

Abstract
Background
The SAMtools utilities comprise a very useful and widely used suite of software for manipulating files and alignments in the SAM and BAM format, used in a wide range of genetic analyses. The SAMtools utilities are implemented in C and provide an API for programmatic access, to help make this functionality available to programmers wishing to develop in the high level Ruby language we have developed bio-samtools, a Ruby binding to the SAMtools library.
Results
The utility of SAMtools is encapsulated in 3 main classes, Bio::DB::Sam, representing the alignment files and providing access to the data in them, Bio::DB::Alignment, representing the individual read alignments inside the files and Bio::DB::Pileup, representing the summarised nucleotides of reads over a single point in the nucleotide sequence to which the reads are aligned.
Conclusions
Bio-samtools is a flexible and easy to use interface that programmers of many levels of experience can use to access information in the popular and common SAM/BAM format.
Keywords: Next-generation sequencing, DNA, High, Throughput, Ruby, Bio, SAM, BAM
Background
High-throughput DNA sequencing in the biological sciences has made it possible for researchers to obtain many millions of sequence reads in single, low-cost experiments. These sequence reads are typically very short compared to the parent genome (reads will usually be in the range of 36 - 200 nucleotides long while genomes are many millions of nucleotides long) and very redundant; many reads may have the same sequence. One widespread use for these sequences is in detecting small differences in the genome sequence of the sample donor, which is achieved by using computational methods to align each short sequence read against a long, reference genome sequence then examining the derived alignments and determining positions at which there are differences. Many programs have been created for alignment including BWA [1], Bowtie [2], SOAP [3], NOVOALIGN [4] and BFAST [5], each implementing different algorithms optimised to address different issues with the alignment problem. Most high-throughput alignment programs produce a standard output file in Sequence Alignment/Map format (SAM) [6], a tab-delimited text-based format for describing alignments. The SAMtools utilities comprise a very useful and widely used suite of software for manipulating files and alignments in the SAM format. The large SAM files can be converted to the binary equivalent BAM files a compressed and indexed variant for random access, which vastly facilitates genetic analyses that rely on high-throughput alignment. The SAMtools utilities are implemented in C and provide an API for programmatic access, for which there are multiple language bindings, notably in Perl [7], Python [8] and Java [9]. Here we describe the Ruby language binding to the SAMtools library, developed for our own work and distributed as a BioRuby plug-in [10].
Implementation
The bio-samtools package is a wrapper around libbam.so (for Linux) and libbam.1.dylib (for Mac OS X), the core shared object library from the SAMtools package. To make it possible for the C functions in libbam to be called from within Ruby code we have used the Ruby Foreign Function Interface (FFI) [11] package as a bridge between the two languages. The flexible FFI package can programatically load dynamic libraries and bind functions without the need to make changes to Ruby itself or to recompile any extensions, so our package can easily run on standard Ruby interpreters without installation and compilation issues beyond that of the normal Ruby gem installation. Importantly, FFI also has useful methods for managing memory, pointers, structs and binary fields are converted to Ruby boolean variables. A further advantage of using FFI is that the binding is compatible with both the standard Ruby interpreter Matz’s Ruby Interpreter (MRI) and the alternative Java implementation of the Ruby language (JRUBY). By wrapping SAMtools in this way the scientist may use the high level easily learned and fast to develop with Ruby that facilitates quick development rather than the native C of SAMtools. bio-samtools hides the low-level API completely making bio-samtools a useful and easily used tool for working with Next-Generation Sequencing data in BAM files. Each .c library from the SAMtools API is represented by a separate Ruby module mapping the C functions (Figure 1), which are unified in the class Sam.
Figure 1.
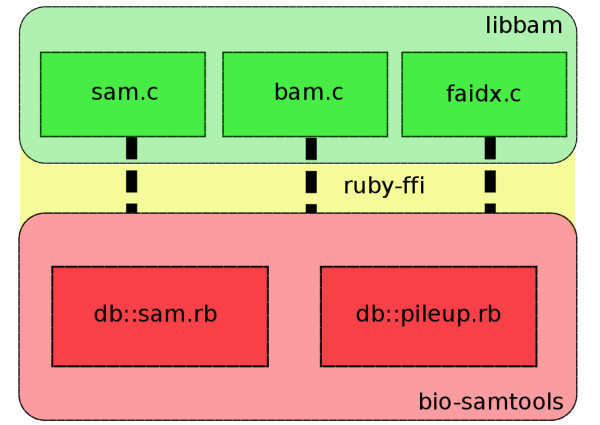
bio-samtools and its relationship to the underlying libbam. Green boxes indicate C source code in libbam, red boxes indicate Ruby files that interact the Ruby FFI represented in yellow.
