

Abstract
We discuss protein post-translational modification (PTM) from an information processing perspective. PTM at multiple sites on a protein creates a combinatorial explosion in the number of potential “mod-forms”, or global patterns of modification. Distinct mod-forms can elicit distinct downstream responses, so that the overall response depends partly on the effectiveness of a particular mod-form to elicit a response and partly on the stoichiometry of that mod-form in the molecular population. We introduce the “mod-form distribution”—the relative stoichiometries of each mod-form—as the most informative measure of a protein’s state. Distinct mod-form distributions may summarise information about distinct cellular and physiological conditions and allow downstream processes to interpret this information accordingly. Such information “encoding” by PTMs may facilitate evolution by weakening the need to directly link upstream conditions to downstream responses. Mod-form distributions provide a quantitative framework in which to interpret ideas of “PTM codes” that are emerging in several areas of biology, as we show by reviewing examples of ion channels, GPCRs, microtubules and transcriptional co-regulators. We focus particularly on examples other than the well known “histone code”, to emphasise the pervasive use of information encoding in molecular biology. Finally, we touch briefly on new methods for measuring mod-form distributions.
Post-translational modification (PTM) is a biochemical mechanism in which aminoacid residues in a protein are covalently modified (106). It is nature’s escape from genetic imprisonment. Gene sequences change on an evolutionary time scale but not on one appropriate for organismal development, adult physiology and the continual battle against disease and disintegration. After exons are chosen and spliced, a protein’s tertiary structure is altered only by conformational fluctuations. PTM allows amino-acid properties to be changed “on the fly”, in response to requirements on a developmental or physiological time scale. Multisite PTM leads to a combinatorial explosion in the number of potential molecular states. Such complexity may provide the foundation for sophisticated forms of cellular information processing that are essential for the emergence of organismal complexity. This information-centric perspective provides the basis for this review.
Reversible phosphorylation as information processing
The ability of PTM to process information can be seen in a simple example of reversible phosphorylation on a single site (Figure 1a). An individual substrate molecule can be either unphosphorylated or phosphorylated. The population of substrate molecules contains a mixture of both molecular states. The state of the population can be summarised in the relative stoichiometry of the phosphorylated state, denoted U in Figure 1b,c. This number varies between 0 (completely unphosphorylated) and 1 (completely phosphorylated). It is easiest to understand the behaviour of U when the system has reached steady state and the rates of phosphorylation and dephosphorylation are equal and opposite. Then, U depends on the relative amounts, or effective levels of activity, of kinase and phosphatase (Figure 1c).
Figure 1. Reversible phosphorylation as information processing.
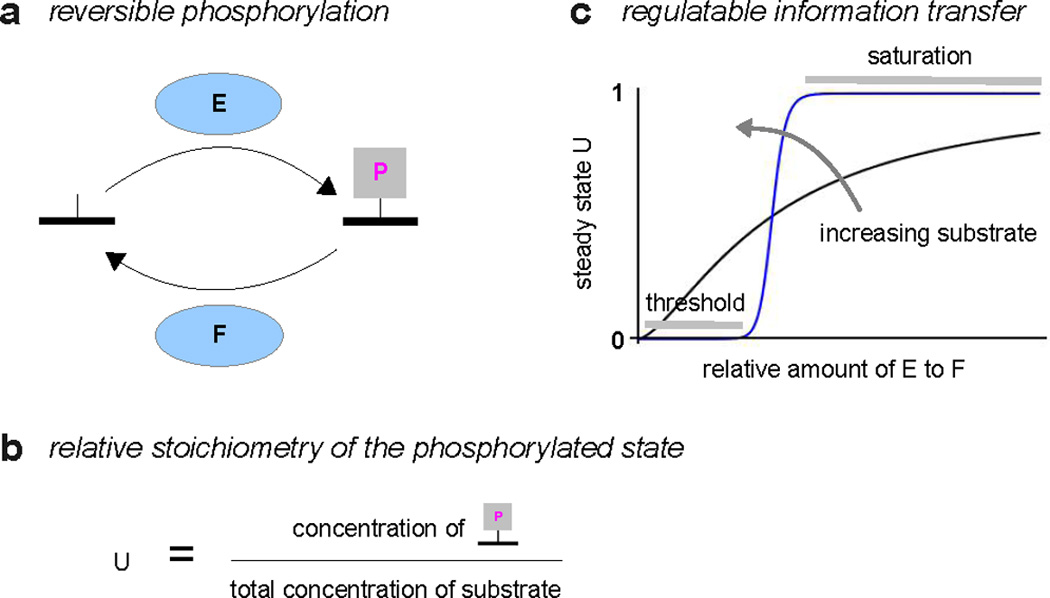
a. A single phosphorylated site on a substrate is dynamically regulated by a forward kinase, E, and a reverse phosphatase, F. Not shown are the donor, ATP, its hydrolysis products, ADP and Pi, and the background metabolic pathways that maintain the ATP “voltage” (see Figure 3a). b. The state of the population of substrate molecules is summarised by the relative stoichiometry of the phosphorylated state, denoted U, and defined by the fraction shown. Note that the denominator may have more contributions than just the free unphosphorylated and phosphorylated states, since, depending on the enzyme mechanisms, substrate may also be bound in enzyme-substrate complexes. c. The steady-state level of U is shown as a function of the relative amounts of kinase and phosphatase. This is a hypothetical, but typical, illustration; the quantitative details depend on the enzyme mechanisms (114). The value of U contains information about the relative amounts of kinase and phosphatase, which can be sensed and utilised by downstream processes. The response curve can exhibit increasing steepness, from nearly hyperbolic (black) to strongly sigmoidal (blue), as the amount of substrate is increased (40), allowing the information processing characteristics to be regulated.
To put it another way, the relative stoichiometry, U, carries information about the amounts or activity levels of the enzymes that are targetting the substrate. If the substrate is itself interacting with other proteins that prefer the phosphorylated state, such as those carrying phospho-specific binding domains, (97), these downstream processes will be able to sense information about the upstream enzymes, indirectly through the value of U. We will see in the course of this review how this idea plays out in intricate ways across a broad range of cellular processes.
PTM information processing is highly regulatable. As shown by Goldbeter and Koshland in a classic mathematical analysis, (40), and later confirmed experimentally, (80), the shape of the U-response curve becomes steeper as the total amount of substrate increases (Figure 1c). If the response is very steep (blue curve), then any changes in enzyme amounts that stay below threshold or above saturation will not be visible through changes in U. The information will have been filtered out. Between threshold and saturation, the dynamic range becomes highly amplified: small changes in enzyme amounts yield large changes in U (“ultrasensitivity”). Such quantitative details matter: if information processing is to be understood, we need to be able to measure relative stoichiometries and to relate their behaviour to the enzyme networks that underlie PTM.
The ability to process information, and to do so in a regulatable way, requires continuous expenditure of energy. This comes from hydrolysis of the donor molecule, in this case ATP, and is a dissipative process: a cell’s core biochemical pathways must continually replenish ATP and maintain the chemical “voltage” that drives phosphorylation. In this sense, PTM behaves like a transistor in electronics, expending energy to encode information. Such functionality becomes vastly enhanced with multiple types and multiple sites of modification. The implications of multisite phosphorylation have been discussed in previous reviews, (19, 47, 94), as has the interplay of different types of modification (115).
Metabolic and polypeptide modifications
Over 200 types of PTM have been identified (54, 106). Several were discovered years ago and their broader significance has emerged only slowly (20). Mass spectrometry has been instrumental in giving a genome-wide and less biased view (54). A recent survey of SwissProt data finds 87,308 experimentally detected modifications of amino-acid residues (58). Phosphorylation on serine/threonine is the most prevalent (Figure 2), although this may reflect the preponderance of phosphorylation studies. Of the other prevalent modifications, some are thought to be irreversible, or, at least, are not known to be reversible. Irreversible modifications have limited information processing capabilities and we focus here only on reversible PTMs (from now on, simply, PTMs), and limit attention further to those occurring in eukaryotes, particularly metazoans (Table 1).
Figure 2. Occurrence of experimentally detected PTMs, as curated from SwissProt, taken from (58, Figure 2A).
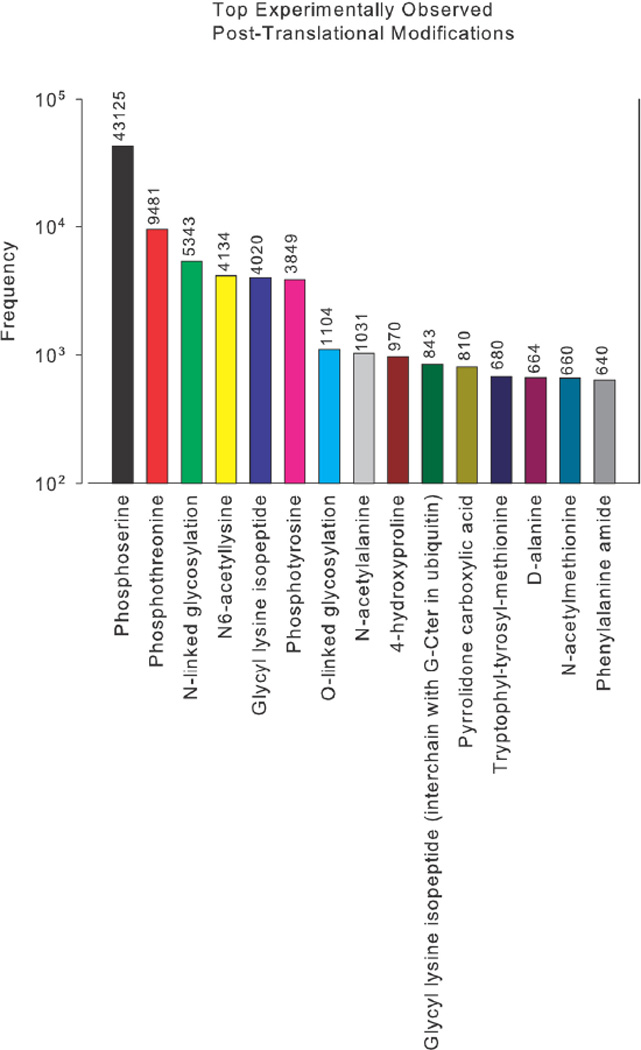
Table 1. Reversible post-translational modifications.
The table shows some of the more widely-studied PTMs in metazoa but is by no means exhaustive. For each PTM, only those residues thought to be most significant are indicated; for more complete details, see (106). The PTMs above the double line are simple modifications, as in Figure 4, while those below are more complex, as in Figure 5. The citations focus on non-histone examples.
| modification | modifier | donor | residues | refs | |
|---|---|---|---|---|---|
| phosphorylation |
|
ATP | S, T, Y1 | 56, 78 | |
| acetylation | CH3CO | AcCoA | K | 17, 121 | |
| GlcNAcylation | C6H12O5(NH)CH3CO | UDP-GlcNAc | S, T | 120 | |
| palmitoylation2 | CH3(CH2)14CO | palmitoyl-CoA | C | 71, 93 | |
| methylation | CH3 | SAM | K3 | 28, 50 | |
| ADP-ribosylation | ADP-ribose | NAD+ | R, K, E4 | 44, 95 | |
| ubiquitin-like | Ub, SUMO, etc | - | K | 96, 113 |
Notes:
Reversible phosphorylation on histidine and aspartate forms the basis for two-component signalling, which is abundant in eubacteria and is also found in plants and fungi (100); acidlabile phosphoramidate attachments to basic residues are also found in eukaryotes (18).
