]. We introduced TAG codons in various positions in plasmid-encoded GFP (Fig. 4). These constructs were then transformed into EcAR7.SEP, EcAR7.supE, BL21 and BL21.L11C.supE. Western blot analysis of GFP with an N-terminal antibody enabled analysis of read-through of the UAG codons. As depicted in Figure 4A, panel 1, no full length GFP is detected in BL21 without a functional UAG suppressor system. The combination of supE and Q94TAG or Q157TAG was selected to examine UAG suppression without changing the amino acid at the position of the UAG in GFP. Using this strategy we clearly showed that while supE can function effectively in BL21, the suppression efficiency is dramatically enhanced in the EcAR7 background (Fig. 4A). After confirming that UAG codons were converted to sense codons in EcAR7 we next examined the efficiency of Sep insertion in this strain. The same GFP variants were expressed in EcAR7.SEP and, while Sep insertion at Q157 was equivalent in expression to WT GFP, Q94TAG was non-permissive to Sep insertion. These results contrast the expression of the same constructs in BL21.L11C.SEP. While Q157TAG was more permissive than Q94TAG in BL21.L11C.SEP, the efficiency of Sep insertion was dramatically reduced and truncated products could easily be detected. These data demonstrate that Sep insertion is competing with RF-1 in BL21.L11C.SEP and enhanced in the RF-1 deficient background of EcAR7.SEP. These data suggest that Sep insertion at Q94TAG might destabilize GFP and result in protein degradation. Similar results have been described in studies that have aimed to introduce unnatural amino acids into GFP [7].
Fig. 4. Recoding of TAG to a sense codon.

(A) BL21 and EcAR7 strains containing either a suppressor tRNA (supE) or the SEP-system (SEP) were transformed with plasmids encoding WT GFP, GFPQ94TAG, GFPQ157TAG or GFPQ94/Q157TAG, respectively. Western blots using the antibody against the N-terminus of GFP are shown (* indicates a nonspecific band). (B) EcAR7.SEP and BL21.L11C.SEP were transformed with plasmids containing GFP-variants encoding TAG at the various positions indicated. A range of site specific truncations that accumulate in BL21.L11C.SEP are indicated by a bracket (* indicates a nonspecific band). (C)EcAR7.SEP and BL21.L11C.SEP were transformed with plasmids harboring WNK4-WT and WNK4-TAG-containing variants as indicated. Western blot against the C-terminal His-tag are shown. (D) MS/MS validation of Sep insertion at E17TAG. Fragment ion spectra ([M+3H]3+ precursor = 897.452) showing characteristic neutral losses consistent with phosphoserine (SP) allowed unambiguous assignment of phosphoserine at position 17 in GFP.
Next, we expanded our screen to find other Sep-permissive sites in GFP (Fig. 4B). We reasoned that glutamate residues in GFP might be more permissive to Sep insertion, but surprisingly found only two of the five glutamate positions tested (E17 and E142) tolerated Sep insertion (Fig. 4B). This result was interesting since glutamate is often used as a phosphoserine mimetic [2], however, we observed glutamate sites that did not permit phosphoserine insertion (E132, E213, and E222). This will be investigated in future studies and could be due to a number of factors that include, protein instability, steric hindrance, or charge incompatibility. Our small scale screen identified 3 out of 10 amino acid positions in GFP that can be efficiently replaced with Sep. Identifying several permissible Sep sites in GFP allowed us to further examine the production of recombinant phosphoproteins in the EcAR7.SEP strain.
Our previous work demonstrated that multiply phosphorylated proteins had extremely low yields (∼1 ug/L) in the BL21.L11C.SEP strains [2]. In contrast, the EcAR7.supE strain showed highly efficient suppression of two in frame UAG codons (Fig. 4A). This suggested that multiply phosphorylated proteins might show a similar translational efficiency in EcAR7.. We found that the combination of two permissible Sep sites (Q157 and E17) was efficiently translated to produce a doubly phosphorylated GFP protein (Fig. 4B). This result was in stark contrast to the very low level of Q157/E17TAG expression in BL21.L11C.SEP. Interestingly, the combination of the non-permissible Y66 and E213 sites with Q157 rendered the entire GFP unstable. We overexpressed and purified various phosphoserine containing GFPs on a larger scale and found the yield was approximately 0.23 g/L culture for E213TAG, 1.1 g/L for E17TAG, 6.6 g/L for E17/Q157TAG and 31 g/L for WT. In general, yields for singly and doubly phosphorylated GFP proteins were 10 fold greater in EcAR7.SEP when compared to yields from the BL21.L11C.SEP strain. Finally, we validated Sep insertion at GFP Q157TAG, E142TAG, and E17TAG (Figure 4C) with mass spectrometry. Sep insertions at all three sites could be unambiguously identified.
