

Abstract
The accumulation of β-amyloid protein (Aβ) is a key risk factor in the development of Alzheimer's disease. The ovarian sex steroid hormones 17β-estradiol (E2) and progesterone (P4) have been shown to regulate Aβ accumulation, although the underlying mechanism(s) remain to be fully elucidated. In this study, we investigate the effects of E2 and P4 treatment on the expression levels of Aβ clearance factors including insulin-degrading enzyme, neprilysin, endothelin-converting enzyme 1 and 2, angiotensin-converting enzyme, and transthyretin, both in primary neuron cultures and female rat brains. Our results show that E2 and P4 affect the expression levels of several Aβ clearance factors in dose- and time-dependent manners. Most notably, expression of insulin-degrading enzyme is significantly increased by both hormones in cultured neurons and in vivo and is inversely associated with the soluble Aβ levels in vivo. These findings further define sex steroid hormone actions involved in regulation of Aβ, a relationship potentially important to therapeutic approaches aimed at reducing risk of Alzheimer's disease.
Alzheimer's disease (AD) is an age-related neurodegenerative disorder that is the leading cause of dementia. Despite the strong association between increasing age and AD risk (1), the changes underlying this relationship are not well defined. One normal age change that has been identified as an AD risk factor is the depletion of sex steroid hormones (2). The relatively abrupt reduction in estrogens and progesterone (P4) at menopause has been theorized to contribute to the increased prevalence (3) and incidence (4, 5) of AD and worsened pathological (6) and clinical presentations (7, 8) of the disease in women. Among postmenopausal women, those with AD are characterized by lower 17β-estradiol (E2) levels in plasma (9), brain (10, 11), and cerebrospinal fluid (12). Further, estrogen-based hormone therapy in postmenopausal women is associated with a reduced risk of AD (13, 14), although this field remains controversial (15, 16) and may require initiation of treatment in middle age (17).
How estrogens and perhaps P4 alter AD risk is not known. The critical molecule in initiating and driving AD neuropathology is widely hypothesized to be β-amyloid protein (Aβ), a peptide that accumulates predominantly in hippocampus and select cerebrocortical regions of brain forming toxic oligomers and extracellular deposits (18). Increasing evidence suggests that sex steroid hormones are significant regulators of Aβ levels. For example, ovariectomy-induced depletion of E2 and P4 can result in elevated Aβ levels in wild-type rodents (19) and transgenic mouse models of AD (11, 20), an effect prevented by treatment with E2 (19, 21) and, in some cases, P4 (22). Peripheral manipulations of E2 do not affect Aβ levels in some animal models (23), suggesting maintained neural levels of E2 by brain-derived mechanisms (11). Normally, Aβ accumulation is prevented by a tightly regulated balance between its production and clearance. Prior work indicates that E2 may reduce Aβ production in part by regulating the processing and/or trafficking of its parent protein, amyloid precursor protein (24, 25).
Less well understood are the roles of E2 and P4 in regulating the clearance of Aβ. An important clearance mechanism is enzyme-mediated degradation of Aβ. Aβ-degrading enzymes are expressed in neurons and are present in the hippocampal and cortical regions of the brain (26). Proteases implicated in Aβ degradation include insulin-degrading enzyme (IDE), neprilysin (NEP), endothelin-converting enzymes-1 (ECE1) and -2 (ECE2), and angiotensin-converting enzyme (ACE) (27). IDE is the most abundant secreted Aβ-degrading enzyme and is significantly involved in degradation of monomeric Aβ (28, 29). Genetic deletion of IDE results in elevated brain levels of Aβ (30), whereas IDE overexpression reduces Aβ deposition (28, 29, 31). NEP plays an important role in the clearance of Aβ (32, 33) but, unlike IDE, NEP is able to degrade oligomeric Aβ (34). Recent evidence suggests NEP may be particularly important to AD because its expression is inversely correlated with levels of both Aβ and cognitive impairment (35). Although IDE and NEP are often regarded as the key Aβ-degrading enzymes, there is compelling evidence that several other factors also may significantly contribute to Aβ degradation. Both ECE1 and ECE2 have been identified as Aβ-cleaving enzymes capable of reducing Aβ levels in in vitro, cell culture, and/or animal paradigms (36, 37). ACE has also been shown to degrade secreted Aβ, and inhibition of ACE activity by either genetic or pharmacological approaches leads to elevated Aβ (38, 39). In addition to Aβ-degrading enzymes, Aβ clearance can be facilitated by other factors such as transthyretin (TTR), which binds Aβ and can lower Aβ burden in transgenic mouse models of AD (26, 40).
To further define the relationship between ovarian hormones and regulation of Aβ, we investigated the effects of E2 and P4 on the expression of IDE, NEP, ECE1, ECE2, ACE, and TTR. Three complementary experimental paradigms were used. First, we performed initial characterization and mechanistic investigation using short-term E2 and P4 treatments in primary neuron cultures. Next, we examined the effects of short-term E2 and P4 treatments in vivo to evaluate the extent to which culture observations extrapolate to the organism level. Finally, we performed long-term studies in vivo to model the effects of extended treatment regimens associated with hormone therapies. Importantly, because hormone therapies in postmenopausal women typically involve combinations of estrogens and progestogens, our long-term in vivo studies compared both individual and combined actions of E2 and P4. In addition, we evaluated brain levels of soluble Aβ in hormone-treated rats to provide insight into the relationships between E2 and P4 regulation of the Aβ clearance factors and the corresponding accumulation of Aβ.
Materials and Methods
Materials
E2 was purchased from Steraloids, Inc. (Newport, RI) and progesterone (P4) was purchased from Acros Organics USA (Morris Plains, NJ). Antiestrogen ICI 182,780, estrogen receptor (ER) α-agonist propylpyrazole triol (PPT), and ERβ agonist 2,3-bis(4-hydroxyphenyl) propionitrile (DPN) were acquired from Tocris (Ellisville, MO). Progesterone receptor (PR) antagonists RU 486 was purchased from Sigma-Aldrich (St. Louis, MO), and Org 31710 was generously provided by N.V. Organon (Oss, The Netherlands). Stock solutions of all compounds were prepared in 100% ethanol.
Animals
For cell culture studies, timed-pregnant female Sprague Dawley rats (Harlan Laboratories, Inc., Livermore, CA) were killed via CO2 inhalation, and the pups were harvested for preparation of neuronal cultures. For in vivo studies, female Sprague Dawley rats were purchased either bilaterally ovariectomized (OVX) or sham-OVX at 3 months of age (Harlan Laboratories, Inc.). All animals were housed individually with ad libitum access to food and water under a 12 h-light, 12-h dark light cycle. All animal procedures were conducted under a protocol that was approved by the University of Southern California and in accordance with National Institute of Health standards.
Primary neuron culture
Neuron-enriched, primary rat cerebrocortical cultures (∼95% neuronal as determined by positive immunoreactivity with the neuron-specific antibody NeuN) were prepared with some modifications of a previously described protocol (41). Briefly, cerebral cortices were dissected from gestational day 17–18 Sprague Dawley rat pups (n ≥ 6 pups per preparation). Cortices were enzymatically dissociated using 0.25% trypsin at 37 C for 5 min. The reaction was quenched using two volumes of DMEM (American Type Culture Collection; Manassas, VA) containing 10% (vol/vol) fetal bovine serum. The tissue was centrifuged at 200 × g and the pellet was resuspended. The resultant cell suspension was mechanically dissociated using flame-polished glass Pasteur pipettes and then filtered through a 40-μm cell strainer (Falcon, Franklin Lakes, NJ). The single-cell suspension was diluted using DMEM containing N2 supplements (without P4) and plated onto poly-l-lysine-coated multiwell plates at a final density of 8 × 105 cells/cm2. Cultures were maintained at 37 C in a humidified incubator supplemented with 5% CO2. All experiments were started after 1–2 d in vitro and were repeated in three to five independent culture preparations. Cultures were treated with ethanol vehicle or various combinations of E2, P4, and ER and PR agonists and antagonists that were diluted from ethanol stock solutions with culture medium to yield a final ethanol concentration of ≥0.01%.
Hormone treatments in animals
In the short-term experiment, rats were randomly assigned to four groups (n = 7/group): sham OVX + vehicle (Sham); OVX + vehicle (OVX); OVX + 17β-estradiol (OVX+E2); and OVX + progesterone (OVX+P4). Treatments were administered via two injections, the first injection 7 d after sham OVX or OVX procedure and the second injection 24 h later. Injections contained either vehicle (100 μl canola oil), 10 μg E2 (100 μl of 100 μg/ml E2 in canola oil), or 500 μg P4 (100 μl of 5 mg/ml P4 in canola oil), doses previously demonstrated to yield physiological levels of E2 and P4 in OVX rats (42). Tissues were collected 24 h after the second injection.