Most lipid modifications are irreversible, S-linked palmitoylation being the exception (67).
What has been said above for phosphorylation holds true for other such PTMs. They are all dissipative mechanisms in which energy is expended to change protein state. However, there are two different kinds of processes that maintain the required “voltages”. One kind of modification is based on small molecular groups—phosphoryl, acetyl, ADP-ribosyl, etc— that are carried by metabolic donors—ATP, acetyl-CoA, NAD, etc—(Table 1). The donor molecules are continuously supplied by the cell’s background metabolic processes, which have the ultimate responsibility for ensuring that the required “voltages” are maintained (Figure 3a). Forward and reverse modifications are each carried out by single enzymes.
Figure 3. Metabolic and polypeptide PTMs.
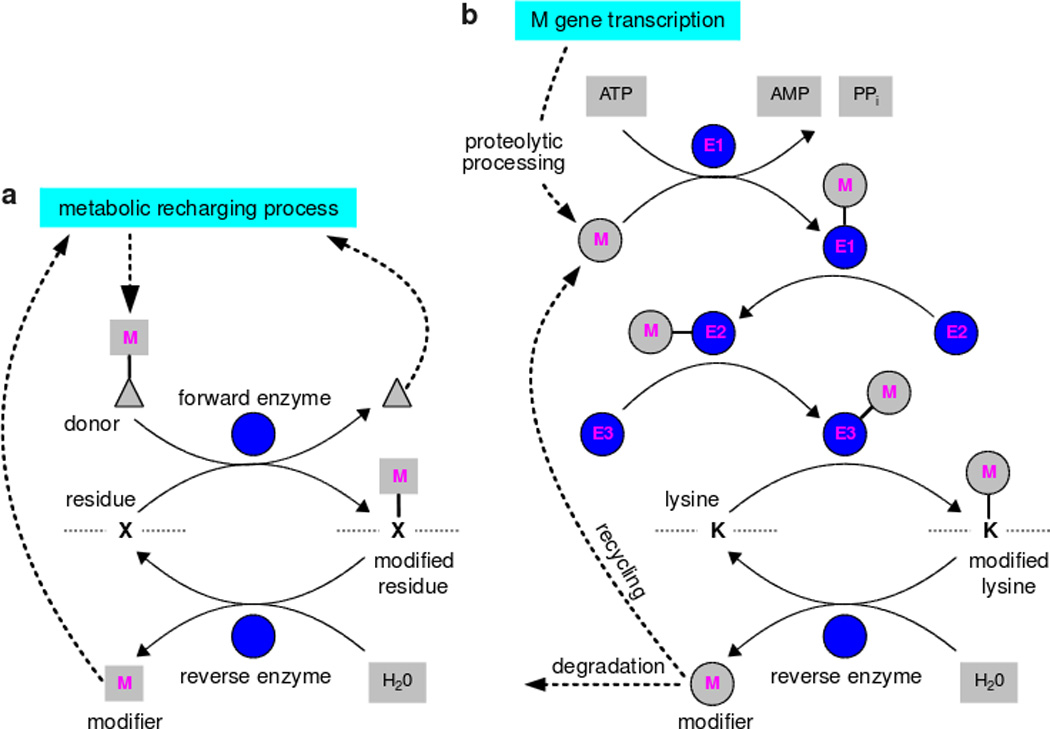
The biochemical details may differ depending on the modification; see (106) for more details. a. Metabolic PTMs. Note that lysine deacetylation by the sirtuins uses NAD+ and releases acetyl-ADP-ribose rather than acetate. b. Polypeptide PTMs. Ubiquitin-like modifiers are synthesised by gene transcription, which, in the case of ubiquitin, yields tandem repeats or fusion proteins. These must be proteolytically cleaved prior to being used for PTM (14). E2 enzymes can sometimes modify substrates independently of E3s; E2 and E3 enzymes often collaborate and E4 elongation factors can join in (46). Assembly of polymeric chains is not fully understood and ubiquitin chains may be preformed prior to substrate ligation (46).
In contrast to small molecule modifications, ubiquitin and ubiqutin-like modifications (SUMO, NEDD, etc) are polypeptide modifications (96). The modifying molecules are made by gene transcription and forward modification is undertaken by a chain of enzymes (Figure 3b). ATP is expended to adenylate the modifier to link it to the first activating enzyme (E1) in the chain. The modifier is passed from the E1 to the second conjugating enzyme (E2). The E2 may sometimes act alone, or in concert with an E3 ligase, or the E3 may act independently, to build an isopeptide linkage between the terminal ε-NH2 group of a lysine residue in the substrate protein and the C-terminal tail of the modifier (88). The modified protein is a branched amino-acid chain and the introduced polypeptide branch can itself become a target for further ubiqutin-like modifications. Single reverse deubiquitinating enzymes cleave the isopeptide linkage and release the modifying polypeptide (4) which may be recycled or degraded. Metabolic processes are not directly involved in maintaining the “voltage”, for which responsibility lies with whatever regulates transcription of the modifier genes and recycling and degradation of the resulting polypeptides (61).
The dissipative character of all PTMs places a burden on the background processes, metabolic or transcriptional, that are responsible for maintaining modifier molecules at the appropriate “voltage”. If such a background process is not homeostatic—if it does not maintain modifier concentration when demand fluctuates—then the efficiency of modification may be compromised, potentially affecting all substrates subject to that modification. In the case of phosphorylation, ATP concentration is remarkably robust even in tissues like skeletal muscle, where demand for ATP can change by over two orders of magnitude (45). Because ATP is so widely used for so many different purposes, there may have been sufficient pressure to evolve the circuitry needed to make its supply robust to fluctuations in demand. This may not be so for other modifications, for which much less is known about modifier homeostasis (16, 61).
Combinatorics of modification
Phosphorylation is a binary modification; a given serine, threonine or tyrosine residue is either phosphorylated or not (Figure 4). The same is true for acetylation on lysine, GlcNA-cylation on serine or threonine and palmitoylation on cysteine. Up to three methyl groups may bind to the ε-NH2 group of lysine, so that a given lysine may be mono-, di- or tri-methylated. For these PTMs, each residue has a small, limited number of discrete modification states (Figure 4).
Figure 4. Simple PTMs.
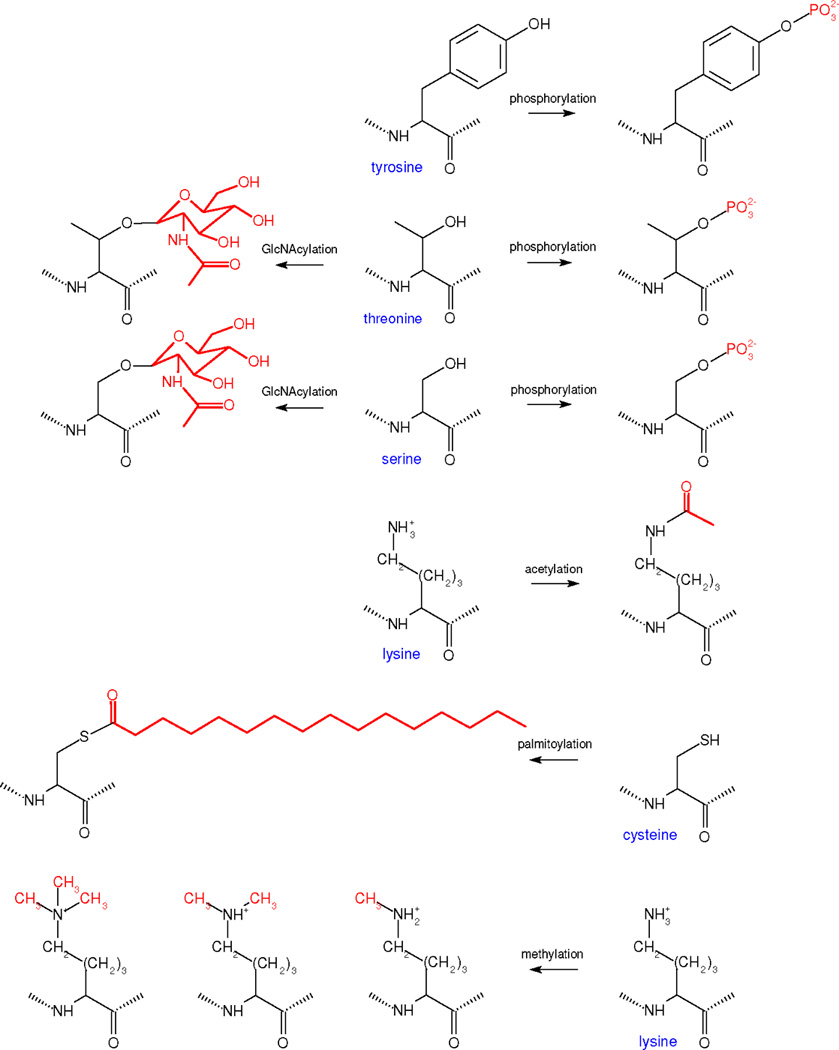
The chemistry of those PTMs above the double line in Table 1, which exhibit a small, limited number of modifications, is shown, with the modifications to each residue in red. Chemical formulas were drawn in BKChem, an open source utility.
The possiblities become more intricate for other modifications, such as ADP-ribosylation (Figure 5). Mono-ADP-ribosylation—the transfer of a single ADP-ribose moiety usually to an arginine residue—was first identified in bacterial toxins which inhibit key cellular processes, such as the GTPase activity of G-proteins (21). In contrast, poly-ADP-ribosylation was first discovered in the DNA damage response, although it is now known to affect a wide range of cellular processes (95). Such “PARsylation” is reversibly catalysed by the PARP and PARG families of enzymes, which can dynamically build, on lysine or glutamate residues, a polymer of ADP-ribose monomers linked by glycosidic bridges. Heterogenous, linear and branched polymers with more than 200 monomers have been found (3). Instead of a simple modification like those in Figure 4, PARsylation offers a potentially unlimited suite of modification structures on a single residue. In-vitro studies show that PAR binding domains can discriminate between polymers of different sizes (30), suggesting that evolution may have been able to exploit this heterogeneity.
Figure 5. Complex PTMs.