Bio::DB::Sam
The main object representing the SAM/BAM file is a Bio::DB::Sam object. Objects of class Sam, have several attributes and methods, summarised in Table 1. Most of the attributes relate to the alignment file type and the location of the BAM file in the file system. The BAM file itself is not held in the object or Ruby memory, rather the Ruby wrapping is used to access the information via the C API. The methods of the Sam object can be divided into utility methods that affect the BAM files, (#sort and #merge), retrieval methods that return objects of other classes representing individual read alignments (#fetch, #fetch_with_function) and summary methods (#average_coverage, #chromosome_coverage and #mpileup,#index_stats).
Table 1.
Attributes and methods of the Bio::DB::Sam object
| binary | denotes whether this is a binary file |
|---|---|
| compressed |
denotes whether this file is compressed |
| fasta_path |
path to the reference FASTA file |
| sam |
path to the associated BAM file |
| chromosome_coverage |
return Ruby Array of coverage over a region |
| fetch |
fetch alignment in a region from a bam file, returning a Ruby Array object |
| fetch_reference |
fetch regions of the reference file returning a String object of the relevant sequence |
| fetch_with_function |
fetch all alignments in a region passing in a Ruby Proc object as a callback, returning an iterator |
| index_stats |
get information about reference and number of mapped reads |
| merge |
merge two or more bam files |
| mpileup |
an iterator that returns Pileup objects representing the reads over a single position |
| sort | sort the BAM file |
Bio::DB::Alignment
The fetch and fetch_with_function method of the Bio::DB::Sam object return individual alignments one at a time from an iterator. The individual alignments represent a single read and its mapping to the reference and are Bio::DB::Alignment objects, whose attributes are described in Table 2. These attributes are derived directly from the SAM format definition [6]. The fetch_with_function method is distinct from fetch in that it allows the user to pass a Ruby Proc object or a block. These are functionally equivalent to closures in other languages and provide advantages in terms of encapsulation and often speed compared to the standard block based equivalent, advanced Ruby programmers are likely to appreciate this feature.
Table 2.
Attributes of the Bio::DB::Alignment object
| calend | nucleotide position of the end of the alignment |
|---|---|
| cigar |
CIGAR string describing the matches/mismatches |
| failed_quality |
this read failed the quality threshold |
| first_in_pair |
first of a pair |
| is_duplicate |
this read is a suspected optical or PCR duplicate |
| is_mapped |
the read was aligned |
| is_paired |
the read is one of a pair |
| isize |
the insert size distance between mapped mates |
| mapq |
the PHRED scaled mapping quality of the alignment |
| mate_strand |
the strand of the mate |
| mate_unmapped |
the mate is unmapped |
| mpos |
start position of the mate on the reference |
| pos |
start position of the alignments |
| primary |
is a primary alignment |
| qlen |
read length |
| qname |
read name |
| qual |
read quality string |
| query_strand |
strand of alignment |
| query_unmapped |
query is unmapped |
| rname |
name of reference to which read mapped |
| second_in_pair |
this is second in the pair |
| seq |
read sequence |
| tags | Bio::DB::Tag object representing the tags for this alignment |
Bio::DB::Pileup
The pileup format is a straightforward way of structuring alignments over single positions for the easy identification of genetic polymorphisms, the format has a long history and has been in use in SAMtools for a while. The original ’pileup’ function has recently been deprecated and removed in favour of ’mpileup’. The output from mpileup is exactly equivalent to the pileup command called without the -c flag set, that is to say the six column format. The class Pileup can parse the old ten column pileup format if an instance is created manually by passing it a raw line from the pileup file. Calling the mpileup method of a SAM object results in the return of a stream of Pileup [12] objects, which have the attributes and methods listed in Table 3. Some of the attributes are related to the ten column format only. Notably, SAMtools will calculate a consensus base call if asked to return a ten column pileup file, so the Pileup class will use SAMtools consensus call if it is available, otherwise it will call a consensus based on a simple majority count.
Table 3.