For the long-term experiment, rats were either sham OVX (Sham, n = 8) or OVX. OVX rats were randomly assigned to one of six groups (n = 8/group): placebo (OVX), continuous E2 (OVX+E2), continuous P4 (OVX+P4cont), cyclic P4 (OVX+P4cyc), continuous E2 with continuous P4 (OVX+E2+P4cont), or continuous E2 with cyclic P4 (OVX+E2+P4cyc). Rats were treated with two consecutive 30-d cycles of hormone treatment (60 d total) initiated 7 d post-OVX and delivered via slow-release sc implants (Innovative Research America, Sarasota, FL). On d 0, Sham and OVX groups were implanted with placebo pellets; each E2-treated rat was implanted with a 0.72-mg E2 90-d release pellet; each continuous P4-treated rat was implanted with a 450-mg P4 90-d release pellet; each cyclic P4 rat was implanted with one 50-mg P4 10-d release pellet at d 20 and a second pellet at d 50. The efficacy of this cyclic P4 regimen has been demonstrated previously (22). After the treatment period, the rats were killed, and each brain was rapidly dissected and bisected midsagitally. The hippocampus from one hemisphere was snap frozen for use in RNA and protein extractions, and the other entire hemisphere was snap frozen for use in β-amyloid ELISA. Uteri were dissected, blotted, and weighed as a bioassay of estrogen levels.
RT-PCR and quantitative PCR
For RNA extractions in all experiments, treated cells and tissues were lysed using TRIzol reagent (Invitrogen, Carlsbad, CA) and processed for total RNA extraction as per manufacturer's protocol. Purified RNA (1–2 μg) was used for reverse transcription using the Superscript First strand synthesis system (Invitrogen) as described previously (43), and the resulting cDNA was used for both standard PCR and real-time quantitative PCR. Quantitative PCR was carried out using DNA Engine Opticon 2 continuous fluorescence detector (MJ Research, Inc., Waltham, MA). The amplification efficiency was estimated from the standard curve for each gene. Relative quantification of mRNA levels from various treated samples was determined by the ΔΔCt method (44). The following primer pairs were used. IDE: forward, 5′-GGAAGCGTTCGCCGAGATCGCA-3′; reverse, 5′-TCTGAATCGACAGCGTTCAC-3′; NEP: forward; 5′-CATTGAACTATGGGGGCATC-3′; reverse, 5′-CCTGAAATTGCCAGGACTGT-3′; ECE1: forward, 5′-GAGCTGACTCATGCTTTC-3′; reverse, 5′-CAGCTCCGTTCTTCTTTA-3′; ECE2: forward, 5′-AGAAAGTTCTCGCTGCCT-3′; reverse, 5′-AGTGGCGACAACAAGAAA-3′; ACE: forward, 5′-GAGCCATCCTTCCCTTTT-3′; reverse, 5′-GGCTGCAGCTCCTGGTAT-3′; TTR: forward, 5′-GGCTCACCACAGATGAGA-3′; reverse, 5′-ACAAATGGGAGCTACTGC-3′; β-actin: forward, 5′-AGCCATGTACGTAGCCATCC-3′; reverse, 5′-CTCTCAGCTGTGGTGGTGAA-3′.
Western blots
For all experiments involving protein analysis by immunoblotting, treated cultures and tissues were lysed using a reducing sample buffer (62.5 mm Tris-HCl, 1% sodium dodecyl sulfate, 2.5% glycerol, 0.5% 2-β-mercaptoethanol), boiled for 5 min at 100 C, and centrifuged at 13,000 × g for 10 min. The resultant supernatants were used for Western blot analysis using a standard protocol previously described (41). Briefly, equal sample amounts were electrophoresed in 10% polyacrylamide gels and transferred onto polyvinylidene difluoride membranes (Millipore Corp., Medford, MA) at constant 100 V for 1 h. The membranes were rinsed in blocking solution (5% BSA in 10 mm Tris, 100 mm NaCl, 0.1% Tween, pH 7.4) for 1 h at room temperature (RT), followed by incubation with primary antibody (α-IDE, Abcam; San Francisco, CA) diluted in blocking solution for 1 h at RT. The membranes were then incubated with corresponding horseradish peroxidase-conjugated secondary antibody for 1 h at RT and detected using ECL (Amersham; Arlington Heights, IL).
β-Amyloid ELISA
Brain levels of soluble Aβ1–42 were determined by ELISA with modifications of a previously described protocol (45). In brief, hemi-brains were processed for Aβ ELISA after extracting soluble protein by homogenization in ice-cold DEA buffer (0.2% diethylamine, 50 mm NaCl; 1 ml/200 mg tissue) with complete protease inhibitor cocktail (Amresco, Solon, OH) using an AHS200 PowerMax polytron. Homogenates were centrifuged at 20,800 × g at 4 C for 30 min, after which the supernatants were collected and neutralized (one tenth volume of 0.5 m Tris-HCl, pH 6.2). Levels of soluble Aβ were then measured via sandwich capture ELISA using a Colorimetric BetaMark β-Amyloid x-42 ELISA kit (Covance Laboratories, Inc., Princeton, NJ).
Statistical analyses
Raw data from all experiments were assessed by ANOVA using GraphPad Prism (version 5.0; GraphPad Software, Inc., San Diego, CA). For analyses showing significant main effects, between-groups comparisons were made using the Tukey honestly significant difference (HSD) test. Effects with P < 0.05 were considered significant.
Results
E2 and P4 regulate mRNA levels of Aβ clearance factors in primary neuron cultures
To investigate the effects of E2 on expression levels of factors involved in Aβ degradation and clearance, primary neuron cultures were treated for 24 h with increasing concentrations of E2 (0–100 nm). RNA was isolated from cultures and processed for PCR using specific primers for IDE, NEP, ACE, ECE1, ECE2, and TTR. Our results qualitatively and quantitatively show that E2 induced a dose-dependent increase in mRNA levels of IDE [F (5, 12) = 6.1, P < 0.01], significantly decreased expression of ACE [F(5,12) = 6.1, P < 0.01] and ECE2 [F(5, 12) = 14.1, P < 0.001], and had no significant effect on mRNA levels of NEP, ECE, and transthyretin (TTR) (Fig. 1, A and B). E2 effects were statistically significant at the 10 nm and 100 nm concentrations. To investigate the time courses of E2 regulatory actions, we treated neuron cultures with 10 nm E2 for increasing periods of time up to 24 h. The E2-induced increase in IDE [F (5, 12) = 22.4, P < 0.001], and decreases in ACE [F(5,12) = 12.7, P < 0.01], and ECE2 [F(5,12) = 11.1, P < 0.01], became apparent within 8 h of treatment and statistically significant by 16 h (Fig. 1, C and D).
Fig. 1.
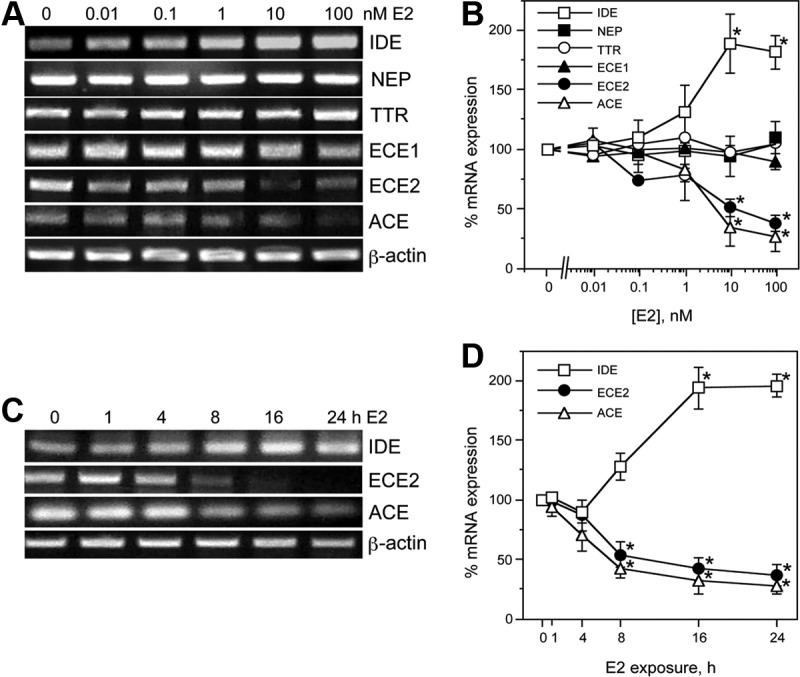
E2 regulates expression of Aβ clearance factors in a dose- and time-dependent manner. Representative agarose gel of RT-PCR products show relative changes in mRNA levels of the Aβ clearance factors TTR, IDE, ACE, ECE1, NEP, and ECE2 induced by 24-h exposure to 0–100 nm E2. β-Actin was used as an internal control. B, The levels of Aβ clearance factors after treatment with increasing E2 concentrations were also determined quantitatively using real-time PCR. Data show the mean (±sem) expression levels, relative to vehicle-treated controls after normalizing with corresponding values of β-actin. Representative agarose gel of RT-PCR (C) and quantitative graph from real-time PCR (D) show changes in levels of IDE, ACE, and ECE2 mRNA induced by 0–24 h exposure to 10 nm E2. Statistical significance is based on analysis of pooled raw data using the Tukey HSD. *, P ≤ 0.05 relative to corresponding vehicle-treated control group.