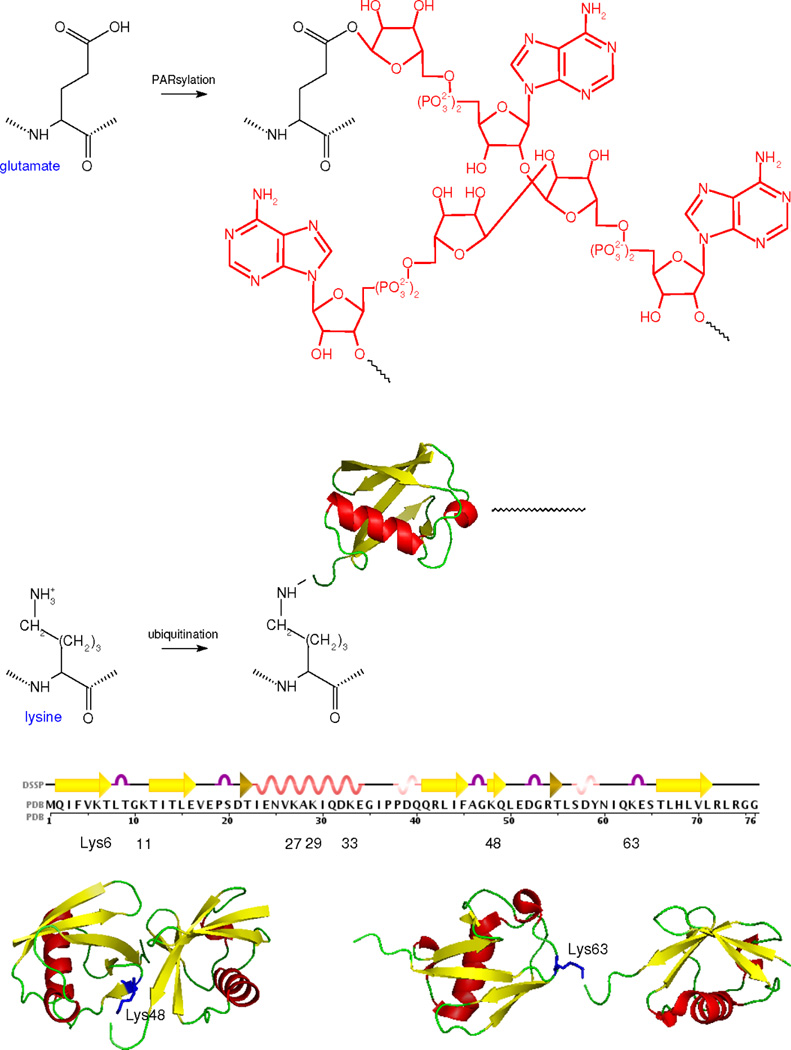
The chemistry of those PTMs below the double line in Table 1, which exhibit potentially unlimited numbers of modifications, is summarised, as in Figure 4. The human ubiquitin sequence was obtained from PDB 1UBI, along with the secondary structure assignment through DSSP. The PDB entries of the ubiquitin structures are 1UBI for the monomer, 1AAR for the Lys48 dimer and 2JF5 for the Lys63 dimer. The structures were oriented and annotated in Open Source PyMol 1.2.X.
Ubiquitin-like modification exhibits even more structural diversity than PARsylation. This is best understood for ubiquitin itself, although SUMO2/3 and NEDD8 are also reported to form polymeric chains (51). Lysine residues may be mono-ubiqutinated or polyubiquitinated, with one of the lysine residues in ubiquitin itself becoming the attachment point for the next ubiquitin monomer (Figure 5). Polymers with over a dozen monomers are reported. Ubiquitin has seven lysines, K6, K11, K27, K29, K33, K48 and K63, each of which may be involved in chain formation in vivo. During log-phase growth of yeast, mass-spectrometry has shown that these lysines occur as attachment points in the respective proportions 8:24:4:3:1:20:8 (113).
Homotypic chains, in which the same position is used for each link, are thought to be most common. For instance, K48-linked chains are associated with proteasomal degradation and may have a compact structure, while K63-linked chains are associated with endocytosis and may have a more open structure (51); see Figure 5. There is evidence for heterotypic linking (2). Forked chains, in which some ubiquitin monomers have more than one lysine to which other monomers are attached, have been constructed in vitro (59) but have not yet been observed in vivo. Significantly, evolution has found a way to discriminate between structural variants. A variety of ubiquitin-binding domains can distinguish not only between different lengths of polymer but also between different linkages (24).
Multiple residues are often modified on the same protein. This may happen through the same type of modification on different sites as well as through different types of modifications on different, or overlapping, sites. If one site has k modification states and another site l, then, in principle, there could be k × l combinatorial states. The possibilities multiply with increasing numbers of sites. If a protein has n sites of phosphorylation then the total number of combinatorial protein states is 2n. Each of these combinatorial states corresponds to a global pattern of modification across the entire protein. If there are also complex modifications, like PARsylation or ubiquitin-like modification, then the number of global patterns increases even faster with n. For ubiquitin, it may be necessary to keep track not only of the size and shape of the polymer but also of the linkages between the components, giving even higher multiplicative possibilities for heterotypic chains. This enormous combinatorial explosion is one of the most characteristic features of PTM and also one of its most perplexing. The “hypothetical computation” of PTM states by Lonard and O’Malley makes the same point (75). Why is so much state needed? What manner of information processing has evolution been able to implement through having such extraordinary complexity at its disposal? To address this question, we build upon the ideas discussed initially to introduce a basic concept for keeping track of global patterns of modification.
The mod-form distribution
From now on, we refer to a global pattern of modification as a “mod-form”. To reiterate the meaning of this, a mod-form is a specific pattern of modifications on all modifiable residues in a protein. Each post-translationally modified protein may have many mod-forms, as discussed above. The customary cartoon depiction of a post-translationally modified protein shows it in one particular mod-form, usually the maximally modified one (Figure 6a). This gives the misleading impression that only one mod-form is present, when, in reality, there are combinatorially many possibilities (Figure 6b). Moreover, there is a population of molecules present and each molecule is in one of the potential mod-forms. It is easy to lose sight of the molecular populations behind the cartoons. For instance, it is often said that two modifications that target the same residue, such as GlcNAcylation and phosphorylation on serine and threonine (Figure 4), are “mutually exclusive”. This is only true of a single molecule. The population may contain both modifications in any proportion.
Figure 6. Mod-form distributions.
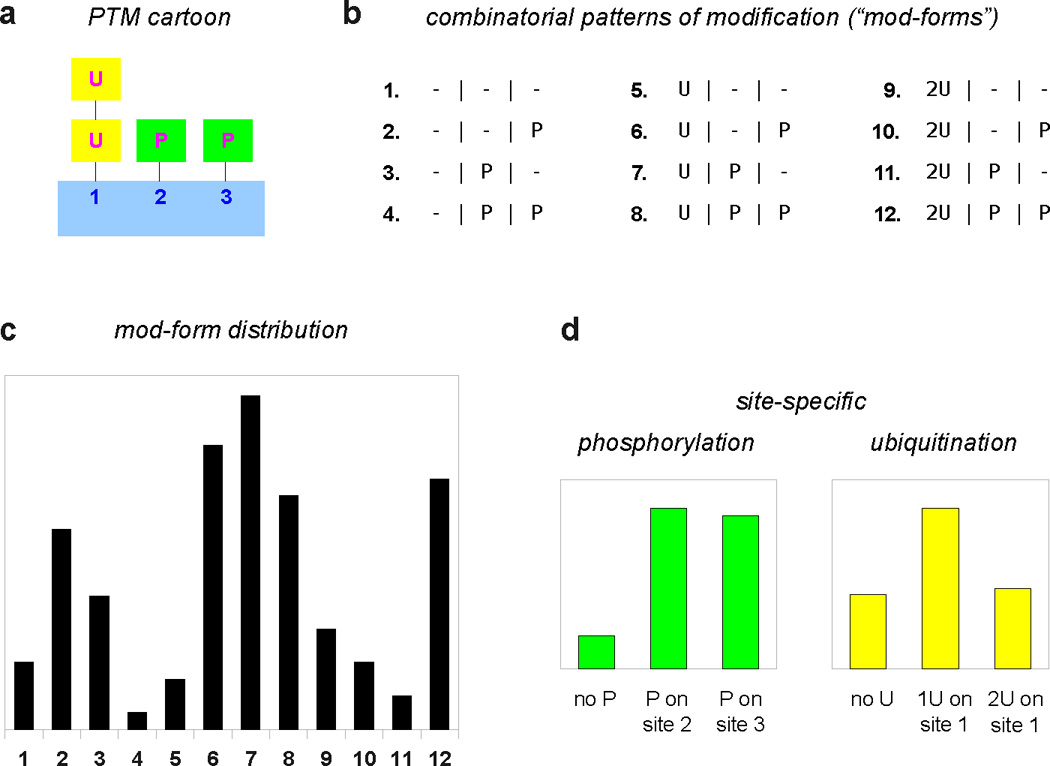
a. Cartoon depiction of a hypothetical substrate with 3 sites of modification; site 1 is ubiquitinated with a chain of up to two monomers; sites 2 and 3 are phosphorylated. b. There are 12 = 3 × 2 × 2 global patterns of modification, enumerated as shown. c. A hypothetical mod-form distribution, showing the proportions in the population of each of the 12 mod-forms, following the numbering used in b. The mod-form distribution can be viewed as a probability distribution, which gives, for each mod-form, the probability of finding a substrate molecule in that mod-form. The vertical scale has been omitted to focus on qualitative aspects. d. In current practice, only limited information may be available. The separate phosphoryl- and ubiquityl-modifications calculated from c are shown, with the phosphoryl-modifications given as site-specific stoichiometries (the proportion of unphosphorylated substrate and of substrate phosphorylated on each site). Such summaries lose considerable information compared to the underlying mod-form distribution, making it harder to infer correlations between modification states and downstream responses.
Of course, not all potential mod-forms may be present in any particular context. The serine/arginine repetitive matrix factor (Srrm2) has over 300 detected phosphorylations, as reported on PhosphoElm (Table 2). Since 2300 exceeds Eddington’s estimate of the number of protons in the Universe, not all mod-forms can ever be present at any one time. However, this only begs the question of which of the many possible mod-forms are present and to what extent.
Table 2. PTM resources.
The table shows online databases of post-translational modifications, focussing on those modifications in Table 1. The list is by no means exhaustive.
| name | url | PTMs | organisms | refs |
|---|---|---|---|---|
| UniProt | www.uniprot.org | many | many | 76 |
| HPRD | www.hprd.org | Ph1 | Hs | 39 |
| Phospho.ELM | phospho.elm.eu.org | Ph | eukaryotes | 25 |
| PhosphoSitePlus | www.phosphosite.org | Ac, Me, Ph, Ub | Hs, Mm | 48 |
| PHOSIDA | www.phosida.com | Ac, Ph | Ce, Dm, Hs, Mm, Sc | 38 |
| PhosphoPep | www.phosphopep.org | Ph | Ce, Dm, Hs, Sc | 11 |
| dbPTM | dbptm.mbc.nctu.edu.tw | many2 | N/A | 65 |
| CPLA | cpla.biocuckoo.org | Ac | N/A | 74 |
| P3DB | www.p3db.org | Ph | At, Bn, Gm, Mt, Os, Zm | 37 |
| PhosPhAt3.0 | phosphat.mpimp-golm.mpg.de | Ph | At | 26 |
| Phosphorylation Site Database | www.phosphorylation.biochem.vt.edu | Ph3 | bacteria, archaea | N/A |
Abbreviations for PTMs: Ph, phosphorylation; Ac, acetylation; Me, methylation, Ub, ubiquitin. Abbreviations for organisms: At, Arabidopsis thaliana; Bn, Brassica napus; Ce, Caenorhabditis elegans; Dm, Drosophila melanogaster; Eu, eukaryotes; Gm, Glycine max; Hs, Homo sapiens; Mm, Mus musculus; Mt, Medicago truncatula; Os, Oryza sativa; Sc, Saccharomyces cerevisiae; Zm, Zea mays. N/A means “not available”.
Notes:
Other PTMs are included but phosphorylation is particularly curated.
Includes irreversible PTMs but focusses on statistics and motifs.
Only phosphorylations on serine, threonine and tyrosine are provided, not histidine and aspartate, as found in two-component signalling.