Attributes and methods of the Bio::DB::Pileup object
| consensus | the consensus nucleotide calculated as the nucleotide with highest count multiple nucleotides returned in a tie |
|---|---|
| coverage |
the number of reads covering this position |
| non_ref_count |
the number of reads that disagree with the reference nucleotide |
| non_ref_count_hash |
a Hash with A,T,G and C as keys and the number each nucleotide appears in the pileup when that nucleotide is not |
| |
the reference |
| pos |
the position in the reference sequence that this pileup represents |
| read_bases |
the read nucleotides covering this position |
| read_quals |
the quality scores of the read nucleotides covering this position |
| ref_base |
the reference sequence nucleotide |
| ref_count |
the number of times the reference nucleotide appears in the read nucleotides covering this position |
| ref_name |
the name of the reference sequence |
| ar1, ar2, ar3 |
the allele calls from pileup |
| consensus1 |
the consensus of the reads according to SAMtools method of calculation |
| consensus_quality1 |
the quality score of the consensus according to SAMtools method of calculation |
| rms_mapq1 |
the root mean square mapping quality at the position |
| snp_quality1 | the SNP quality at the position |
1ten column format only.
Results and discussion
Using bio-samtools: a brief tutorial
bio-samtools in use is straightforward, here are a few examples of interacting with BAM files with the package. More information on specific functions is provided in the RubyDoc documentation and in the files bioruby-samtools/doc/tutorial.html and bioruby-samtools/doc/tutorial.pdf. The location of the bio-samtools installation folder can be found by typing ’gem which bio-samtools’ at the command-line.
Installation
bio-samtools is easily installed from a machine with an internet connection and a Ruby installation with the straightforward Gem invocation ’gem install bio-samtools’. bio-samtools automatically downloads the original libbam C source code and compiles it for Linux or OSX as appropriate. The new version of the library is kept locally to the bio-samtools code to avoid conflicts with other installations of the library.
Loading a BAM file
A SAM object represents the alignments in the BAM file, and is very straightforward to create, you will need a sorted BAM file, to access the alignments and a reference sequence in FASTA format to use the reference sequence. The object can be created and opened as follows:require 'bio-samtools'bam=Bio::DB::Sam.new(:bam=>"my_sorted.bam", :fasta=>'ref.fasta')bam.openbam.close
Opening the file needs only to be done once for multiple operations on it, access to the alignments is random so you don’t need to loop over all the entries in the file, as you would with a manual SAM file parse.
Getting summary information
The length of reference sequences and the number of reads mapped to each can be obtained with the index_stats function. A Hash object, keyed by reference name and with a Hash at each value is returned. The Hash at the value has keys :length, :mapped_reads and :unmapped_reads and values for each of these. The index_stats function wraps the SAMtools idxstats command.sam.index_stats# returns { "chr_1"=> {:length=>69930,:mapped_reads=>1000,:unmapped_reads=>0 }, }
Retrieving reference sequence
Retrieving the reference can only be done if the reference has been loaded, which isn’t done automatically in order to save memory. Reference need only be loaded once, and is accessed using reference name, start, end in 1-based co-ordinates. A standard Ruby String object is returned. In this example a 500 nucleotide region from the start of the sequence is returned.bam.load_referenceseq = bam.fetch_reference("Chr1", 1, 500)
Retrieving alignments in a region
Alignments in a region of interest can be obtained one at a time by giving the region to the fetch() function.bam.fetch("Chr1", 3000, 4000).each do | alignment |puts alignment.qname #do something with the alignment objectend
Get a summary of coverage in a region
It is easy to get the total depth of reads at a given position, the chromosome_coverage function is used. This differs from the previous functions in that a start position and length (rather than end position) are passed to the function. An array of coverages is returned, eg [26,26,27 .. ]. The first position in the array gives the depth of coverage at the given start position in the genome, the last position in the array gives the depth of coverage at the given start position plus the length given.coverages = bam.chromosome_coverage("Chr1", 3000, 1000)Similarly, average (arithmetic mean) of coverage can be retrieved, also with start and length parametersav_cov = bam.average_coverage("Chr1", 3000, 1000)
Getting pileup information
Pileup format represents the coverage of reads over a single base in the reference. Getting a Pileup over a region is very easy. Note that this is done with mpileup and NOT the now deprecated and removed from SAMTools pileup function. Calling the mpileup method creates an iterator that yields a Pileup object for each base.bam.mpileup do |pileup|puts pileup.consensusend
The mpileup function takes a range of parameters to allow SAMTools level filtering of reads and alignments. They are specified as key, value pairs. In this example a region is specified by :r and a minimum per base quality score is specified by :Q.bam.mpileup(:r => "Chr1:1000-2000", :Q => 50) do |pileup|puts pileup.coverageend
Not all the options SAMTools allows you to pass to mpileup are supported, those that cause mpileup to return Binary Variant Call Format (BCF) [13] are ignored. Specifically these are g,u,e,h,I,L,o,p. Table 4 lists the SAMTools flags supported and the symbols you can use to call them in the mpileup command.
Table 4.