In parallel experiments, we similarly evaluated the effects of P4 on expression of IDE, NEP, ACE, ECE1, ECE2, and TTR. Treatment of neuron cultures for 24 h with P4 concentrations of 3 nm, 30 nm, and 300 nm resulted in statistically significant, approximately 2-fold increases of IDE [F(5, 12) = 9.6, P < 0.001], ACE [F(5,12) = 7.0, P < 0.01], and TTR [F(5,12) = 52.4, P < 0.001] mRNA levels (Fig. 2, A and B). Time course analyses after treatment with 30 nm P4 revealed that ACE mRNA levels were significantly elevated within 4 h and began to decline by 24 h [F(5,12) = 67.7, P < 0.001], whereas mRNA levels of both TTR [F(5,12) = 12.3, P < 0.01] and IDE [F(5,12) = 15.1, P < 0.01] increased more gradually, reaching statistical significance by 8 h and 16 h, respectively (Fig. 2, C and D).
Fig. 2.

P4 regulates expression of Aβ clearance factors in a dose- and time-dependent manner. A, Representative agarose gel of RT-PCR products show relative changes in mRNA levels of the Aβ clearance factors TTR, IDE, ACE, ECE1, NEP, and ECE2 induced by 16-h exposure to 0–300 nm P4. β-Actin was used as an internal control. B, The levels of Aβ clearance factors after treatment with increasing P4 concentrations were also determined quantitatively using real-time PCR. Data show the mean (±sem) expression levels, relative to vehicle-treated controls after normalizing with corresponding values of β-actin. Representative agarose gel (C) and quantitative graph from real-time PCR (D) show changes in levels of IDE, ACE, and TTR mRNA induced by 0–24 h exposure to 30 nm P4. Statistical significance is based on analysis of pooled raw data using the Tukey HSD. *, P ≤ 0.05 relative to corresponding vehicle-treated control group.
The role of hormone receptors in E2 and P4 regulation of Aβ clearance factors
To investigate the contributions of estrogen receptors (ER) to the observed E2 regulation of IDE, ACE, and ECE2 expression, we first evaluated the effect of the antiestrogen ICI 182,780 (46). Neuron cultures were pretreated with 1 μm ICI 182,780 for 1 h followed by 10 nm E2 or vehicle treatments for 24 h. Cultures were harvested for RNA followed by qualitative and quantitative PCR. Our results demonstrate ICI 182,780 completely blocks the E2-mediated increase in IDE mRNA, suggesting an ER-dependent mechanism (Fig. 3, A and B). In contrast, the E2-induced down-regulation of ACE and ECE2 mRNA levels remained unchanged in the presence of ICI 182,780, suggesting that these effects occur via either ER-independent pathways or nongenomic mechanisms that are ER dependent but insensitive to ICI 182,780 (Fig. 3, A and B). To gain further insight into the role of ER, we treated neuron cultures for 24 h with increasing concentrations (0–100 nm) of PPT (47) and DPN (48), agonists that are relatively selective for ERα and ERβ, respectively. PCR analyses indicate that both PPT [F(3,8) = 12.3, P < 0.01] and DPN [F(3,8) = 11.7, P < 0.01], significantly increased IDE mRNA levels, but neither ER agonist significantly altered ACE [F(3,8) = 1.0, P = 0.43; F(3,8) = 0.3, P = 0.81] and ECE2 [F(3, 8) = 0.4, P = 0.78; F(3,8) = 1.5, P = 0.30] mRNA expression (Fig. 3, C–F).
Fig. 3.

Effects of ER agonists and ER and PR antagonists on E2 and P4 regulation of Aβ clearance factors. A, Representative agarose gel of RT-PCR products and (B) quantitative real-time PCR data show the effect of the ER-antagonist ICI 182,780 on E2-mediated changes in the mRNA levels of IDE, ACE, and ECE2. β-Actin was used as an internal control. Representative agarose gel (panel C) and quantitative real-time PCR data (panel D) show the effect of 0–100 nm PPT, an ERα-agonist, on the levels of IDE, ACE, and ECE2 mRNA. Representative agarose gel (panel E) and quantitative real-time PCR data (panel F) show the effect of DPN, an ERβ-agonist, on the mRNA levels of IDE, ACE, and ECE2. (G) Representative agarose gel (panel G) and quantitative real-time PCR graph (panel H) show the effects of two PR antagonists, RU486 and Org 31710, on the mRNA levels of IDE, ACE, and TTR. Data show the mean (±sem) expression levels, relative to vehicle-treated controls after normalizing with corresponding values of β-actin. Statistical significance is based on analysis of pooled raw data using the Tukey HSD. *, P ≤ 0.05 relative to corresponding vehicle-treated control group. ICI, ICI 182,780; Org, Org 31710; Ru, RU486.
To evaluate the role of PR on P4-induced regulation of IDE, ACE, and TTR mRNA, we used the PR antagonists RU486 (49) and Org 31710 (50). Neuron cultures were pretreated with vehicle, 50 nm RU486, or 1 μm Org 31710 followed by 16-h exposure to 30 nm P4 and then harvested for RNA isolation. Our PCR results show that P4-induced increases in mRNA levels of IDE and ACE mRNA levels were not significantly altered in the presence of the PR antagonists. Conversely, both RU486 and Org 31710 effectively blocked the up-regulation of TTR mRNA levels by P4 (Fig. 3, G and H).
E2 and P4 regulate levels of Aβ clearance factors in rat brain
As an initial step to determine whether significant neuronal culture observations extrapolate to brain, we examined the effects of short-term exposures of E2 and P4 on mRNA levels of IDE, ACE, ECE2, and TTR in female rat brain. Young adult, female Sprague Dawley rats were sham OVX or OVX to deplete endogenous E2 and P4. One week after surgery, rats were injected twice with vehicle (canola oil), 10 μg E2, or 500 μg P4, once at time 0 h and again 24 h later. Rats were killed 24 h after the second injection, at which time the brains were immediately collected, frontal cortices were dissected, and RNA was isolated for PCR analyses. Analysis of uterine weight confirmed efficacy of the OVX procedure in depleting endogenous estrogens and the efficacy of E2 treatment in restoring uterine mass (Table 1). We observed statistically significant effects of the hormone manipulations on mRNA levels of IDE [F(3,8) = 11.0, P < 0.01], ACE [F(3,8) = 4.9, P = 0.03], and ECE2 [F(3,8) = 9.9, P < 0.01], but not on TTR levels [F(3,8) = 0.6, P = 0.66]. In comparison with sham controls, the OVX group showed nonsignificant trends of reduced IDE mRNA and increased ACE mRNA (Fig 4, A and B) and a statistically significant increase in ECE2 mRNA (Fig. 4, A, C, and D). The OVX+E2 group showed significantly elevated levels of IDE and reduced levels of ACE and ECE2 relative to the OVX group (Fig. 4, A–D). Similarly, the OVX+P4 group was associated with significantly increased IDE and decreased ECE2 mRNA in comparison with the OVX group, but P4 treatment exhibited no significant effect on ACE levels (Fig. 4, A–D).
Table 1.
Uterine weights across treatment groups
| Treatment group | Study length | Uterine weight (g) |
|---|---|---|
| Sham OVX | Short term | 0.249 ± 0.022 |
| OVX | Short term | 0.094 ± 0.015a |
| OVX+E2 | Short term | 0.234 ± 0.007 |
| OVX+P4 | Short term | 0.096 ± 0.004a |
| Sham OVX | Long term | 0.503 ± 0.022 |
| OVX | Long term | 0.099 ± 0.003b |
| OVX+E2 | Long term | 0.567 ± 0.043 |
| OVX+P4co | Long term | 0.090 ± 0.008b |
| OVX+P4cy | Long term | 0.087 ± 0.006b |
| OVX+E2+P4co | Long term | 0.645 ± 0.036 |
| OVX+E2+P4cy | Long term | 0.520 ± 0.037 |
Data expressed as means ± sem.