This is a matter of biochemical dynamics. The pattern of mod-forms in the population is dynamically regulated by the cognate forward and reverse enzymes working collectively. It is sometimes thought that forward and reverse enzymes work in sequence, with the former being activated first to create the modifications and the latter being activated next to down-regulate them. This may be useful in some contexts to create a tightly focussed mod-form distribution but any stochastic (noisy) fluctuation in the forward enzyme will precipitate irreversible modification, suggesting that this is not a robust mechanism in general. It is more usually the case that opposing enzymes are constitutively present (1, 118). PTM is a highly dynamic business.
For a single site, as discussed initially, enzyme activities can be regulated to set the relative stoichiometry of phosphorylation anywhere between 0 and 1 and can do so in either a graded or ultrasensitive manner (Figure 1c). The situation becomes more complicated with multiple sites (41) or complex enzyme mechanisms (114). A kinase may operate processively, phosphorylating a substrate on multiple sites without releasing it (7); intermediate mod-forms may not then appear. Enzyme action may depend on the prior existence of certain mod-forms, as in “primed” or hierarchical phosphorylation (32); some mod-forms may only appear at certain times and with the right sequence of enzymes. In short, the pattern of mod-forms and the way this changes over time depend on the mechanistic details of the network of enzymes that target the substrate. New mathematical methods developed by one of our labs have the potential to analyse such combinatorially complex dynamical behaviour (42, 102).
Downstream processes typically (but not always—see the next paragraph) interact with a modified substrate by sampling all the substrate molecules in the population. They are therefore influenced by whichever mod-forms are present. As we shall see, distinct mod-form may exert distinct effects on downstream processes. The overall response will depend both on how much effect each mod-form exerts and on how much of each mod-form is present. If a mod-form has high effect but is present only at low stoichiometry, it may have less impact than one of low effect but high stoichiometry. The stoichiometry to which each mod-form is present, or the proportion of total substrate in each mod-form, determines the substrate’s “mod-form distribution”. This is a histogram over all the mod-forms that lists, for each mod-form, the effective probability of finding the protein in that state (Figure 6c). The overall effect of a given downstream process can be quantified as an average over the mod-form distribution of the effect of each individual mod-form. The mod-form distribution provides the most comprehensive and quantitative accounting of the combinatorial possibilities.
The role of the substrate population can vary with context. For instance, the carboxy terminal domain (CTD) of the largest subunit of RNA Pol II consists of tandem hexapeptide repeats, 52 in humans, whose differential phosphorylation correlates with the progress of transcription (27). Here, transcription of a particular gene is influenced only by the CTD of the Pol II that is transcribing that gene; the population of CTDs is not sampled. In such cases, the frequency with which different patterns appear over time on an individual molecule may play a similar role to the relative stoichiometry in a population. However, stochastic fluctuations, which are not discussed here, may become very significant at the single-molecule level.
Measurement of mod-form distributions remains challenging. Current techniques may only be able to detect certain modifications, such as the phosphoryl-forms or the ubiquityl-forms, and, even then, it may not be feasible to distinguish all the combinatorial possibilities (Figure 6d). In practice, we may only see a limited summary of the actual mod-form distribution. This may involve substantial information loss (Figure 6c,d), which may weaken the correlation between the observed modification states and the downstream responses. In this respect, we are in the position of the blind men trying to work out an elephant, with one seeing a pillar and the other a snake. The mod-form distribution is the elephant in the room. It helps not to lose sight of it if we are to ground experimental observations in biochemical mechanisms. We touch on how mod-form distributions can be measured after first reviewing their significance in different biological settings.
Different mod-forms elicit different responses
The differential behaviour of different mod-forms is well known in special cases, particularly in the regulation of gene transcription, as in the histone “code” (55, 105) and the CTD “code” of RNA Pol II (27). We focus here on other contexts to show that these are not exceptional situations but, rather, instances of a broad theme relevant to many areas of biology.
Membrane signalling proteins
Ion channels are integral membrane proteins that maintain ionic balance in all cell types. They play a particularly significant role in the electrical excitability of neurons, which is the basis for organismal memory and learning. Post-translational modification of ion channels, particularly phosphorylation, is a key mechanism underlying such plasticity (68).
Potassium channels help repolarise cells to their resting membrane potential of around −60mV, by permitting selective passage of K+ ions down their concentration gradient. K+ concentration is higher inside cells than outside (in contrast to Na+) so that potassium channels allow K+ efflux, making membrane potential more negative. The Kv2.1 channel is a homo-tetramer composed of four 98kDa α subunits, each with six membrane-spanning domains and a large cytoplasmic C-terminus. The channel’s conductance is “voltage gated”: the channel pore opens with increasing probability as the membrane potential rises above its resting level (ie: becomes more positive). The membrane potential at which whole-cell conductance is half maximal has a range of levels, depending on cell type (82). This particular channel is “delayed”, with a high threshold for activation and slow kinetics. Rather than directly controlling the action potential, which requires less sluggish kinetics, Kv2.1 plays a more homeostatic role in controlling rapid spiking and over-excitability (101) and may protect against the excito-toxicity arising during epilepsy or cerebral ischemia or hypoxia.
The flexibility required for this complex physiological role is partly implemented by PTMs. Quantitative mass-spectrometry originally revealed 16 phosphorylated sites on the α subunit of Kv2.1, nearly all serines in the C-terminal cytoplasmic region (86). (More recent studies have found several more (15)). Single-cell patch-clamp recording and mutational analysis identified 8 sites with an impact on voltage gating. Importantly, the effect on gating of multiple mutations depended on both the number and the positions of the mutations, with increasing amounts of phosphorylation causing graded increases in half-maximal potential (86). Electrical activity causes a rise in intracellular Ca2+, which activates the calcium-and calmodulin-dependent phosphatase, calcineurin, which can dephosphorylate some of the Kv2.1 sites, increasing the sensitivity of the channel to depolarisation.
More recently, an unique sumoylation site has been found on the α subunit which also influences voltage gating (89). Single-molecule photobleaching has shown that up to two subunits in the channel tetramer can be sumoylated with correspondingly graded changes in half-maximal conductance. (This presumably also confirms mono-sumoylation at each site.) Distinct phosphoryl- and sumoyl-forms of the channel therefore have distinct effects on the relationship between membrane voltage and conductance. No data are available yet on the interaction between the two modifications, if any, but cross talk, or “switching”, between phosphorylation and lysine-attached modifications has been found elsewhere (33, 77). Here, the combinatorial possibilities provided by PTM potentially allow highly nuanced modulation of channel sensitivity and responsiveness, depending on cell type and physiological conditions.
Distinct electrophysiological behaviour of distinct mod-forms has also been observed for tyrosine phosphorylation of the Kv1.3 channel (29) and for serine/threonine phosphorylation of the cystic fibrosis transmembrane conductance regulator (CFTR) chloride channel (36).
G-protein coupled receptors (GPCRs) are the largest signalling superfamily in the human genome, with at least 800 members (34). GPCRs play a central role in organismal physiology, responding to a huge variety of stimulants—light, hormones, neurotransmitters, cytokines, odorants—-and providing targets for a substantial number of the drugs in current clinical use (12, 66). Each GPCR is monomeric with seven transmembrane helices, three cytosolic loops and a cytosolic C-terminus. GPCRs were originally characterised by their eponymous signalling through heterotrimeric GTP-binding proteins (G-proteins) but the classical allosteric two-state model of GPCR activation has evolved, at least for non-visual GPCRs, into an intricate picture of functional selectivity (“protean agonism”, “collateral efficacy”, etc), in which different stimulations can elicit different patterns of downstream signalling in a cell context-dependent manner (57).
Multisite phosphorylation is emerging as a key mechanism underlying this plasticity (104). GPCRs are downregulated in part by recruitment to the receptor of G-protein coupled receptor kinases (GRKs), which phosphorylate cytosolic receptor segments, thereby enhancing binding of β-arrestins and severing the G-protein interaction. β-arrestins can then act as scaffolds for multiple signalling pathways. Mass-spectrometric analysis of the β2 adrenergic receptor reveals 13 serine/threonine phosphorylation sites on the receptor’s third intracellular loop and C-terminal tail, which are phosphorylated by multiple kinases (84). GRK2 and GRK6 target distinct subsets of sites and each kinase elicits a distinct β-arrestin conformation and a distinct pattern of downstream signalling. Related analysis of the M3 muscarinic acetylcholine receptor reveals 15 serine/threonine phosphorylation sites in the third intracellular loop, for which 2D-gels and phospho-specific antibodies show different patterns of phosphorylation in response to stimulation in different cell types and tissues and, following heterologous expression in CHO cells, in response to different ligands (13). These studies provide suggestive evidence for a phosphorylation “barcode” that can be written in distinct ways by distinct ligands and/or cellular contexts and interpreted in distinct ways by multiple downstream signalling pathways (70). Such barcodes are best seen quantitatively as phosphoryl-form distributions (see below).
Visual GPCRs, such as rhodopsin, have a more restricted function than their chemically-liganded cousins, responding only to photons in a single cell type. They are more akin to ion channels, whose sensitivity and responsiveness must adapt to varying environmental conditions. Here too, distinct phosphoryl-form distributions underlie this process of adaptation (6).
Microtubules
Microtubules are fundamental structural elements in eukaryotic cells. They form the mitotic spindle and midbody during cell division, the highways along which organelles and cargos are trafficked (over distances of one metre in some neurons), the axonemes of cilia and flagella and the centrioles of centrosomes and basal bodies. Despite this variety, microtubules are made up of polymers in which the repeating unit is a heterodimer of α- and β-tubulin. Multiple tubulin isoforms are present in some organisms but it remains unclear to what extent they contribute to functional diversity (103, 108). Microtubules are also subject to a host of PTMs that may reflect the cellular context in which the microtubules appear and thereby orchestrate distinct responses.
In addition to familiar PTMs, like acetylation of α-tubulin, microtubules are also modified in unusual, almost unique, ways. The C-terminal tails of both α- and β-tubulin are highly acidic, unstructured and exposed on the cytoplasmic face of microtubules (52). The C-terminal tyrosine of alpha-tubulin may be removed by detyrosination and, more surprisingly, reversibly replaced. Specific glutamate residues may be modified by glutamylation or glycylation, in which glutamate or glycine, respectively, is joined to the γ-carboxyl of the glutamate residue by an iso-peptide linkage. Polyglutamylases and polyglycylases can elongate these mono-modifications into linear polypeptide polymers, yielding complex modifications similar to those in Figure 5. The forward enzymes are members of the TTLL (tubulin tyrosine ligase like) superfamily, with the uncommon ability to make polypeptides without RNA. The reverse enzymes have not all been identified but a family of cytosolic carboxypeptidases have recently been found to be deglutamylases (60, 92).
Mutational studies in genetically-tractable systems, like the ciliate Tetrahymena, have shown that motility and cytokinesis defects depend on the number and the position of polyglycylated glutamate residues in β-tubulin, of which there are five in the wild type. Distinct glycylated-forms exhibit distinct phenotypes (112). Neuronal microtubules must not only organise distinct cellular regions—growth cones, axons, dendrites, dendritic spines, etc—but also contribute to the remodelling and plasticity underlying long-term potentiation and other forms of synapse-specific cellular memory. Distinct detyrosinyl/acetyl mod-forms may demarcate distinct regions and these mod-forms may in turn differentially influence the behaviour of motor proteins and microtubule binding proteins (53). The flagellar axonemes of Chlamydomonas exhibit a decreasing gradient of polyglutamyl/polyglycyl mod-forms, which may similarly act as molecular signposts (108). Here, in a manner similar to non-visual GPCRs, distinct cellular contexts appear to “encode” distinct information on microtubule PTMs, which can then be interpreted in distinct ways by downstream processes.