One important application of our Sep-expression system is to study physiologically relevant protein phosphorylation [2]. Furthermore, we wondered if phosphoserine insertions at naturally phosphorylated sites might be more permissible and contrast to the general impermissibility of phosphoserine insertion across GFP (Fig. 4A and B). To examine this directly we expressed an important switch domain of the mouse serine/threonine kinase WNK4. WNK4 acts as a molecular switch that can vary the balance between NaCl reabsorption and K+ secretion to control blood pressure in humans [23-26]. Two phosphoserine residues have been implicated in the physiological regulation of WNK4 and a system to produce this phosphoprotein could enable new types of research into its function. We expressed recombinant WNK4 proteins containing either one or two phosphoserine sites and observed variable expression levels in BL21.SEP and very low levels of multiply phosphorylated protein (Fig. 4C). In contrast, singly and multiply phosphorylated proteins were produced at very similar levels in EcAR7.SEP. However, expression of the WT protein was higher in BL21.SEP and, in general, the level of phosphoserine containing protein was lower. This shows that addition of chaperones or further genetic modifications to increase fitness might be required for some recombinant proteins.
4. Discussion
4.1 Efficiency of phosphoserine incorporation increases in a RF-1 knockout strain
In this study we aimed to enhance the incorporation efficiency of phosphoprotein production. Several methods for RF-1 knockout have been published recently [7,8,10,11] and we sought to combine these advances with newly developed methods of genome engineering (i.e., MAGE) to reassign the previously described essential TAG stop codons to synonymous TAA codons [10,12]. Our results show that a RF-1 deletion strain prevents premature stopping at the TAG codon and improves yields of phosphoprotein production in strains that harbor deficiencies in both fitness and viability.
In the resulting strain EcAR7 we were able to produce large amounts of phosphoprotein in GFP and the eukaryotic WNK4 phosphoprotein compared to the previously used strains BL21.SEP and BL21.L11C.SEP. Specifically, phosphoserine incorporation at a single TAG site yielded a 48 fold increase in protein yield (e.g. 1.2 mg/L culture for GFP E17TAG compared to 25 μg MEK1SEP /Liter culture in BL21.L11C.SEP) [2]. In WNK4, no reduction in protein yield was observed upon introduction of one or two TAG sites, respectively. The newly created strain EcAR7.SEP is thus a highly useful tool in phosphoprotein production and could be employed to investigate the function of single phosphoserine sites (e.g., WNK4) as well as other unnatural amino acids [22].
4.2 Phosphoserine is incorporated into the natural proteome and leads to reduced cell fitness and viability
While we were able to improve overall phosphoprotein yield up to 120 fold, the deletion of RF-1 and especially the introduction of our phosphoserine incorporation system had a major effect on strain viability (see Fig. 2). Interestingly, some of the growth phenotype of the release factor deletion could be rescued upon introduction of a natural suppressor tRNA, which allows for incorporation of glutamine in response to UAG, as previously described [11]. However, Sep incorporation had a deleterious effect on the host cell. Taken together, these results suggest that the striking loss in cell fitness is most likely the result of Sep-induced synthetic peptide chain extension at native UAG sites, as demonstrated by the incorporation of phosphoserine into the final peptide of LuxS. It is possible that some proteins may be able to tolerate extensions with glutamine incorporation, while the significant negative charge that accompanies phosphoserine may lead to misfolded or dysfunctional proteins. While the reassignment of seven TAG stop codons to TAA codons permitted the deletion of RF-1, more codon reassignments will be required to further increase phosphoserine incorporation and to compensate for the introduction of an unnatural amino acid (e.g., highly negatively charged phosphoserine) at a dedicated TAG sense codon without suffering loss of cellular fitness [16].
Supplementary Material
Highlights.
We produced an improved system for production of recombinant phosphoproteins in E. coli.
Genome modification and strain engineering of E. coli enabled RF1 deletion.
Single and double phosphoserine residues were incorporated into recombinant proteins at enhanced efficiency.
Phosphoserine insertion at native UAG sites reduces cellular fitness and viability.
Acknowledgments
This work was supported by grants from the National Institute of General Medical Sciences and the National Science Foundation (to D.S.), DARPA CLIO Contract #N66001-12C-4020 (to F.I.), and NIDDK (K01DK089006) (to J.R.). I.U.H. was supported from the Deutsche Forschungsgemeinschaft (HE5802/1-1). We thank Patrick O'Donoghue and Laure Prat for helpful discussions.