P < 0.05 relative to Sham OVX (short term);
P < 0.05 relative to Sham OVX (long term)
Fig. 4.
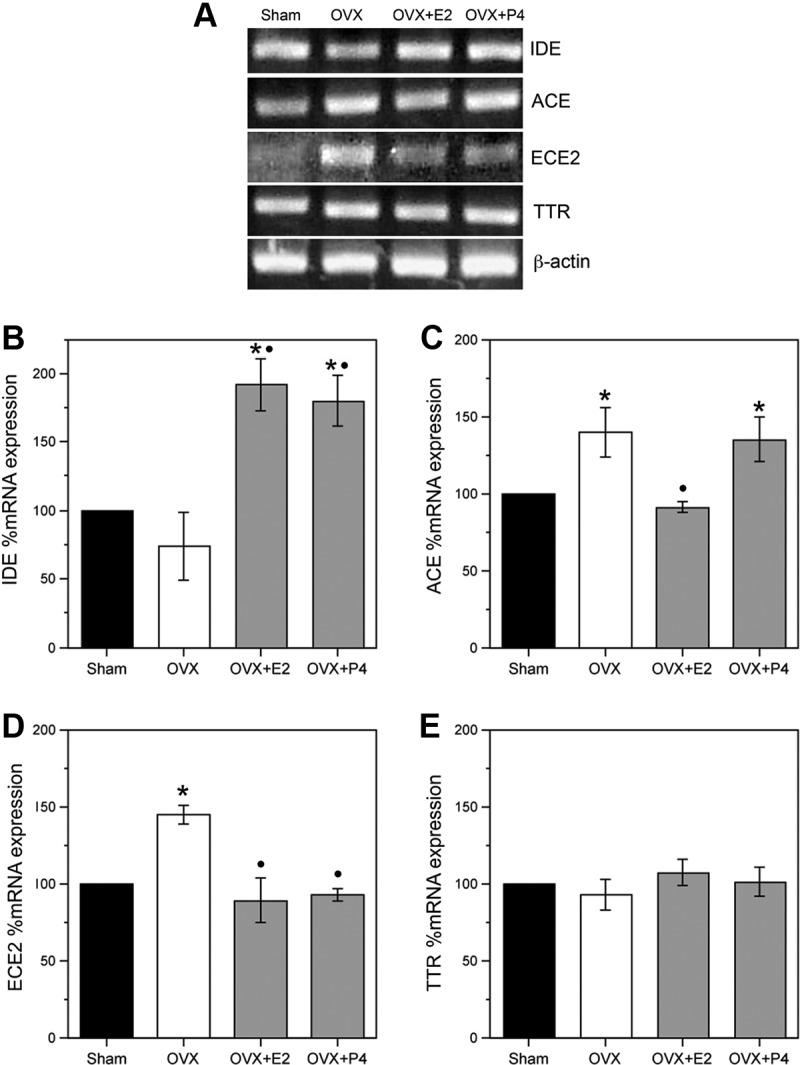
Effects of short-term in vivo hormone treatments on levels of Aβ clearance factors. A, Representative agarose gel of RT-PCR products shows the relative levels of IDE, ACE, ECE2, and TTR mRNA in sham OVX (Sham), vehicle-treated OVX (OVX), and OVX rats after short-term treatment with E2 (OVX+E2) or P4 (OVX+P4). B–E, Quantitative real-time PCR data show the mean (±sem) expression levels compared with the Sham control group for IDE, ACE, ECE2, and TTR mRNA, respectively. All data are normalized with corresponding β-actin values. Statistical significance is based on analysis of pooled raw data using the Tukey HSD. *, P < 0.01 relative to the vehicle-treated Sham group. •, P < 0.01 relative to the OVX group.
We next evaluated the individual and combined effects of E2 and P4 over an extended treatment period. Consistent with our previously established protocol (22), we exposed OVX female rats to two consecutive, 30-d cycles of hormone treatment consisting of continuous E2, continuous P4, cyclic P4 (10 d/cycle), or combinations of E2 with continuous or cyclic P4. As with the short-term experiment, uterine weights were significantly reduced by OVX, an effect reversed in OVX groups treated with E2 (Table 1). Expression of IDE mRNA significantly differed across groups [F(6,21) = 15.3, P < 0.001]. Neocortical IDE mRNA was significantly decreased in the OVX group, an effect that was prevented by treatment of OVX rats with continuous E2 (OVX+E2) and cyclic P4 (OVX+Pcyc) but not by continuous P4 (OVX+P4cont). Notably, the effect of combining E2 and P4 depended upon the delivery regimen of P4. The up-regulation of IDE mRNA by continuous E2 was blocked by cotreatment with continuous P4 (OVX+E2+P4cont) but not by cyclic P4 (OVX+E2+P4cyc) (Fig 5, A and B). There were also significant treatment effects on mRNA levels of ACE [F(6,21) = 10.9, P < 0.001]. In comparison with the Sham group, OVX was associated with a significant increase in ACE mRNA that was significantly attenuated in the OVX+E2 group. Cyclic P4 (OVX+Pcyc) did not significantly reduce ACE mRNA, but continuous P4 (OVX+P4cont) had an intermediate effect yielding ACE mRNA levels that were not significantly different from either Sham OVX or OVX (Fig. 5, A and C). Both P4 treatments significantly inhibited the effect of E2 on ACE mRNA (Fig. 5, A and C). There was no statistically significant main effect of treatment group on ECE2 mRNA levels [F(6,14) = 1.5, P = 0.24] (Fig. 5, A and D). Levels of TTR mRNA levels significantly differed across groups [F(6,21) = 5.5, P < 0.01]. Although there were no significant effects of OVX or E2 treatment, there was a modest increase in TTR mRNA in the OVX+P4cyc group relative to both Sham and OVX groups (Fig. 5, A and E).
Fig. 5.

Effects of long-term in vivo hormone treatments on levels of Aβ clearance factors. A, Representative agarose gel of RT-PCR products qualitatively shows changes in the levels of IDE, ACE, ECE2, and TTR mRNA across the following treatment groups (n = 8/group): vehicle-treated Sham OVX (Sham), vehicle-treated OVX (OVX), and OVX treated with continuous E2 (OVX+E2), continuous P4 (OVX+P4co), cyclic P4 (OVX+P4 cy), or continuous E2 combined with either continuous P4 (OVX+E2+P4co) or cyclic P4 (OVX+E2+P4cy). B–E, Quantitative real-time PCR data show the mean (±sem) expression levels compared with the Sham OVX control group (solid bar) for IDE, ACE, ECE2, and TTR mRNA, respectively. All data are normalized with corresponding β-actin values. Statistical significance is based on analysis of pooled raw data using the Tukey HSD. *, P < 0.01 relative to the vehicle-treated Sham OVX group. •, P < 0.01 relative to the OVX group.
E2 and P4 regulate IDE protein in vitro and in vivo
The only Aβ clearance factor that was positively regulated at the mRNA level by E2 and/or P4 across all of our cell culture and in vivo paradigms was IDE. To confirm that the observed up-regulation of IDE mRNA by E2 and P4 yielded increased protein levels of IDE, we conducted Western blots in both cell culture and brain samples. In neuronal cultures, E2 increased IDE protein in a dose-dependent manner by up to 2-fold with statistically significant effects apparent at 0.1 nm (Fig. 6, A and F) [F(5,12) = 92.6, P < 0.001]. In cultures treated with 10 nm E2, significant increases in IDE protein occurred within 8 h and were retained across the 48-h experimental period [F(4,10) = 142.1, P < 0.001] (Fig. 6, B and G). Similarly, P4 induced increased IDE protein with significant effects observed between concentrations of 0.3 nm and 300 nm [F(5,12) = 33.4, P < 0.001] (Fig. 6, C and H) and at exposure times between 8 h and 48 h [F(4,10) = 23.9, P < 0.001] (Fig. 6, D and I). In female rats, IDE levels were significantly affected by treatment [F(3,8) = 20.4, P < 0.001]. We observed that the OVX group exhibited a nonsignificant trend of reduced IDE protein relative to the Sham group. Short-term treatment of OVX rats with E2 (OVX+E2) or P4 (OVX+P4) yielded significant increases in IDE protein levels, with the OVX+E2 group increasing IDE levels significantly higher than the Sham group (Fig. 6, E and J).
Fig. 6.