Many other cytoskeletal components are subject to PTM and one has particular clinical significance. The microtubule associated protein, tau, is the primary constituent of the neurofibrillary tangles found in the brains of Alzheimer’s disease patients, where it is hyperphosphorylated on as many as forty sites. Distinct phosphoryl-forms have distinct molecular properties and are associated with distinct stages of the disease (5, 8). It is conceivable that buildup of specific phosphoryl-forms, either by chance or through gradual destabilisation of kinases or phosphatases, could trigger onset of the fatal aggregation process. Competition between serine/threonine phosphorylation and GlcNAcylation, via the “yin-yang” mechanism (107), has been suggested as a potential therapeutic strategy against Alzheimer’s (119). Determining the corresponding mod-form distributions may offer key insights into this devastating disease (64).
Transcriptional effectors
Steroid receptor co-regulators (SRCs) were first identified as transcriptional adaptors for nuclear hormone receptors (85). The latter are transcription factors that bind lipophilic hormones—estrogen, retinoic acid, thyroxine, etc—that can reach the nucleus without triggering intervening signal transduction. SRCs help assemble a transcriptional complex that includes fellow effectors, such as the acetyltransferase CBP/p300 and the arginine methyltransferase, CARM1, which in turn help reorganise chromatin structure and regulate gene expression. It is now understood that these coregulators participate in many physiological responses and interact with a wide range of transcription factors (75, 116).
This functional plasticity may be implemented in part by a zoo of post-translational modifications, including phosphorylation, ubiqutination, sumoylation, acetylation and (possibly irreversible) arginine methylation (69). Studies of SRC-3 phosphorylation have been particularly informative (111). Mass spectrometry and phospho-specific antibodies uncovered six phosphorylated residues—Thr24, Ser505, Ser543, Ser857, Ser860 and Ser867. Different patterns of phosphorylation are observed for different upstream stimulations and different sets of upstream kinases and phosphatases are able to differentially phosphorylate SRC-3 both in vitro and in cell culture. Mutational studies suggest that differential phosphorylation elicits distinct downstream responses, such as different patterns of cytokine expression. To confirm these results in a physiological setting, a knockin mouse was constructed, SRC-3Δ/Δ, in which SRC-3’s central four phosphorylation sites were mutated to alanine (117). Interestingly, these mice showed opposite growth phenotypes—increased body weight, increased fat mass, increased plasma IGF, etc—to what was found in SRC-3−/− knockout mice, suggesting that the inability to produce certain SRC-3 mod-forms was specifically affecting major metabolic growth pathways.
Sumoylation of SRC-3 on three lysines—Lys723, Lys786 and Lys1194—is inversely correlated with phosphorylation and directly correlated with transcriptional attenuation at responsive genes (109). In contrast, ubiquitination of SRC-3 depends on prior phosphorylation at Ser505, which leads initially to mono-ubiquitination on two of the three lysines—Lys723 and Lys786—and transcriptional enhancement. This is subsequently followed by poly-ubiquitination and proteasome-mediated degradation which may set the time-scale of transcriptional activation (110). Arginine methylation on Arg1171 encourages disassembly of the transcriptional complex (31), which may be part of the cyclical re-assembly of the transcriptional complex at estrogen-responsive genes (81). The apparent lack of arginine demethylases makes it unclear how methylated SRC-3 can be recycled and it may simply be degraded through the proteasome.
These studies paint a vivid picture of distinct mod-forms orchestrating dynamic and flexible gene regulation to yield a “pleiotropic rheostat” (116). Similar kinds of plasticity have been found to be implemented by PTM on several transcription factors (91, 99), of which the most spectacular in scope and complexity is p53 (79, 83).
Encoding by mod-form distributions
The examples discussed above cover a wide range of cellular processes but reveal a broadly similar type of functional behaviour. In each case, modification of a single substrate serves to summarise, or “encode”, the prevailing physiological and cellular conditions in distinct mod-form distributions (Figure 7a). An example of how such an encoding might work was given in our initial discussion of phosphorylation of a single-site substrate (Figure 1). The “conditions” that are encoded may reflect different cell types or tissues, cell state (such as the level of excitability of a neuron), location within a cell, or the extent of physiological stimulation or exogenous ligands. The mechanisms through which the conditions determine the mod-forms may be many and varied. For instance, different cell types may express different balances of forward and reverse enzymes, scaffolding may bring different components together in different locations and external stimulations may alter conformations or activate enzymes to different degrees. Crucially, the “conditions” usually reflect the states of many cellular components, which are encoded by PTM into the state of a single component. Engineers refer to this kind of many-to-one mapping as “fan-in”.
Figure 7. Encoding of information by mod-form distributions.
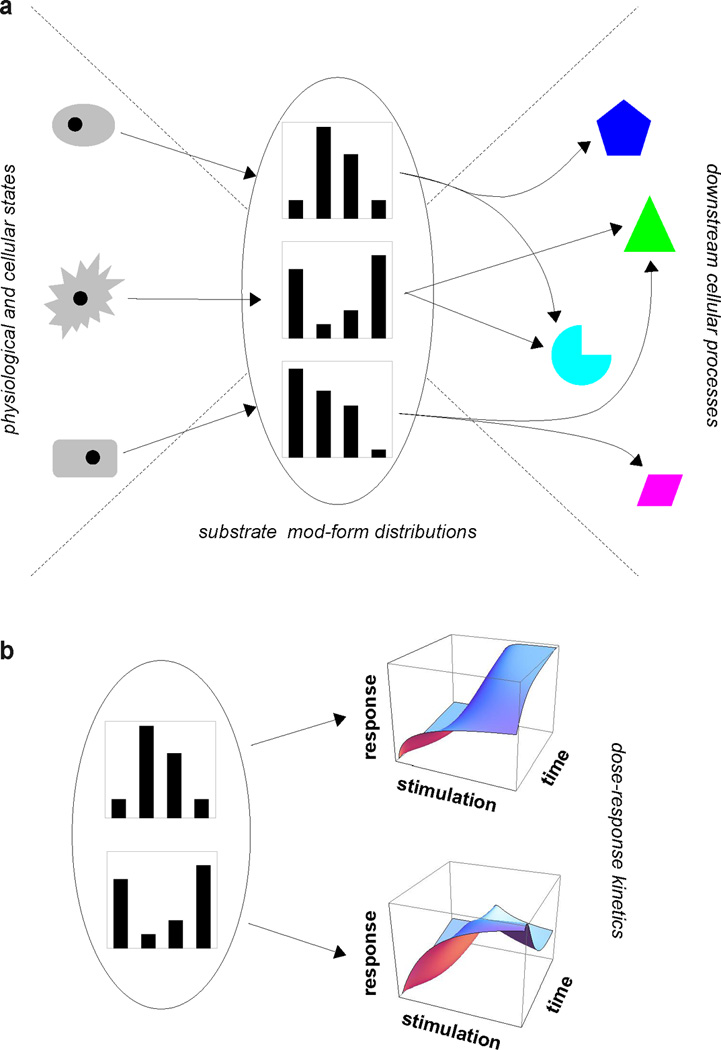
a. A bow-tie architecture describes the behaviour of many of the examples discussed here. Distinct physiological and cellular states on the left can be represented (“encoded”) by distinct mod-form distributions of a single substrate at the centre of the bow-tie (“fan-in”). Each mod-form distribution can then orchestrate its own mix of downstream cellular processes, as on the right (“fan-out”). Figure 1 of (75) reflects a similar architecture for the particular case of SRC-3. The mod-form distribution plays a central role here as the quantitative representation of the encoded information. b. The mod-form distribution can also affect the behaviour of the substrate itself, as in the example of ion channels. This is shown here by the hypothetical dose-response kinetics on the right, whose characteristics—threshold sensitivity, steepness, saturation level, latency, etc—may be modulated by changes in the mod-form distribution.
Such encoding can have varied consequences. It may directly influence the behaviour of the substrate itself, as seen for ion channels like Kv2.1, which alter their threshold activation depending on the number and position of phosphorylated sites. Channels like this can have quantitative dose-response kinetics with many features—threshold sensitivity, steepness, saturation level, latency—that can potentially be mod-form dependent, as illustrated hypothetically in Figure 7b. The mechanisms through which this could be implemented are not well understood (77).
Another possibility, which may not be exclusive of the first, is that distinct mod-forms orchestrate distinct downstream responses by activating different subsets of downstream cellular processes (Figure 7a). This seems to be the case for GPCR barcoding, microtubule signposting and SRC-3 coregulation. There are many potential mechanisms through which it could occur. For instance, distinct mod-forms could recruit distinct modification-specific binding domains (97). Also, for the β2 adrenergic receptor, β-arrestin assumes distinct conformations, and thereby triggers distinct downstream responses, when binding to distinct receptor phosphoryl-forms. Here, a single component with multiple PTMs can potentially influence many downstream processes, an instance of one-to-many “fan-out”.
Viewed differently, downstream responses may be said to “interpret” (or “translate”) the mod-form distribution. It is conceivable that the same mod-form distribution gives rise to different interpretations, depending on the downstream context. The “code” may mean different things in different situations.
The combination of fan-in and fan-out, centering on a single component, gives a “bow-tie” architecture (Figure 7a). Analogies with engineered systems have suggested that such designs offer a trade-off between robustness and flexibility (23, 63). Whether or not this is so, the biological implication of a bow-tie is that it relaxes the need for direct linkage between the cellular conditions and the downstream processes. Downstream processes can acquire information about cellular state indirectly, by interacting only with the modified substrate, without having to directly sense cellular conditions. As Kirschner and Gerhart have persuasively argued, such “weak regulatory linkage” creates neutral spaces that facilitate evolution (62). Exploitation of combinatorial PTMs in this way may have been a pre-requisite for the evolution of multicellular complexity. By the same token, the profusion of post-translational modification sites may reflect continuing exploratory creation of new variants that could offer future possibilities for selection and evolution. Indeed, it is often pointed out that enzymes are promiscuous and that some modifications lack apparent function. The evolutionary perspective suggests that such neutral variation is to be expected.
The capability shown in Figure 7a can be described as “state-based encoding”. The mod-form distribution reflects, or tracks, the cellular conditions. If the conditions are repeated, the mod-form distribution should be the same. An alternative possibility is “history-based encoding”, in which the same cellular condition can give rise to distinct mod-forms depending on the prior history through which that condition was reached. This kind of encoding requires multistability, or hysteresis, on the part of the enzymes involved, with different histories giving rise to different stable states. This amounts to a form of activity-dependent memory that can persist despite molecular turnover. The suggestion that post-translational modification could be the basis for such a persistent cellular memory goes back to Crick, (22), and Lisman (72). More recent theoretical work has identified PTM networks with specific memory capacities, (87, 102), and experimental studies have implicated the calcium- and calmodulin-dependent protein kinase II, CaMKII, in implementing a particular form of persistent activity-dependent memory, “long-term potentiation”, at individual neuronal post-synaptic densities (73). It would be interesting if similar memory circuits were to be found in cells other than neurons.