SAMtools options recognised by the Bio::DB:Sam#mpileup method and the symbols used to invoke them
| SAMTools option | description | short symbol | long symbol | default |
|---|---|---|---|---|
| r |
limit retrieval to a region |
:r |
:region |
all positions |
| 6 |
assume Illumina scaled quality scores |
:six |
:illumina_quals |
FALSE |
| A |
count anomalous read pairs scores |
:A |
:count_anomalous |
FALSE |
| B |
disable BAQ computation |
:B |
:no_baq |
FALSE |
| C |
parameter for adjusting mapQ |
:C |
:adjust_mapq |
0 |
| d |
max per-BAM depth to avoid excessive memory usage |
:d |
:max_per_bam_depth |
250 |
| E |
extended BAQ for higher sensitivity but lower specificity |
:E |
:extended_baq |
FALSE |
| G |
exclude read groups listed in FILE |
:G |
:exclude_reads_file |
FALSE |
| l |
list of positions (chr pos) or regions (BED) |
:l |
:list_of_positions |
FALSE |
| M |
cap mapping quality at value |
:M |
:mapping_quality_cap |
60 |
| R |
ignore RG tags |
:R |
:ignore_rg |
FALSE |
| q |
skip alignments with mapping quality smaller than value |
:q |
:min_mapping_quality |
0 |
| Q | skip bases with base quality smaller than value | :Q | :imin_base_quality | 13 |
Conclusions
Ruby is an easily written and understood high-level language, ideal for beginners or those wishing to develop analysis scripts and prototype applications in short timeframes. A major advantage of scripting in Ruby for biologists is the BioRuby project that provides a lot of classes and functionality for dealing with common biological data types and file formats. bio-samtools is a BioRuby plugin which extends the original BioRuby framework by providing a useful and flexible interface for Ruby coders who wish to have programmatical access to the data in BAM and SAM files without losing performance, the C API is very much quicker than a pure Ruby implementation would be and wrapping it provides the best of both languages. The interface we provide gives access to all the API components of the SAMtools core library libbam.so and extends with some useful high level methods. The open class system of Ruby means that the SAM class which encapsulates the functionality of SAMtools can easily be extended at run-time by the user. These features together mean that bio-samtools can be an extremely useful tool for scientists wishing to examine the results of next-generation sequencing alignments.
Availability and requirements
Project name: bio-samtools Project home page:http://rubygems.org/gems/bio-samtoolsOperating systems: Linux and Mac OS X Programming language: Ruby Other requirements: none License: as BioRuby Any restrictions to use by non-academics: none
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RRG wrote the binding, tests, co-wrote the documentation and co-wrote the manuscript, RB created and organised the Gem and contributed to the binding and tests and co-wrote the manuscript and DM conceived of the binding, contributed to the binding and tests, tested the implementations with sample data and co-wrote the manuscript. RRG and RB contributed equally to this work. All authors read and approved the final manuscript.
Contributor Information
Ricardo H Ramirez-Gonzalez, Email: ricardo.ramirez-gonzalez@tgac.ac.uk.
Raoul Bonnal, Email: bonnal@ingm.org.
Mario Caccamo, Email: mario.caccamo@tgac.ac.uk.
Daniel MacLean, Email: dan.maclean@tsl.ac.uk.
Acknowledgements
RHRG and MC are supported by the BBSRC and DM is supported by The Gatsby Charitable Foundation. RHRG and RB contributed equally to this work.
References
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–60. doi: 10.1093/bioinformatics/btp324. [ http://bioinformatics.oxfordjournals.org/cgi/content/full/25/14/1754?view=long&pmid=19451168] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [ http://genomebiology.com/content/10/3/R25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–4. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- Novocraft. [ http://www.novocraft.com]
- Homer N, Merriman B, Nelson SF. BFAST: an alignment tool for large scale genome resequencing. PLoS ONE. 2009;4(11):e7767. doi: 10.1371/journal.pone.0007767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. Subgroup GPDP. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25(16):2078–9. doi: 10.1093/bioinformatics/btp352. [ http://bioinformatics.oxfordjournals.org/cgi/content/full/25/16/2078] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bio-SamTools. [ http://search.cpan.org/lds/Bio-SamTools/]
- pysam. [ http://code.google.com/p/pysam/]
- Picard. [ http://picard.sourceforge.net/index.shtml]
- BioRuby plugins. [ http://bioruby.open-bio.org/wiki/Plugins]
- Ruby Foreign Function Interface. [ https://github.com/ffi/ffi/wiki]
- Pileup Format. [ http://samtools.sourceforge.net/pileup.shtml]
- Variant Call Format. [ http://www.1000genomes.org/node/101]