Effects of E2 and P4 on IDE expression levels. Representative Western blots show regulation of IDE protein levels by E2 and P4 treatment in cultured neurons and OVX rats. A, Dose-dependent regulation of IDE by 0–100 nm E2 in cultured neurons is demonstrated by a representative Western blot (upper panel) and quantification of combined data across experiments (lower panel). B, Time-dependent regulation of IDE in cultured neurons by 10 nm E2 over 0–48 h is demonstrated by a representative Western blot (upper panel) and quantification of combined data across experiments (lower panel). C, Dose-dependent regulation of IDE by 0–300 nm P4 in cultured neurons is demonstrated by a representative Western blot (upper panel) and quantification of combined data across experiments (lower panel). D, Time-dependent regulation of IDE in cultured neurons by 30 nm P4 over 0–48 h is demonstrated by a representative Western blot (upper panel) and quantification of combined data across experiments (lower panel). Relative amounts of IDE protein levels were determined by densitometric scanning of Western blots from three independent experiments. E, Effect of OVX and short-term treatment of OVX rats with E2 (OVX+E2) or P4 (OVX+P4) on IDE protein levels is shown by a representative Western blot (upper panel) and quantification of Western blot data from all animals (n = 7/group) (lower panel). Data are represented as a mean (±sem) percentage of control values. Statistical significance is based on analysis of pooled raw data using the Tukey HSD. *, P < 0.01 relative to vehicle-treated control groups or Sham OVX group. •, P < 0.01 relative to the OVX group.
Long-term regulation of soluble Aβ levels by E2 and P4
The significant positive regulation of the Aβ degrading enzyme IDE by both E2 and P4 across culture and in vivo paradigms suggests a possible role in regulating brain levels of Aβ. To begin investigating this possibility, we homogenized one hemi-brain from each of the rats in the extended hormone treatment experiment to analyze soluble Aβ levels by ELISA. Our results indicate a statistically significant effect of treatment on Aβ42 levels [F(3,28) = 3.3, P = 0.01]. Ovarian hormone depletion associated with OVX resulted in a significant, approximately 2-fold increase in Aβ that was largely prevented by continuous E2 treatment (OVX+E2) (Fig. 7). Treatment with P4 alone delivered either continuously (OVX+P4cont) or cyclically (OVX+P4cyc) did not significantly lower Aβ levels relative to the OVX group. Interestingly, the regimen of cyclic P4 in combination with E2 (OVX+E2+P4cyc) showed the lowest Aβ42 levels.
Fig. 7.
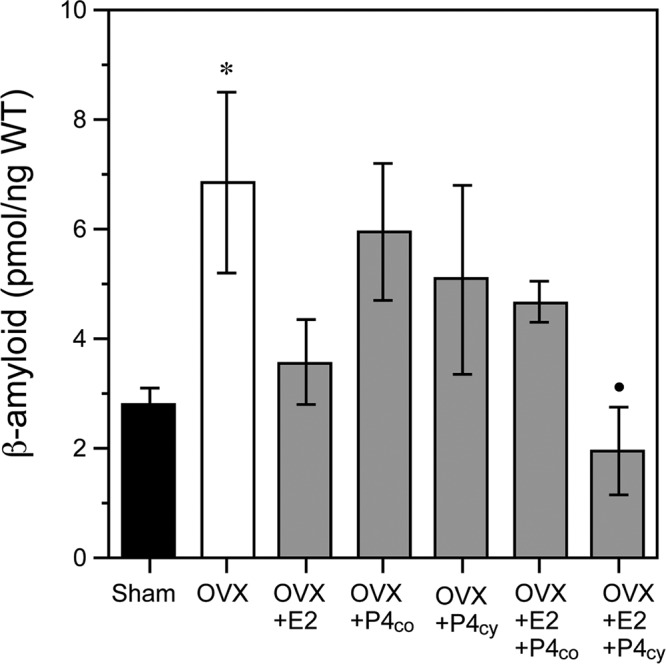
Effects of long-term hormonal treatments on soluble Aβ levels in vivo. Levels of soluble Aβ in vivo were determined by Aβ ELISA after long-term hormone manipulations. Data show mean (±sem) Aβ1–42 levels from hemi-brain lysates of adult female rats under the following treatment groups (n = 8/group): vehicle-treated sham OVX (Sham; solid bar), vehicle-treated OVX (OVX; open bar), and OVX treated with continuous E2 (OVX+E2), continuous P4 (OVX+P4co), cyclic P4cy (OVX+P4 cy) or continuous E2 combined with either continuous P4 (OVX+E2+P4co) or cyclic P4 (OVX+E2+P4cy). Statistical significance is based on analysis of pooled raw data using the Tukey HSD. *, P ≤ 0.05 relative to Sham OVX group. •, P ≤ 0.05 relative to the OVX group. WT, Wet tissue.
Discussion
Although there are no definitive approaches for preventing and treating AD, clinical studies have demonstrated that the risk of AD in women can be significantly reduced by hormone therapy. Because accumulation of Aβ is widely theorized to initiate AD pathogenesis (51), optimizing hormone therapy will likely require thorough understanding of how estrogens and progestagens regulate Aβ accumulation. Prior work has clearly linked E2 with regulation of Aβ production by affecting APP metabolism and trafficking (52). Recent work has demonstrated that E2 and P4 may also play a role in Aβ clearance (2). Our results suggest that E2 and P4 are able to affect mRNA expression of some but not all Aβ clearance factors. Most notably, data in neuron cultures show that E2 affects IDE, ACE, and ECE2 mRNA in dose- and time-dependent manners whereas P4 regulates the expression levels of IDE, ACE, and TTR mRNA. In rat brain, both E2 and P4 are also shown to regulate IDE expression. Importantly, we also assess the relationship of expression changes with endogenous soluble brain levels of Aβ, demonstrating an inverse association between the levels of IDE and soluble Aβ.
Our data demonstrate that E2 regulates expression of several factors involved in Aβ clearance. The most robust effect across paradigms was the E2-induced increase in IDE expression, which was observed at both the mRNA and protein levels. The data show that the approximately 2-fold increase in IDE mRNA is ER dependent because it is blocked by an ER antagonist but mimicked by agonists for both ERα and ERβ. This observation is consistent with the finding that uterine IDE level and activity decrease during low E2 phase and increase during high E2 phase of the estrous cycle (53). In addition, a recent independent study done by our group found that E2 increased neuronal IDE expression in a mouse model of AD (54).
In contrast to increasing IDE expression, we observed that E2 did not alter or reduce expression of several other Aβ clearance factors. E2 significantly decreased expression of ACE mRNA in neuron culture as well as in rat brain. This observation is consistent with several prior studies showing that E2 reduces ACE levels in various tissues, including heart, lung, kidney, and brain (55–58). Similar results have been obtained in studies of postmenopausal women in which estrogen-based hormone therapy is associated with decreased ACE (59, 60). In addition, we found that E2 decreased ECE2 mRNA expression but had no significant effect on ECE1 transcript levels. E2 regulation of ECE1 and ECE2 in brain has not been previously reported, but these enzymes are known to be regulated by sex steroid hormones (61) with E2 shown to reduce ECE1 expression (62). We observed no significant effect of E2 treatment on either NEP or TTR mRNA expression. These data are in contrast to some prior reports. In the case of NEP, E2 has been linked to increased expression in uterus (63) as well as in rat brain and a neuroblastoma cell line (64–66). E2 is also associated with increased TTR expression in choroid plexus (67, 68) and in an AD transgenic mouse model (69). The reason for the disparity in findings is unclear but may reflect paradigm differences. For example, because neurons are not fully differentiated in our culture paradigm, it is possible that qualitatively different hormone responses could be observed in neurons with an adult phenotype. Because IDE is the only Aβ clearance factor positively regulated by E2 in our models, it appears to be the strongest candidate for contributing to the established ability of E2 to reduce Aβ levels.
Although recent evidence indicates a role for P4 (22) and P4 metabolites (70, 71) in reducing Aβ, there has been limited investigation of its potential regulation of Aβ clearance factors. Our neuron culture data show that P4 increases by approximately 2-fold the mRNA levels of IDE, ACE, and TTR but does not significantly alter expression of NEP, ECE1, and ECE2 mRNA. Upon short-term and extended P4 exposure in vivo, IDE mRNA continued to exhibit strong up-regulation by P4 but TTR mRNA was only modestly increased and ACE expression was not significantly affected. This incomplete concordance in findings between in vitro and in vivo paradigms suggests that although P4 and E2 have the potential to regulate numerous genes in simple culture systems, only a subset of these effects are manifested at significant levels in vivo owing to the influence of multiple tissue-specific and systems-wide interactions. The lack of significant P4 regulation of ACE mRNA in vivo is consistent with a prior observation in uterine artery (72). There are no previous reports of P4 regulation of IDE or the ECE. Some evidence indicates that P4 can increase NEP expression in human endometrium (73) and TTR in rat choroid plexus (67). Similar to our observations with E2, the strongest effect of P4 on Aβ clearance factors across paradigms was increased expression of IDE.