Encoding suggests that information about cellular conditions is “written” in the mod-form distribution and subsequently “read out”. Such analogical language conceals more than it reveals. Readers and writers may not be so clearly separated; what gets written may depend on what was previously read. The encoding process may operate at many different time scales without reaching a steady state, so that there may be no clear window of time in which the information can be said to be fully “present”. If the information is present, then how much is there? What is the information capacity of the system? In the case of the static DNA code, it is well understood that its information capacity is 2 bits per base pair. If we really understood PTM encoding, we should be able to make a similar estimate. So far, this has only been possible for simple models of history-based encoding (87, 102). As yet, we have no idea what biochemical mechanisms are minimally required for robust and reproducible state-based encoding and what kind of information and how much of it can be encoded in this way. While the concept of information encoding provides a powerful organising principle, and the mod-form distribution gives a quantitative basis for it, the biochemical details have not yet been worked out for any PTM code. The absence of a mechanistic basis for coding underlies discussions in the literature as to whether or not something is a code and, if so, what that means experimentally (10, 98). Formulating an experimental-verifiable mechanistic basis for information encoding remains a fundamental problem.
Measuring mod-form distributions
Unravelling PTM encoding will rely on accurate measurement of mod-forms. Lack of space precludes a detailed discussion here, for which we refer to our recent paper (90) and its accompanying citations.
Modification-specific antibiodies have been the backbone of PTM detection. They have many advantages, especially high sensitivity and single-cell resolution via immunostaining. They also have two significant disadvantages. First, they miss correlations between modifications that are widely separated on a protein and so cannot detect combinatorial mod-forms. Second, antigen recognition may be highly context-dependent so that antibodies may give quantitatively unreliable results in the face of combinatorial modifications (35, 90). The implications of this have not been fully appreciated.
Mass spectrometry can, in principle, circumvent these difficulties, especially new protein-based methods that retain correlations between widely-separated modifications (Figure 8). The mod-form distributions of histone tails (below 10kDa) have been enumerated but it remains challenging to apply such methods to typical cellular proteins (around 50kDa) with widely-separated sites, multiple types of modification and mod-forms of low abundance. Nevertheless, new instruments and methods are being developed that promise significant advances. Nuclear magnetic resonance spectroscopy is a complementary method, that is more limited in scope than MS but has real-time and even in-vivo potential. Studies of tau phosphoryl-forms by NMR, undertaken by one of our labs, suggest what can be achieved for a large unstructured protein with complex phosphorylation patterns (64).
Figure 8. Measurement of a four-site phosphoryl-form distribution.
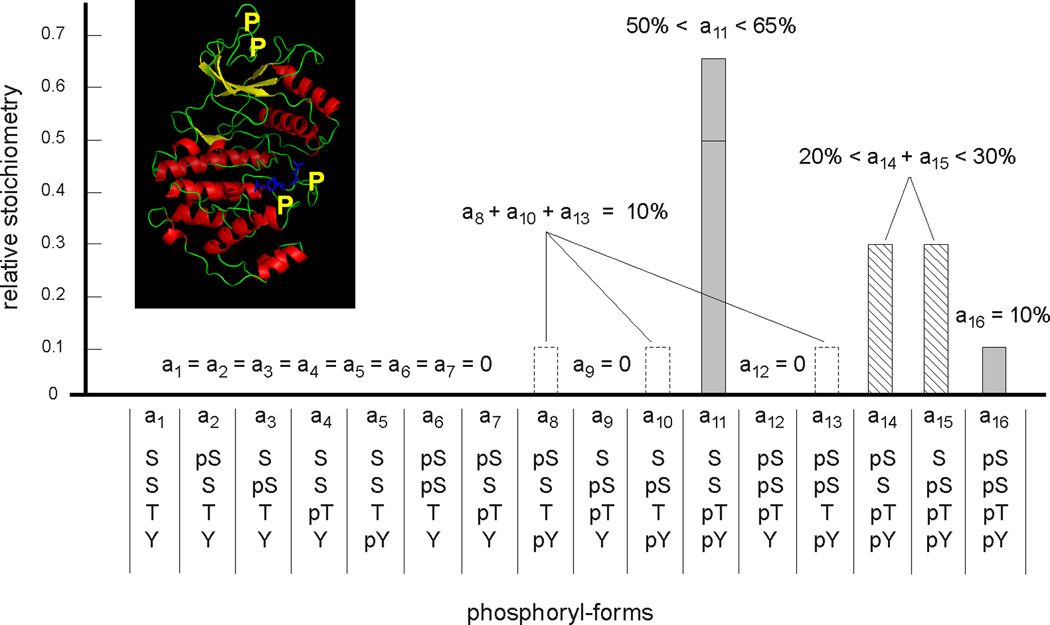
The phosphoryl-form distribution of the MAP kinase Erk2 is shown, after in-vitro preparation to generate four sites of phosphorylation, two canonical ones, Thr and Tyr, on the activation loop and two novel ones, both Ser, on the N-terminal tail. Their approximate positions are marked by yellow Ps on the ribbon diagram of PDB-2ERK in the inset. Note, in particular, that these residues are not all on the same tryptic peptide. A combination of peptide-based and protein-based mass spectrometry, confirmed by nuclear magnetic resonance spectroscopy, was used to measure the relative stoichiometry of the sixteen potential phosphoryl-forms, not all of which could be individually determined. Adapted from Figure 6B of (90), with the inset created in Open Source PyMol 1.2.X.
Summary and conclusions
We have tried to make the following points in this review. First, it is the combinatorial patterns of modification across an entire protein—its “mod-forms”—that provide the most complete information about a protein’s state. Second, distinct mod-forms can elicit distinct downstream responses. We have assembled evidence from several biological contexts to confirm that this is a general feature of PTMs. Third, the overall response to a modified protein depends both on the effectiveness of a given mod-form to trigger the response and on the relative stoichiometry of that mod-form in the population. The latter quantities can be enumerated in the protein’s “mod-form distribution” (Figure 6). Mass spectrometry offers new capabilities for measuring mod-form distributions on typical cellular proteins (Figure 8). Fourth, the mod-form distribution provides the quantitative basis for interpreting PTM “codes”, as suggested for ion-channels, GPCRs, microtubules and transcriptional co-regulators. This leads to the kind of “bow-tie” architectures that provide weak regulatory linkage (Figure 7) and enhance evolvability. Fifth, the shape of the mod-form distribution is dynamically regulated by the collective actions of the cognate forward and reverse enzymes and depends on the details of their biochemistry.
The mechanistic basis of PTM encoding presents an intriguing and challenging scientific problem, whose solution will require quantitative measurement, new experimental capabilities and new mathematical insights. Such “systems biology” is not a new-fangled trend but an old strategy, well known to the biochemists and physiologists of the premolecular era (43). We will need to recapture these ways of thinking to unravel the remarkable information processing capabilities of post-translational modification.
Acknowledgements
We are grateful to the following colleagues for their thoughtful comments on this paper but must stress that any omission or errors that remain are entirely our responsibility: Xavier Hanoulle, Carsten Janke, Isabelle Landrieu, Robert Lefkowitz, Kathryn Lilley, Denis Michel, Bert O’Malley, Anna Philpott, Valérie Schreiber, Caroline Smet, James Trimmer, Andrew Tobin and Christopher T. Walsh. We had to focus on a limited number of examples for reasons of space and we apologise to colleagues whose work we were unable to cite. The research underlying this review was supported by the US NIH under R01-GM081578 (SP, HS, JG) and by the French CNRS (GL).
References
- 1.Acuto O, Di Bartolo v, Michel F. Tailoring T-cell receptor signals by proximal negative feedback mechanisms. Nat. Rev. Immunol. 2008;8:699–712. doi: 10.1038/nri2397. [DOI] [PubMed] [Google Scholar]
- 2.Al-Hakim AK, Zagorska A, Chapman L, Deak M, Peggie M, Alessi DR. Control of AMPK-related kinases by USP9X and atypical Lys29/Lys33-linked polyubiquitin chains. Biochem. J. 2008;411:249–260. doi: 10.1042/BJ20080067. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez-Gonzalez R, Jacobson MK. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry. 1987;26:3218–3224. doi: 10.1021/bi00385a042. [DOI] [PubMed] [Google Scholar]
- 4.Amerik AY, Hochstrasser M. Mechanism and function of deubiquitinating enzymes. Biochim. Biophys. Acta. 2004;1695:189–207. doi: 10.1016/j.bbamcr.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Amniai L, Barbier P, Sillen A, Wieruszeski J-M, Peyrot v, Lippens G, Landrieu I. Alzheimer disease specific phosphoepitopes of Tau interfere with assembly of tubulin but not binding to microtubules. FASEB J. 2009;23:1146–1152. doi: 10.1096/fj.08-121590. [DOI] [PubMed] [Google Scholar]
- 6.Arshavsky VY. Rhodopsin phosphorylation: from terminating single photon responses to photoreceptor dark adaptation. Trends Neurosci. 2002;25:124–126. doi: 10.1016/s0166-2236(00)02094-4. [DOI] [PubMed] [Google Scholar]
- 7.Aubol BE, Chakrabarti S, Ngo J, Shaffer J, Nolen B, Fu X-D, Ghosh G, Adams JA. Processive phosphorylation of alternative splicing factor/splicing factor-2. Proc. Natl. Acad. Sci. USA. 2003;100:12601–12606. doi: 10.1073/pnas.1635129100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Augustinack JC, Schneider A, Mandelkow E-M, Hyman BT. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimers disease. Acta Neuropathol. 2002;103:26–35. doi: 10.1007/s004010100423. [DOI] [PubMed] [Google Scholar]
- 9.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol. Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–411. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 11.Bodenmiller B, Campbell D, Gerrits B, Lam H, Jovanovic M, Picotti P, Schlapbach R, Aebersold R. PhosphoPep—a database of protein phosphorylation sites in model organisms. Nat. Biotechnol. 2008;26:1339–1340. doi: 10.1038/nbt1208-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bridges TM, Lindsley CW. G-protein coupled receptors: from classical models of modulation to allosteric mechanisms. ACS Chem. Biol. 2008;19:530–541. doi: 10.1021/cb800116f. [DOI] [PubMed] [Google Scholar]
- 13.Butcher AJ, Prihandoko R, Kong KC, McWilliams P, Edwards JM, Bottrill A, Mistry S, Tobin AB. Differential G-protein coupled receptor phosphorylation provides evidence for a signaling bar code. J. Biol. Chem. 2011;286:11507–11518. doi: 10.1074/jbc.M110.154526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Catic A, Ploegh HL. Ubiquitin—conserved protein or selfish gene? Trends Biochem. Sci. 2005;30:600–604. doi: 10.1016/j.tibs.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 15.Cerda O, Baek J-H, Trimmer JS. Mining recent brain proteomic databases for ion channel phosphosite nuggets. J. Gen. Physiol. 2010;137:3–16. doi: 10.1085/jgp.201010555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen P-C, Bhattacharayya BJ, Hanna J, Minkel H, Wilson JA, Finley D, Miller RJ, Wilson SM. Ubiquitin homeostasis is critical for synaptic development and function. J. Neurosci. 2011;31:17505–17513. doi: 10.1523/JNEUROSCI.2922-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 18.Cieśla J, Fra̧czyk T, Rode W. Phosphorylation of basic amino acid residues in proteins: important but easily missed. Acta Biochim. Pol. 2011;58:137–147. [PubMed] [Google Scholar]
- 19.Cohen P. The regulation of protein function by multisite phosphorylation - a 25 year update. Trends Biochem. Sci. 2000;25:596–601. doi: 10.1016/s0968-0004(00)01712-6. [DOI] [PubMed] [Google Scholar]