Abbreviations
- GFP
green fluorescent protein
- RF-1
release factor-1 (prfA)
- RF-2
release factor-2 (prfB)
- SepRS
phosphoseryl-tRNA synthetase
- WNK4
Serine/threonine-protein kinase WNK4
- Sep
phosphoserine
- MAGE
Multiplex automated genome engineering
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tarrant MK, Cole PA. The chemical biology of protein phosphorylation. Annu Rev Biochem. 2009;78:797–825. doi: 10.1146/annurev.biochem.78.070907.103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park HS, et al. Expanding the genetic code of Escherichia coli with phosphoserine. Science. 2011;333:1151–4. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eggertsson G, Söll D. Transfer ribonucleic acid-mediated suppression of termination codons in Escherichia coli. Microbiol Rev. 1988;52:354–74. doi: 10.1128/mr.52.3.354-374.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, Brock A, Herberich B, Schultz PG. Expanding the genetic code of Escherichia coli. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
- 5.Ryden SM, Isaksson LA. A temperature-sensitive mutant of Escherichia coli that shows enhanced misreading of UAG/A and increased efficiency for some tRNA nonsense suppressors. Mol Gen Genet. 1984;193:38–45. doi: 10.1007/BF00327411. [DOI] [PubMed] [Google Scholar]
- 6.Gerdes SY, et al. Experimental determination and system level analysis of essential genes in Escherichia coli MG1655. J Bacteriol. 2003;185:5673–84. doi: 10.1128/JB.185.19.5673-5684.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson DB, et al. RF1 knockout allows ribosomal incorporation of unnatural amino acids at multiple sites. Nat Chem Biol. 2011;7:779–86. doi: 10.1038/nchembio.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson DB, Wang C, Xu J, Schultz MD, Schmitz RJ, Ecker JR, Wang L. Release Factor One Is Nonessential in Escherichia coli. ACS Chem Biol. 2012 doi: 10.1021/cb300229q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukai T, Hayashi A, Iraha F, Sato A, Ohtake K, Yokoyama S, Sakamoto K. Codon reassignment in the Escherichia coli genetic code. Nucleic Acids Res. 2010;38:8188–95. doi: 10.1093/nar/gkq707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mukai T, et al. Genetic-code evolution for protein synthesis with non-natural amino acids. Biochem Biophys Res Commun. 2011;411:757–61. doi: 10.1016/j.bbrc.2011.07.020. [DOI] [PubMed] [Google Scholar]
- 11.Ohtake K, Sato A, Mukai T, Hino N, Yokoyama S, Sakamoto K. Efficient decoding of the UAG triplet as a full-fledged sense codon enhances the growth of a prfA-deficient strain of Escherichia coli. J Bacteriol. 2012;194:2606–13. doi: 10.1128/JB.00195-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460:894–8. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeVito JA. Recombineering with tolC as a selectable/counter-selectable marker: remodeling the rRNA operons of Escherichia coli. Nucleic Acids Res. 2008;36:e4. doi: 10.1093/nar/gkm1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isaacs FJ, Dwyer DJ, Ding C, Pervouchine DD, Cantor CR, Collins JJ. Engineered riboregulators enable post-transcriptional control of gene expression. Nature Biotechnol. 2004;22:841–7. doi: 10.1038/nbt986. [DOI] [PubMed] [Google Scholar]
- 15.Lutz R, Bujard H. Independent and tight regulation of transcriptional units in Escherichia coli via the LacR/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 1997;25:1203–10. doi: 10.1093/nar/25.6.1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isaacs FJ, et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science. 2011;333:348–53. doi: 10.1126/science.1205822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2(2006):0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jahn M, Rogers MJ, Söll D. Anticodon and acceptor stem nucleotides in tRNA(Gln) are major recognition elements for E. coli glutaminyl-tRNA synthetase. Nature. 1991;352:258–60. doi: 10.1038/352258a0. [DOI] [PubMed] [Google Scholar]
- 19.Schattner P, Brooks AN, Lowe TM. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005;33:W686–9. doi: 10.1093/nar/gki366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayashi K, et al. Highly accurate genome sequences of Escherichia coli K-12 strains MG1655 and W3110. Mol Syst Biol. 2006;2(2006):0007. doi: 10.1038/msb4100049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, Russell WK, Wan W, Pai PJ, Russell DH, Liu W. A convenient method for genetic incorporation of multiple noncanonical amino acids into one protein in Escherichia coli. Mol Biosyst. 2010;6:683–6. doi: 10.1039/b920120c. [DOI] [PubMed] [Google Scholar]
- 22.Neumann H. Rewiring translation - Genetic code expansion and its applications. FEBS Lett. 2012;586:2057–64. doi: 10.1016/j.febslet.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 23.Yang CL, Zhu X, Wang Z, Subramanya AR, Ellison DH. Mechanisms of WNK1 and WNK4 interaction in the regulation of thiazide-sensitive NaCl cotransport. J Clin Invest. 2005;115:1379–87. doi: 10.1172/JCI22452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rozansky DJ, et al. Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest. 2009;119:2601–12. doi: 10.1172/JCI38323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ring AM, Leng Q, Rinehart J, Wilson FH, Kahle KT, Hebert SC, Lifton RP. An SGK1 site in WNK4 regulates Na+ channel and K+ channel activity and has implications for aldosterone signaling and K+ homeostasis. Proc Natl Acad Sci USA. 2007;104:4025–9. doi: 10.1073/pnas.0611728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin DH, Yue P, Rinehart J, Sun P, Wang Z, Lifton R, Wang WH. Protein phosphatase 1 modulates the inhibitory effect of With-no-Lysine kinase 4 on ROMK channels. Am J Physiol Renal Physiol. 2012;303:F110–9. doi: 10.1152/ajprenal.00676.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.