Because E2 and P4 often exert interactive effects on tissues and both are present endogenously and typically coadministered in postmenopausal hormone therapy (HT), it is important to understand their combined effects. To address this issue, we compared delivery of P4 in continuous vs. cyclic manners, alone and in combination with E2. Our data show that neural IDE mRNA expression in OVX rats is increased by treatment with E2, cyclic P4, and the combination of E2 and cyclic P4. In the same animals, the lowest levels of soluble Aβ were observed in the E2 and E2+cyclic P4 groups. Notably, continuous P4 delivered either alone or in combination with E2 neither increased IDE mRNA expression nor reduced Aβ levels. One limitation of this model is the use of sc hormone delivery pellets, which can result in supraphysiological levels of hormones (74). However, our findings of significant IDE regulation by E2 and P4 across three paradigms with different hormone delivery regimens argue that the observed relationships are significant.
The data indicate an inverse relationship between IDE expression and Aβ levels that is consistent with the possibility that regulation of IDE expression by E2 and P4 may contribute to their Aβ-lowering actions. In addition, the results support conclusions of prior studies from our laboratory and others demonstrating that cyclic P4 treatment is generally more beneficial than continuous P4 treatment either alone or in combination with E2 (22, 75, 76). For example, choline acetyltransferase activity was observed to be higher in OVX female rats treated with E2 with cyclic P4 than with E2 alone, but lowest in rats treated with E2 and continuous P4 (75). In data particularly relevant to this study, cyclic but not continuous P4 reduced Aβ accumulation in 3xTg-AD mice, whereas continuous P4 but not cyclic P4 prevented the Aβ-lowering action of E2 (22). The differential effects of continuous vs. cyclic P4 may reflect broad differences in gene expression profiles that vary according to hormone regimens (76). Unclear is whether delivery of E2 in a cyclic manner may offer further benefits, although the near-maximal effects of the used continuous E2 delivery relative to OVX alone suggests limited opportunity for improvement.
The mechanism underlying the regulation of Aβ-degrading enzymes by E2 and P4 is not well defined. Some evidence suggests roles of the estrogen receptors ERα and ERβ in E2-mediated changes (54, 65, 67). We show that the E2 regulation of IDE mRNA is blocked by the antiestrogen ICI 182,780, implicating ER-dependent signaling. We further demonstrate that both ERα and ERβ may be important in the E2-mediated regulation of IDE because both ERα and ERβ agonists up-regulated IDE transcript expression in a dose-dependent manner. E2 could directly increase the IDE mRNA expression via classic genomic signaling in which ER bind to estrogen-response elements (ERE) on the target gene for transcriptional regulation. Consistent with this possibility, analysis of the promoter region of rat IDE gene using MatInspector (77) reveals four putative canonical ERE (GGTCAnnnTGACC). Alternatively, E2 may increase IDE expression indirectly via activation of one or more of the cell-signaling pathways. For example, E2 is known to activate phosphatidylinositol-3 kinase (78), which in turn is implicated in the insulin-dependent up-regulation of IDE (79). The role of PR in regulating IDE is unclear. Although the PR antagonists RU486 and Org 31710 inhibited P4 regulation of TTR mRNA, the antagonists failed to block the P4-mediated increase in IDE mRNA levels in neuron culture, suggesting that neither of the two PR isoforms A and B mediates this P4 response. Future work will determine whether the mechanism involves a nonclassical mediator of P4 action (e.g. PR membrane component 1) or perhaps P4 metabolites (e.g. allopregnanolone). Further investigation of the roles of ER and PR is required to completely understand the mechanism underlying the observed regulation of these Aβ clearance factors.
Our study provides novel insight into the roles of individual and interactive effects of E2 and P4 in regulating Aβ by analyzing their effects on the expression of Aβ clearance factors. The most significant observation is that both E2 and P4 increase IDE mRNA expression in vitro and in vivo. This, taken together with the inverse relationship in vivo between IDE expression and Aβ levels, suggests another possible mechanism by which E2 and P4 can affect Aβ accumulation. Continued investigation of the interactions between E2 and P4 in regulating Aβ production and degradation is essential for optimizing hormone-based strategies for the prevention and/or treatment of AD.
Acknowledgments
This work was supported by National Institutes of Health Grant AG026572.
Disclosure Summary: The authors have nothing to disclose and no conflicts of interest with the information presented in this manuscript including any personal, financial, or other conflicts.
Footnotes
- Aβ
- β-amyloid protein
- ACE
- angiotensin-converting enzyme
- AD
- Alzheimers disease
- DPN
- 2,3-dis(4-hydroxyphenyl) propionitrile
- E2
- 17β-estradiol
- ECE
- endothelin-converting enzyme
- ER
- estrogen receptor
- HSD
- honestly significant difference
- IDE
- insulin-degrading enzyme
- NEP
- neprilysin
- OVX
- ovariectomized
- P4
- progesterone
- PPT
- propylpyrazole triol
- PR
- progesterone receptor
- RT
- room temperature
- TTR
- transthyretin.
References
- 1. Launer LJ, Andersen K, Dewey ME, Letenneur L, Ott A, Amaducci LA, Brayne C, Copeland JR, Dartigues JF, Kragh-Sorensen P, Lobo A, Martinez-Lage JM, Stijnen T, Hofman A. 1999. Rates and risk factors for dementia and Alzheimer's disease: results from EURODEM pooled analyses. EURODEM Incidence Research Group and Work Groups. European Studies of Dementia. Neurology 52:78–84 [DOI] [PubMed] [Google Scholar]
- 2. Pike CJ, Carroll JC, Rosario ER, Barron AM. 2009. Protective actions of sex steroid hormones in Alzheimer's disease. Front Neuroendocrinol 30:239–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bachman DL, Wolf PA, Linn R, Knoefel JE, Cobb J, Belanger A, D'Agostino RB, White LR. 1992. Prevalence of dementia and probable senile dementia of the Alzheimer type in the Framingham Study. Neurology 42:115–119 [DOI] [PubMed] [Google Scholar]
- 4. Andersen K, Nielsen H, Lolk A, Andersen J, Becker I, Kragh-Sørensen P. 1999. Incidence of very mild to severe dementia and Alzheimer's disease in Denmark: the Odense Study. Neurology 52:85–90 [DOI] [PubMed] [Google Scholar]
- 5. Sohrabji F. 2008. Premenopausal oophorectomy and the risk for dementia. Womens Health (Lond Engl) 4:127–131 [DOI] [PubMed] [Google Scholar]
- 6. Corder EH, Ghebremedhin E, Taylor MG, Thal DR, Ohm TG, Braak H. 2004. The biphasic relationship between regional brain senile plaque and neurofibrillary tangle distributions: modification by age, sex, and APOE polymorphism. Ann NY Acad Sci 1019:24–28 [DOI] [PubMed] [Google Scholar]
- 7. Henderson VW, Buckwalter JG. 1994. Cognitive deficits of men and women with Alzheimer's disease. Neurology 44:90–96 [DOI] [PubMed] [Google Scholar]
- 8. Barnes LL, Wilson RS, Bienias JL, Schneider JA, Evans DA, Bennett DA. 2005. Sex differences in the clinical manifestations of Alzheimer disease pathology. Arch Gen Psychiatry 62:685–691 [DOI] [PubMed] [Google Scholar]
- 9. Manly JJ, Merchant CA, Jacobs DM, Small SA, Bell K, Ferin M, Mayeux R. 2000. Endogenous estrogen levels and Alzheimer's disease among postmenopausal women. Neurology 54:833–837 [DOI] [PubMed] [Google Scholar]