- 20.Cohen P. The origins of protein phosphorylation. Nat. Cell Biol. 2002;4:E127–E130. doi: 10.1038/ncb0502-e127. [DOI] [PubMed] [Google Scholar]
- 21.Corda D, di Girolamo M. Functional aspects of protein mono-ADPribosylation. EMBO J. 2003;22:1953–1958. doi: 10.1093/emboj/cdg209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crick F. Memory and molecular turnover. Nature. 1984;312:101. doi: 10.1038/312101a0. [DOI] [PubMed] [Google Scholar]
- 23.Csete ME, Doyle JC. Bow ties, metabolism and disease. Trends Biotechnol. 2004;22:446–450. doi: 10.1016/j.tibtech.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 24.Dikic I, Wakatsuki S, Walters KJ. Ubiquitin-binding domains—from structures to function. Nat. Rev. Mol. Cell Biol. 2009;10:659–671. doi: 10.1038/nrm2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dinkel H, Chica C, Via A, Gould CM, Jensen LJ, Gibson TJ, Diella F. Phospho.ELM: a database of phosphorylation sites—update 2011. Nucleic Acids Res. 2011;39:D261–D267. doi: 10.1093/nar/gkq1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Durek P, Schmidt R, Heazlewood JL, Jones A, MacLean D, Nagel A, Kersten B, Schulze WX. PhosPhAt: the Arabidopsis thaliana phosphorylation site database. an update. Nucleic Acids Res. 2010;38:D828–D834. doi: 10.1093/nar/gkp810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Egloff S, Murphy S. Cracking the RNA polymerase II CTD code. Trends Genet. 2008;24:260–268. doi: 10.1016/j.tig.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Erce MA, Pang CNI, Hart-Smith G, Wilkins MR. The methylproteome and the intracellular methylation network. Proteomics. 2012 doi: 10.1002/pmic.201100397. [DOI] [PubMed] [Google Scholar]
- 29.Fadool DA, Holmes TC, Berman K, Daga D, Levitan IB. Tyrosine phosphorylation modulates current amplitude and kinetics of a neuronal voltage-gated potassium channel. J. Neurophysiol. 1997;78:1563–1573. doi: 10.1152/jn.1997.78.3.1563. [DOI] [PubMed] [Google Scholar]
- 30.Fahrer J, Popp O, Malanga M, Beneke S, Markovitz DM, Ferrando-May E, Bürkle A, Kappes F. High-affinity interaction of poly(ADP-ribose) and the human DEK oncoprotein depends upon chain length. Biochemistry. 2010;49:7119–7130. doi: 10.1021/bi1004365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feng Q, Yi P, Wong J, O’Malley BW. Signaling with a coactivator complex: methylation of SRC-3/AIB1 is a molecular switch for complex disassembly. Mol. Cell Biol. 2006;26:7846–7857. doi: 10.1128/MCB.00568-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fiol CJ, Roach PJ. Hierarchical phosphorylation of proteins. In: Marks F, editor. Protein Phosphorylation. VCH Verlagsgesellschaft, Federal Republic of Germany; 1996. [Google Scholar]
- 33.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–479. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 34.Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 35.Fuchs SM, Krajewski K, Baker RW, Miller VL, Strahl BD. Influence of combinatorial histone modifications on antibody and effector protein recognition. Current Biol. 2011;21:53–58. doi: 10.1016/j.cub.2010.11.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 1999;79:S77–S107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- 37.Gao J, Agrawal GK, Thelen JJ, Xu D. P3DB: a plant protein phosphorylation database. Nucleic Acids Res. 2009;37:D960–D962. doi: 10.1093/nar/gkn733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gnad F, Gunawardena J, Mann M. PHOSIDA 2011: the posttranslational modification database. Nucleic Acids Res. 2011;39:D253–D260. doi: 10.1093/nar/gkq1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goel R, Harsha HC, Pandey A, Prasad TS. Human Protein Reference Database and Human Proteinpedia as resources for phosphoproteome analysis. Mol. Biosyst. 2012;8:453–463. doi: 10.1039/c1mb05340j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldbeter A, Koshland DE. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl. Acad. Sci. USA. 1981;78(11):6840–6844. doi: 10.1073/pnas.78.11.6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gunawardena J. Multisite protein phosphorylation makes a good threshold but can be a poor switch. Proc. Natl. Acad. Sci. USA. 2005;102:14617–14622. doi: 10.1073/pnas.0507322102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gunawardena J. A linear framework for time-scale separation in nonlinear biochemical systems. PLoS ONE. 2012;7:e36321. doi: 10.1371/journal.pone.0036321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gunawardena J. Some lessons about models from Michaelis and Menten. Mol. Biol. Cell. 2012;23:517–519. doi: 10.1091/mbc.E11-07-0643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hassa PO, Haenni SS, Elser M, Hottiger MO. Nuclear ADP-ribosylation reactions in mammalian cells: where are we today and where are we going? Microbiol. Mol. Biol. Rev. 2006;70:789–829. doi: 10.1128/MMBR.00040-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hochachka PW, McClelland GB. Cellular metabolic homeostasis during large-scale change in ATP turnover rates in muscles. J. Exp. Biol. 1997;200:381–386. doi: 10.1242/jeb.200.2.381. [DOI] [PubMed] [Google Scholar]
- 46.Hochstrasser M. Lingering mysteries of ubiquitin-chain assembly. Cell. 2006;124:27–34. doi: 10.1016/j.cell.2005.12.025. [DOI] [PubMed] [Google Scholar]
- 47.Holmberg CI, Tran SEF, Eriksson JE, Sistonen L. Multisite phosphorylation provides sophisticated regulation of transcription factors. Trends Biochem. Sci. 2002;27:619–627. doi: 10.1016/s0968-0004(02)02207-7. [DOI] [PubMed] [Google Scholar]
- 48.Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: a bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- 49.Hsu J-M, Chen C-T, Chou C-K, Kuo H-P, Li L-Y, Lin Chun-Yi, Lee H-J, Wang Y-N, Liu M, Liao H-W, Shi B, Lai C-C, Bedford MT, Tsai C-H, Hung C. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nature Cell Biol. 2011;13:174–181. doi: 10.1038/ncb2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huang J, Berger SL. The emerging field of dynamic lysine methylation of non-histone proteins. Curr. Opin. Genet. Dev. 2008;18:1–7. doi: 10.1016/j.gde.2008.01.012. [DOI] [PubMed] [Google Scholar]
- 51.Ikeda F, Dikic I. Atypical ubiquitin chains: new molecular signals. EMBO Rep. 2008;9:536–542. doi: 10.1038/embor.2008.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat. Rev. Mol. Cell Biol. 2011;12:773–786. doi: 10.1038/nrm3227. [DOI] [PubMed] [Google Scholar]
- 53.Janke C, Kneussel M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010;33:363–372. doi: 10.1016/j.tins.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 54.Jensen ON. Interpreting the protein language using proteomics. Nat. Rev. Mol. Cell Biol. 2006;7:391–403. doi: 10.1038/nrm1939. [DOI] [PubMed] [Google Scholar]
- 55.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 56.Johnson LN. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009;37:627–641. doi: 10.1042/BST0370627. [DOI] [PubMed] [Google Scholar]
- 57.Kenakin T. New concepts in drug discovery: collateral efficacy and permissive antagonism. Nature Rev. Drug Discov. 2005;4:919–927. doi: 10.1038/nrd1875. [DOI] [PubMed] [Google Scholar]
- 58.Khoury GA, Baliban RC, Floudas CA. Proteome-wide post-translational modification statistics: frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011;1 doi: 10.1038/srep00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, Goldberg AL. Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J. Biol. Chem. 2007;282:17375–17386. doi: 10.1074/jbc.M609659200. [DOI] [PubMed] [Google Scholar]
- 60.Kimura Y, Kurabe N, Ikegami K, Tsutsumi K, Konishi Y, Kaplan OI, Kunitomo H, Iino Y, Blacque OE, Setou M. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs) J. Biol. Chem. 2010;285:22936–22941. doi: 10.1074/jbc.C110.128280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kimura Y, Tanaka K. Regulatory mechanisms involved in the control of ubiquitin homeostasis. J. Biochem. 2010;147:793–798. doi: 10.1093/jb/mvq044. [DOI] [PubMed] [Google Scholar]
- 62.Kirschner M, Gerhart J. The theory of facilitated variation. Proc. Natl. Acad. Sci. USA. 2007;104:8582–8589. doi: 10.1073/pnas.0701035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kitano H. Biological robustness. Nat. Rev. Genet. 2004;5:826–837. doi: 10.1038/nrg1471. [DOI] [PubMed] [Google Scholar]
- 64.Landrieu I, Leroy A, Smet-Nocca C, Huvent I, Amniai L, Hamdane M, Sibille N, Buée L, Wieruszeski J-M, Lippens G. NMR spectroscopy of the neuronal tau protein: normal function and implication in alzheimers disease. Biochem. Soc. Trans. 2010;38:1006–1011. doi: 10.1042/BST0381006. [DOI] [PubMed] [Google Scholar]
- 65.Lee TY, Huang HD, Hung JH, Huang HY, Yang YS, Wang TH. dbPTM: an information repository of protein post-translational modification. Nucleic Acids Res. 2006;34:D622–D627. doi: 10.1093/nar/gkj083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lefkowitz RJ. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004;25:413–422. doi: 10.1016/j.tips.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 67.Levental I, Grzybek M, Simons K. Greasing their way: lipid modifications determine protein association with membrane rafts. Biochemistry. 2010;49:6305–6316. doi: 10.1021/bi100882y. [DOI] [PubMed] [Google Scholar]
- 68.Levitan I. Modulation of ion channels by protein phosphorylation and dephosphorylation. Annu. Rev. Physiol. 1994;56:193–212. doi: 10.1146/annurev.ph.56.030194.001205. [DOI] [PubMed] [Google Scholar]
- 69.Li S, Shang Y. Regulation of SRC family coactivators by post-translational modifications. Cell Signal. 2007;19:1101–1112. doi: 10.1016/j.cellsig.2007.02.002. [DOI] [PubMed] [Google Scholar]
- 70.Liggett SB. Phosphorylation barcoding as a mechanism for directing GPCR signaling. Sci. Signal. 2011;4:pe36. doi: 10.1126/scisignal.2002331. [DOI] [PubMed] [Google Scholar]
- 71.Linder ME, Deschenes RE. Palmitoylation: policing protein stability and traffic. Nature Rev. Mol. Cell Biol. 2007;8:74–84. doi: 10.1038/nrm2084. [DOI] [PubMed] [Google Scholar]
- 72.Lisman JE. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc. Natl. Acad. Sci. USA. 1985;82:3055–3057. doi: 10.1073/pnas.82.9.3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lisman JE, Schulman H, Kline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- 74.Liu Z, Cao J, Gao X, Zhou Y, Wen L, Yang X, Yao X. CPLA 1.0: an integrated database of protein lysine acetylation. Nucleic Acids Res. 2011;39:D1029–D1034. doi: 10.1093/nar/gkq939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lonard DM, O’Malley BW. Nuclear receptor coregulators: judges, juries and executioners of cellular regulation. Mol. Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 76.Magrane M UniProt Consortium. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011;2011 doi: 10.1093/database/bar009. bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mandikian D, Cerda O, Sack JT, Trimmer JS. A SUMO-phospho tag team for wrestling with potassium channel gating. J. Gen. Physiol. 2011;137:435–439. doi: 10.1085/jgp.201110648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mann M, Ong S-E, Grønberg M, Steen H, Jensen ON, Pandey A. Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends Biotechnol. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 79.Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harbor Perspect. Biol. 2009;1 doi: 10.1101/cshperspect.a000950. a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Meinke MH, Bishop JS, Edstrom RD. Zero-order ultrasensitivity in the regulation of glycogen phosphorylase. Proc. Natl. Acad. Sci. USA. 1986;83:2865–2868. doi: 10.1073/pnas.83.9.2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Métivier R, Penot G, Hübner MR, Reid G, Brand H, Koš M, Gannon F. Estrogen receptor-α directs ordered, cyclical and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 82.Mohapatra DP, Park K-S, Trimmer JS. Dynamic regulation of the voltage-gated Kv2.1 potassium channel by multisite phosphorylation. Biochem. Soc. Trans. 2007;35:1064–1068. doi: 10.1042/BST0351064. [DOI] [PubMed] [Google Scholar]
- 83.Murray-Zmijewski F, Shue EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat. Rev. Mol. Cell Biol. 2008;9:702–712. doi: 10.1038/nrm2451. [DOI] [PubMed] [Google Scholar]
- 84.Nobles KM, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang T-Y, Bressler EA, Hara MR, Shenoy SK, gygi SP, Lefkowitz RJ. Distinct phosphorylation sites on the β2 adrenergic receptor establish a barcode that encodes differential functions ofβ-arrestin. Sci. Signal. 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.O’Malley BW. A life-long search for the molecular pathways of steroid hormone action. Mol. Endocrinol. 2005;19:1402–1411. doi: 10.1210/me.2004-0480. [DOI] [PubMed] [Google Scholar]
- 86.Park K-S, Mohapatra DP, Misonou H, Trimmer JS. Graded regulation of the Kv2.1 potassium channel by variable phosphorylation. Science. 2006;313:976–979. doi: 10.1126/science.1124254. [DOI] [PubMed] [Google Scholar]
- 87.Pi HJ, Lisman JE. Coupled phosphatase and kinase switches produce the tristability required for long-term potentiation and long-term depression. J. Neurosci. 2008;28:13132–13138. doi: 10.1523/JNEUROSCI.2348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pickart CM. Mechanisms underlying ubiquitination. Annu. Rev. Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- 89.Plant LD, Dowdell EJ, Dementieva IS, Marks JD, Goldstein SAN. SUMO modification of cell surface Kv2.1 potassium channels regulates the activity of rat hippocampal neurons. J. Gen. Physiol. 2011;137:441–454. doi: 10.1085/jgp.201110604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Prabhakaran S, Everley RA, Landrieu I, Wieruszeski JM, Lippens G, Steen H, Gunawardena J. Comparative analysis of erk phosphorylation suggests a mixed strategy for measuring phospho-form distributions. Mol. Syst. Biol. 2011;7:482. doi: 10.1038/msb.2011.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Pufall MA, Lee GM, Nelson ML, Kang H-S, Velyvis A, Kay LE, McIntosh LP, Graves BJ. Variable control of Ets-1 DNA binding by multiple phosphates in an unstructured region. Science. 2005;309:142–145. doi: 10.1126/science.1111915. [DOI] [PubMed] [Google Scholar]
- 92.Rogowski K, van Dijk J, Magiera MM, Bosc C, Deloulme J-C, Bosson A, Peris L, Gold ND, Lacroix B, Grau MB, Bec N, Larroque C, Desagher S, Holzer M, Andrieux A, Moutin M-J, Janke C. A family of protein-deglutamylating enzymes associated with neurodegeneration. Cell. 2010;143:564–578. doi: 10.1016/j.cell.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 93.Roth AF, W J, Bailey AO, Sun B, Kuchar JA, Green WN, Phinney BS, Yates JR, III, Davis NG. Global analysis of protein palmitoylation in yeast. Immunol. Cell Biol. 2006;125:1003–1013. doi: 10.1016/j.cell.2006.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Salazar C, Höfer T. Multisite protein phosphorylation—from molecular mechanisms to kinetic models. FEBS J. 2009;276:3177–3198. doi: 10.1111/j.1742-4658.2009.07027.x. [DOI] [PubMed] [Google Scholar]
- 95.Schreiber v, Dantzer F, Amé J-C, de Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 96.Schwartz DC, Hochstrasser M. A superfamily of protein tags: ubiquitin, SUMO and related modifiers. Trends Biochem. Sci. 2003;28:321–328. doi: 10.1016/S0968-0004(03)00113-0. [DOI] [PubMed] [Google Scholar]
- 97.Seet BT, Dikic I, Zhou M-M, Pawson T. Reading protein modifications with interaction domains. Nat. Rev. Mol. Cell Biol. 2006;7:473–483. doi: 10.1038/nrm1960. [DOI] [PubMed] [Google Scholar]
- 98.Sims RJ, Reinberg D. Is there a code embedded in proteins that is based on post-translational modification? Nat. Rev. Mol. Cell Biol. 2008;9:815–820. doi: 10.1038/nrm2502. [DOI] [PubMed] [Google Scholar]
- 99.Springer M, Wykoff DD, Miller N, O’Shea EK. Partially phosphorylated Pho4 activates transcription of a subset of phosphate-responsive genes. PLOS Biology. 2003;1:261–270. doi: 10.1371/journal.pbio.0000028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Stock AM, Robinson VL, Goudreau PN. Two component signal transduction. Annu. Rev. Biochem. 2000;69:183–215. doi: 10.1146/annurev.biochem.69.1.183. [DOI] [PubMed] [Google Scholar]
- 101.Surmeier DJ, Foehring R. A mechanism for homeostatic plasticity. Nat. Neurosci. 2004;7:691–692. doi: 10.1038/nn0704-691. [DOI] [PubMed] [Google Scholar]
- 102.Thomson M, Gunawardena J. Unlimited multistability in multisite phosphorylation systems. Nature. 2009;460:274–277. doi: 10.1038/nature08102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tischfield MA, Baris HN, Wu C, Rudolph L, Van Maldergem G, He W, Chan W-M, Andrews C, Demer JL, Robertson RL, Mackey DA, Ruddle JB, Bird TD, Gottlob I, Pieh C, Traboulsi EI, Pomeroy SL, Hunter DG, Soul JS, Newlin A, Sabol LJ, Doherty EJ, de Uzcátegui CE, de Uzcátegui N, Collins MLZ, Sener EC, Wabbels B, Hellebrand H, Meitinger T, de Berardinis T, Magli A, Schiavi C, Pastore-Trossello M, Koc F, Wong AM, Levin AV, Geraghty MT, Descartes M, Flaherty M, Jamieson RV, Mller HU, Meuthen I, Callen DF, Kerwin J, Lindsay S, Meindl A, Gupta ML, Jr, Pellman D, Engle EC. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell. 2010;140:74–87. doi: 10.1016/j.cell.2009.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tobin AB. G-protein coupled receptor phosphorylation: where, when and by whom. Br. J. Pharmacol. 2008;153:S167–S176. doi: 10.1038/sj.bjp.0707662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Turner B. Cellular memory and the histone code. Cell. 2002;111:285–291. doi: 10.1016/s0092-8674(02)01080-2. [DOI] [PubMed] [Google Scholar]
- 106.Walsh CT. Posttranslational Modification of Proteins. Englewood, Colorado: Roberts and Company; 2006. [Google Scholar]
- 107.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc. Natl. Acad. Sci. USA. 2008;105:13793–13798. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Westermann S, Weber K. Post-translational modifications regulate tubulin function. Nat. Rev. Mol. Cell Biol. 2003;4:938–947. doi: 10.1038/nrm1260. [DOI] [PubMed] [Google Scholar]
- 109.Wu H, Sun L, Zhang Y, Chen Y, Shi B, Li R, Wang Y, Liang J, Fan D, Wu G, Wang D, Li S, Shang Y. Coordinated regulation of AIB1 transcriptional activity by sumoylation and phosphorylation. J. Biol. Chem. 2006;281:21848–21856. doi: 10.1074/jbc.M603772200. [DOI] [PubMed] [Google Scholar]
- 110.Wu RC, Feng Q, Lonard DM, O’Malley BW. SRC 3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129:1125–1140. doi: 10.1016/j.cell.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 111.Wu RC, Qin J, Yi P, Wong J, Tsai SY, Tsai MJ, O’Malley BW. Selective phosphorylations of the SRC-3/AIB1 coactivator integrate genomic responses to multiple cellular signaling pathways. Mol. Cell. 2004;15:937–949. doi: 10.1016/j.molcel.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 112.Xia L, Hai B, Gao Y, Burnette D, Thazhath R, Duan J, Br M-H, Levilliers N, Gorovsky MA, Gaertig J. Polyglycylation of tubulin is essential and affects cell motility and division in Tetrahymena thermophila. J. Cell Biol. 2000;149:1097–1106. doi: 10.1083/jcb.149.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Xu P, Peng J. Dissecting the ubiquitin pathway by mass spectrometry. Biochim. Biophys. Acta. 2006;1764:1940–1947. doi: 10.1016/j.bbapap.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu Y, Gunawardena J. Realistic enzymology for post-translational modification: zero-order ultrasensitivity revisited. Submitted. 2011 doi: 10.1016/j.jtbi.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yang X-J. Multisite protein modification and intramolecular signaling. Oncogene. 2005;24:1653–1662. doi: 10.1038/sj.onc.1208173. [DOI] [PubMed] [Google Scholar]
- 116.York B, O’Malley BW. Steroid receptor coactivator (SRC) family: masters of systems biology. J. Biol. Chem. 2010;285:38743–38750. doi: 10.1074/jbc.R110.193367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.York B, Yu C, Sagen JV, Liu Z, Nikolai BC, Wu R-C, Finegold M, Xu J, O’Malley BW. Reprogramming the posttranslational code of SRC-3 confers a switch in mammalian systems biology. Proc. Natl. Acad. Sci. USA. 2010;107:11122–11127. doi: 10.1073/pnas.1005262107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yudushkin IA, Schleifenbaum A, Kinkhabwala A, Neel BG, Schultz C, Bastiaens PIH. Live-cell imaging of enzyme-substrate interaction reveals spatial regulation of PTP1B. Science. 2007;315:115–119. doi: 10.1126/science.1134966. [DOI] [PubMed] [Google Scholar]
- 119.Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, Vocadlo DJ. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008;4:483–490. doi: 10.1038/nchembio.96. [DOI] [PubMed] [Google Scholar]
- 120.Zachara NE, Hart GW. The emerging significance of O-GlcNAc in cellular regulation. Chem. Rev. 2002;102:431–438. doi: 10.1021/cr000406u. [DOI] [PubMed] [Google Scholar]
- 121.Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, Li Y, Shi J, An W, Hancock SM, He F, Qin L, Chin J, Yang P, Chen X, Lei Q, Xiong Y, Guan K-L. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]