- 10. Rosario ER, Chang L, Head EH, Stanczyk FZ, Pike CJ. 2011. Brain levels of sex steroid hormones in men and women during normal aging and in Alzheimer's disease. Neurobiol Aging 32:604–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yue X, Lu M, Lancaster T, Cao P, Honda S, Staufenbiel M, Harada N, Zhong Z, Shen Y, Li R. 2005. Brain estrogen deficiency accelerates Aβ plaque formation in an Alzheimer's disease animal model. Proc Natl Acad Sci USA 102:19198–19203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lewczuk P, Kornhuber J, Vanderstichele H, Vanmechelen E, Esselmann H, Bibl M, Wolf S, Otto M, Reulbach U, Kölsch H, Jessen F, Schröder J, Schönknecht P, Hampel H, Peters O, Weimer E, Perneczky R, Jahn H, Luckhaus C, Lamla U, Supprian T, Maler JM, Wiltfang J. 2008. Multiplexed quantification of dementia biomarkers in the CSF of patients with early dementias and MCI: a multicenter study. Neurobiol Aging 29:812–818 [DOI] [PubMed] [Google Scholar]
- 13. Kawas C, Resnick S, Morrison A, Brookmeyer R, Corrada M, Zonderman A, Bacal C, Donnell Lingle D, Metter E. 1997. A prospective study of estrogen replacement therapy and the risk of developing Alzheimer's disease: the Baltimore longitudinal study of aging. Neurology [Erratum (1998) 51:564]48:1517–1521 [DOI] [PubMed] [Google Scholar]
- 14. Zandi PP, Carlson MC, Plassman BL, Welsh-Bohmer KA, Mayer LS, Steffens DC, Breitner JC. 2002. Hormone replacement therapy and incidence of Alzheimer disease in older women: the Cache County Study. JAMA 288:2123–2129 [DOI] [PubMed] [Google Scholar]
- 15. Shumaker SA, Legault C, Rapp SR, Thal L, Wallace RB, Ockene JK, Hendrix SL, Jones BN, III, Assaf AR, Jackson RD, Kotchen JM, Wassertheil-Smoller S, Wactawski-Wende J. 2003. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women's Health Initiative Memory Study: a randomized controlled trial. JAMA 289:2651–2662 [DOI] [PubMed] [Google Scholar]
- 16. Singh M, Sumien N, Kyser C, Simpkins JW. 2008. Estrogens and progesterone as neuroprotectants: what animal models teach us. Front Biosci 13:1083–1089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Whitmer RA, Quesenberry CP, Zhou J, Yaffe K. 2011. Timing of hormone therapy and dementia: the critical window theory revisited. Ann Neurol 69:163–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Selkoe DJ. 1994. Amyloid beta-protein precursor: new clues to the genesis of Alzheimer's disease. Curr Opin Neurobiol 4:708–716 [DOI] [PubMed] [Google Scholar]
- 19. Petanceska SS, Nagy V, Frail D, Gandy S. 2000. Ovariectomy and 17β-estradiol modulate the levels of Alzheimer's amyloid β peptides in brain. Exp Gerontol 35:1317–1325 [DOI] [PubMed] [Google Scholar]
- 20. Levin-Allerhand JA, Lominska CE, Wang J, Smith JD. 2002. 17Alpha-estradiol and 17β-estradiol treatments are effective in lowering cerebral amyloid-beta levels in AβPPSWE transgenic mice. J Alzheimers Dis 4:449–457 [DOI] [PubMed] [Google Scholar]
- 21. Carroll JC, Rosario ER, Chang L, Stanczyk FZ, Oddo S, LaFerla FM, Pike CJ. 2007. Progesterone and estrogen regulate Alzheimer-like neuropathology in female 3xTg-AD mice. J Neurosci 27:13357–13365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Carroll JC, Rosario ER, Villamagna A, Pike CJ. 2010. Continuous and cyclic progesterone differentially interact with estradiol in the regulation of Alzheimer-like pathology in female 3xTransgenic-Alzheimer's disease mice. Endocrinology 151:2713–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heikkinen T, Kalesnykas G, Rissanen A, Tapiola T, Iivonen S, Wang J, Chaudhuri J, Tanila H, Meittinen R, Puoliväli J. 2004. Estrogen treatment improves spatial learning in APP+PS1 mice but does not affect β amyloid accumulation and plaque formation. Exp Neurol 187:105–117 [DOI] [PubMed] [Google Scholar]
- 24. Jaffe AB, Toran-Allerand CD, Greengard P, Gandy SE. 1994. Estrogen regulates metabolism of Alzheimer amyloid β precursor protein. J Biol Chem 269:13065–13068 [PubMed] [Google Scholar]
- 25. Xu H, Gouras GK, Greenfield JP, Vincent B, Naslund J, Mazzarelli L, Fried G, Jovanovic JN, Seeger M, Relkin NR, Liao F, Checler F, Buxbaum JD, Chait BT, Thinakaran G, Sisodia SS, Wang R, Greengard P, Gandy S. 1998. Estrogen reduces neuronal generation of Alzheimer β-amyloid peptides. Nat Med 4:447–451 [DOI] [PubMed] [Google Scholar]
- 26. Choi SH, Leight SN, Lee VM, Li T, Wong PC, Johnson JA, Saraiva MJ, Sisodia SS. 2007. Accelerated Aβ deposition in APPswe/PS1δE9 mice with hemizygous deletions of TTR (transthyretin). J Neurosci 27:7006–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Leissring MA. 2008. The AβCs of Aβ-cleaving proteases. J Biol Chem 283:29645–29649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mukherjee A, Song E, Kihiko-Ehmann M, Goodman JP, Jr, Pyrek JS, Estus S, Hersh LB. 2000. Insulysin hydrolyzes amyloid β peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J Neurosci 20:8745–8749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ. 2000. Neurons regulate extracellular levels of amyloid β-protein via proteolysis by Insulin-degrading enzyme. J Neurosci 10:1657–1665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. 2003. Amyloid-β peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA 100:6221–6226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Leissring MA, Farris W, Chang AY, Walsh DM, Wu X, Sun X, Frosch MP, Selkoe DJ. 2003. Endoplasmic reticulum-localized amyloid β-peptide is degraded in the cytosol by two distinct degradation pathways. Neuron 40:1087–109314687544 [Google Scholar]
- 32. Shirotani K, Tsubuki S, Iwata N, Takaki Y, Harigaya W, Maruyama K, Kiryu-Seo S, Kiyama H, Iwata H, Tomita T, Iwatsubo T, Saido TC. 2001. Neprilysin degrades both amyloid β peptides 1–40 and 1–42 most rapidly and efficiently among thiorphan- and phosphoramidon-sensitive endopeptidases. J Biol Chem 276:21895–21901 [DOI] [PubMed] [Google Scholar]
- 33. Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. 2001. Metabolic regulation of brain Aβ by neprilysin. Science 292:1550–1552 [DOI] [PubMed] [Google Scholar]
- 34. Kanemitsu H, Tomiyama T, Mori H. 2003. Human neprilysin is capable of degrading amyloid β peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett 350:113–116 [DOI] [PubMed] [Google Scholar]
- 35. Wang S, Wang R, Chen L, Bennett DA, Dickson DW, Wang DS. 2010. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer's brain. J Neurochem 115:47–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eckman EA, Reed DK, Eckman CB. 2001. Degradation of the Alzheimer's amyloid β peptide by endothelin-converting enzyme. J Biol Chem 276:24540–24548 [DOI] [PubMed] [Google Scholar]
- 37. Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. 2003. Alzheimer's disease β-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem 278:2081–2084 [DOI] [PubMed] [Google Scholar]
- 38. Hemming ML, Selkoe DJ. 2005. Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem 280:37644–37650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zou K, Yamaguchi H, Akatsu H, Sakamoto T, Ko M, Mizoguchi K, Gong JS, Yu W, Yamamoto T, Kosaka K, Yanagisawa K, Michikawa M. 2007. Angiotensin-converting enzyme converts amyloid β-protein 1–42 (Aβ(1–42)) to Aβ(1–40), and its inhibition enhances brain Aβ deposition. J Neurosci 27:8628–8635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Buxbaum JN, Ye Z, Reixach N, Friske L, Levy C, Das P, Golde T, Masliah E, Roberts AR, Bartfai T. 2008. Transthyretin protects Alzheimer's mice from the behavioral and biochemical effects of Aβ toxicity. Proc Natl Acad Sci USA 105:2681–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pike CJ. 1999. Estrogen modulates neuronal Bcl-xL expression and β-amyloid-induced apoptosis: relevance to Alzheimer's disease. J Neurochem 72:1552–1563 [DOI] [PubMed] [Google Scholar]
- 42. Figueiredo HF, Ulrich-Lai YM, Choi DC, Herman JP. 2007. Estrogen potentiates adrenocortical responses to stress in female rats. Am J Physiol Endocrinol Metab 292:E1173–E1182 [DOI] [PubMed] [Google Scholar]
- 43. Jayaraman A, Pike CJ. 2009. Progesterone attenuates oestrogen neuroprotection via downregulation of oestrogen receptor expression in cultured neurones. J Neuroendocrinol 21:77–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 45. Beckett TL, Niedowicz DM, Studzinski CM, Weidner AM, Webb RL, Holler CJ, Ahmed RR, LeVine H, III, Murphy MP. 2010. Effects of nonsteroidal anti-inflammatory drugs on amyloid-β pathology in mouse skeletal muscle. Neurobiol Dis 39:449–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wakeling AE, Bowler J. 1992. ICI 182,780, a new antioestrogen with clinical potential. J Steroid Biochem Mol Biol 43:173–177 [DOI] [PubMed] [Google Scholar]
- 47. Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, Katzenellenbogen BS, Katzenellenbogen JA. 2000. Pyrazole ligands: structure-affinity/activity relationships and estrogen receptor-α-selective agonists. J Med Chem 43:4934–4947 [DOI] [PubMed] [Google Scholar]
- 48. Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. 2001. Estrogen receptor-β potency-selective ligands: structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem 44:4230–4251 [DOI] [PubMed] [Google Scholar]
- 49. Schreiber JR, Hsueh AJ, Baulieu EE. 1983. Binding of the anti-progestin RU-486 to rat ovary steroid receptors. Contraception 28:77–85 [DOI] [PubMed] [Google Scholar]
- 50. Kloosterboer HJ, Schoonen WG, Deckers GH, Klijn JG. 1994. Effects of progestagens and Org OD14 in in vitro and in vivo tumor models. J Steroid Biochem Mol Biol 49:311–318 [DOI] [PubMed] [Google Scholar]
- 51. Hardy J, Selkoe DJ. 2002. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297:353–356 [DOI] [PubMed] [Google Scholar]
- 52. Gandy S, Petanceska S. 2001. Regulation of Alzheimer β-amyloid precursor trafficking and metabolism. Adv Exp Med Biol 487:85–100 [DOI] [PubMed] [Google Scholar]
- 53. Udrisar DP, Wanderley MI, Porto RC, Cardodo CL, Barbosa MC, Canberos MC, Cresto JC. 2005. Androgen- and estrogen-dependent regulation of insulin-degrading enzyme in subcellular fractions of rat prostate and uterus. Exp Biol Med (Maywood) 230:479–486 [DOI] [PubMed] [Google Scholar]
- 54. Zhao L, Yao J, Mao Z, Chen S, Wang Y, Brinton RD. 2010. 17β-Estradiol regulates insulin-degrading enzyme expression via an ERβ/PI3-K pathway in hippocampus: relevance to Alzheimer's prevention. Neurobiol Aging 32:1949–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brosnihan KB, Hodgin JB, Smithies O, Maeda N, Gallagher P. 2008. Tissue-specific regulation of ACE/ACE2 and AT1/AT2 receptor gene expression by oestrogen in apolipoprotein E/oestrogen receptor-α knock-out mice. Exp Physiol 93:658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gallagher PE, Li P, Lenhart JR, Chappell MC, Brosnihan KB. 1999. Estrogen regulation of angiotensin-converting enzyme mRNA. Hypertension 33:323–328 [DOI] [PubMed] [Google Scholar]
- 57. Seltzer A, Pinto JE, Viglione PN, Correa FM, Libertun C, Tsutsumi K, Steele MK, Saavedra JM. 1992. Estrogens regulate angiotensin-converting enzyme and angiotensin receptors in female rat anterior pituitary. Neuroendocrinology 55:460–467 [DOI] [PubMed] [Google Scholar]
- 58. Dean SA, Tan J, O'Brien ER, Leenen FH. 2005. 17β-Estradiol downregulates tissue angiotensin-converting enzyme and ANG II type 1 receptor in female rats. Am J Physiol Regul Integr Comp Physiol 288:R759–R766 [DOI] [PubMed] [Google Scholar]
- 59. Proudler AJ, Cooper A, Whitehead M, Stevenson JC. 2003. Effects of oestrogen-only and oestrogen-progestogen replacement therapy upon circulating angiotensin I-converting enzyme activity in postmenopausal women. Clin Endocrinol (Oxf) 58:30–35 [DOI] [PubMed] [Google Scholar]
- 60. Sanada M, Higashi Y, Nakagawa K, Sasaki S, Kodama I, Sakashita T, Tsuda M, Ohama K. 2001. Estrogen replacement therapy in postmenopausal women augments reactive hyperemia in the forearm by reducing angiotensin converting enzyme activity. Atherosclerosis 158:391–397 [DOI] [PubMed] [Google Scholar]
- 61. Keator CS, Mah K, Ohm L, Slayden OD. 2011. Estrogen and progesterone regulate expression of the endothelins in the rhesus macaque endometrium. Hum Reprod 26:1715–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rodrigo MC, Martin DS, Eyster KM. 2003. Vascular ECE-1 mRNA expression decreases in response to estrogens. Life Sci 73:2973–2983 [DOI] [PubMed] [Google Scholar]
- 63. Neves LA, Chappell MC, Ferrario CM, Gallagher PE, Ganten D, Brosnihan KB. 2006. Effect of estrogen on neprilysin expression in uterus and kidney of Sprague-Dawley normotensive and heterozygous (mRen2)27-transgenic hypertensive rats. Peptides 27:2912–2918 [DOI] [PubMed] [Google Scholar]
- 64. Huang J, Guan H, Booze RM, Eckman CB, Hersh LB. 2004. Estrogen regulates neprilysin activity in rat brain. Neurosci Lett 367:85–87 [DOI] [PubMed] [Google Scholar]
- 65. Liang K, Yang L, Yin C, Xiao Z, Zhang J, Liu Y, Huang J. 2010. Estrogen stimulates degradation of β-amyloid peptide by up-regulating neprilysin. J Biol Chem 285:935–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xiao ZM, Sun L, Liu YM, Zhang JJ, Huang J. 2009. Estrogen regulation of the neprilysin gene through a hormone-responsive element. J Mol Neurosci 39:22–26 [DOI] [PubMed] [Google Scholar]
- 67. Quintela T, Gonçalves I, Baltazar G, Alves CH, Saraiva MJ, Santos CR. 2009. 17β-Estradiol induces transthyretin expression in murine choroid plexus via an oestrogen receptor dependent pathway. Cell Mol Neurobiol 29:475–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tang YP, Haslam SZ, Conrad SE, Sisk CL. 2004. Estrogen increases brain expression of the mRNA encoding transthyretin, an amyloid β scavenger protein. J Alzheimers Dis 6:413–420; discussion 443–449 [DOI] [PubMed] [Google Scholar]
- 69. Amtul Z, Wang L, Westaway D, Rozmahel RF. 2010. Neuroprotective mechanism conferred by 17β-estradiol on the biochemical basis of Alzheimer's disease. Neuroscience 169:781–786 [DOI] [PubMed] [Google Scholar]
- 70. Wang JM, Singh C, Liu L, Irwin RW, Chen S, Chung EJ, Thompson RF, Brinton RD. 2010. Allopregnanolone reverses neurogenic and cognitive deficits in mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 107:6498–6503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen S, Wang JM, Irwin RW, Yao J, Liu L, Brinton RD. 2011. Allopregnanolone promotes regeneration and reduces β-amyloid burden in a preclinical model of Alzheimer's disease. PLoS One 6:e24293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Gao H, Yallampalli U, Yallampalli C. 2012. Protein restriction to pregnant rats increases the plasma levels of angiotensin II and expression of angiotensin II receptors in uterine arteries. Biol Reprod 86:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Casey ML, Smith JW, Nagai K, Hersh LB, MacDonald PC. 1991. Progesterone-regulated cyclic modulation of membrane metalloendopeptidase (enkephalinase) in human endometrium. J Biol Chem 266:23041–23047 [PubMed] [Google Scholar]
- 74. Strom JO, Theodorsson A, Theodorsson E. 2009. Dose-related neuroprotective versus neurodamaging effects of estrogens in rat cerebral ischemia: a systematic analysis. J Cereb Blood Flow Metab 29:1359–1372 [DOI] [PubMed] [Google Scholar]
- 75. Gibbs RB. 2000. Effects of gonadal hormone replacement on measures of basal forebrain cholinergic function. Neuroscience 101:931–938 [DOI] [PubMed] [Google Scholar]
- 76. Zhao L, Morgan TE, Mao Z, Lin S, Cadenas E, Finch CE, Pike CJ, Mack WJ, Brinton RD. 2012. Continuous versus cyclic progesterone exposure differentially regulates hippocampal gene expression and functional profiles. PLoS One 7:e31267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. 2005. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics 21:2933–2942 [DOI] [PubMed] [Google Scholar]
- 78. Mannella P, Brinton RD. 2006. Estrogen receptor protein interaction with phosphatidylinositol 3-kinase leads to activation of phosphorylated Akt and extracellular signal-regulated kinase 1/2 in the same population of cortical neurons: a unified mechanism of estrogen action. J Neurosci 26:9439–9447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhao L, Teter B, Morihara T, Lim GP, Ambegaokar SS, Ubeda OJ, Frautschy SA, Cole GM. 2004. Insulin-degrading enzyme as a downstream target of insulin receptor signaling cascade: implications for Alzheimer's disease intervention. J Neurosci 24:11120–11126 [DOI] [PMC free article] [PubMed] [Google Scholar]