

Abstract
This review focuses on the vascular smooth muscle cells present in the medial layer of the blood vessels wall in the fully differentiated state (dVSMCs). The dVSMC contractile phenotype enables these cells to respond in a highly regulated manner to changes in extracellular stimuli. Through modulation of vascular contractile force and vascular compliance dVSMCs regulate blood pressure and blood flow. The cellular and molecular mechanisms by which vascular smooth muscle contractile functions are regulated are not completely elucidated. Recent studies have documented a critical role for actin polymerization and cytoskeletal dynamics in the regulation of contractile function. Here we will review the current understanding of actin cytoskeletal dynamics and focal adhesion function in dVSMCs in order to better understand actin cytoskeleton connections to the extracellular matrix and the effects of cytoskeletal remodelling on vascular contractility and vascular stiffness in health and disease.

Rina Yamin (left) is currently a research Assistant Professor in the Health Sciences Department at Boston University. She obtained her PhD from the Hebrew University of Jerusalem, Israel, and did her post-doctoral training at MGH/Harvard Medical School Boston, MA. USA. Her research interest focuses on arterial stiffness with age and more specifically looking at structure and dynamics of the cytoskeleton in focal adhesions of differentiated vascular smooth muscle cells. Kathleen Morgan (right) is currently Professor and Chair of the Department of Health Sciences at Boston University. Her work focuses on the cytoskeleton and signalling pathways of smooth muscle cells in the context of cardiovascular disease and pre-term labour.
Introduction
Vascular smooth muscle cells can exist in a contractile or a proliferating phenotype. Both phenotypes are of interest. Proliferating VSMCs are necessary for embryogenesis and angiogenesis but accelerated proliferation also plays an integral role in pathophysiological states such as atherosclerosis and vascular tumours. On the other hand, the differentiated, contractile phenotype is present in the normal healthy adult blood vessel and is necessary for maintaining normal blood flow and blood pressure. However, when diseased, its function contributes to hypertension, stroke and vascular dementia, heart failure, kidney disease and shock. Here we will use the term VSMC to refer to both phenotypes but dVSMC to refer to the differentiated, contractile phenotype.
Proliferating VSMCs do not contract in the traditional sense of the term and dVSMCs do not proliferate. However, dVSMCs are not terminally differentiated. They can change their phenotype from a quiescent contractile phenotype to a proliferative phenotype in response to inflammatory mediators and growth factors which then, due to transcriptional regulation of specific genes, cause a phenotype change that contributes to the pathogenesis of vascular disease (Beamish et al. 2010; Long et al. 2011). For more details on the functions and mechanisms of proliferative VSMCs see Nishimura et al. (2006). Several excellent reviews have been published covering the consequences of changes from the differentiated to the proliferative phenotype and can be consulted for further details (House et al. 2008; Beamish et al. 2010; Wang et al. 2010). It is worth pointing out that, because of the facility of studying proliferative VSMCs in culture, we know far more about their function than about the cells of the contractile phenotype.
We will review here the current understanding of the actin cytoskeleton in contractile VSMC, its connection to the extra cellular matrix, and the effects of cytoskeletal remodelling on vascular contractility and vascular stiffness in health and disease.
The cytoskeletal components of contractile VSMCs
Definitions
The major types of filaments in cytoskeletal networks are: intermediate filaments, microtubules, myosin and actin.
Intermediate filaments
The intermediate filament network helps to maintain the three-dimensional integrity of the VSMCs. In dVSM the intermediate filaments are found in bundles associated with and often surrounding the dense bodies (DBs) (Berner et al. 1981). They are the dominant filaments during development, but decrease in number as the cells mature (Berner et al. 1981). In dVSMCs intermediate filaments are composed of two proteins, vimentin and desmin (Berner et al. 1981). Vimentin, which is typically expressed in mesenchymally derived non-muscle cells, is the major intermediate filament in smooth muscle in the large arteries. A gradient in the vimentin/desmin expression ratio is found in human and some animal arteries from proximal to distal, with larger arteries containing a high ratio and smaller arteries a low ratio (Frank & Warren, 1981; Gabbiani et al. 1981; Johansson et al. 1997). Tang (2008) reported that in airway smooth muscle the vimentin filament network undergoes spatial rearrangement upon contractile stimulation in response to protein phosphorylation; however, others who looked for phosphorylation-induced regulation of the intermediate filament network in dVSM have produced negative results (Marganski et al. 2005).
Microtubules
Although there is no doubt that dVSM contains abundant tubulin, the degree to which tubulin polymerizes into microtubules and the possible function of microtubules in contractile dVSM is not clear. Many high-resolution micrographs of dVSM show no obvious microtubules (Somlyo, 1980) but the ability to detect microtubules may well depend on the smooth muscle tissue type as well as the staining method since it is notoriously difficult to adequately stain microtubules in dense contractile tissues. Different studies describing microtubular staining in dVSMCs provide contradicting reports on the role of microtubules in contractility of dVSMCs (Paul et al. 2000; Zhang et al. 2000, 2004). One study has shown staining of microtubules with a morphology not very different from that of actin filaments in freshly enzymatically isolated dVSM, but high concentrations of antitubulin antibodies were used and negative controls would have made the results more convincing. Colchicine (15 μm) dimmed the tubulin signal but produced no detectable effect on contractility as measured by shortening velocity (Zhang et al. 2004). Others have also come to similar conclusions based on pharmacological studies (Paul et al. 2000), though some have seen a modest increase in contractility with microtubule depolymerizing drugs (Zhang et al. 2000). There are also reports of endothelial effects of microtubular depolymerization to block relaxation of underlying dVSM (Brum Cde et al. 2005) rather than a direct effect on dVSM contractility.
Smooth muscle myosin II
Smooth muscle myosin contains two heavy chains and two types of light chains, MLC20 and MLC17. MLC20 phosphorylation regulates smooth muscle contraction in response to agonist or electrical depolarization stimuli and is reviewed in Pfitzer (2001). Of note is the fact that individual smooth muscle myosin filaments are surrounded by about fifteen actin filaments (Devine & Somlyo, 1971; Murphy et al. 1974), whereas striated muscle contractile filaments have an actin/myosin filament ratio of 6–8 (Somlyo, 1980). Non-muscle myosin II is also expressed in dVSM but its function is still a matter of controversy (Lofgren et al. 2003; Yuen et al. 2009).
Actin filaments
In dVSM, actin makes up ∼20% of total protein content (Kim et al. 2008b). To date most reported evidence about actin filaments in dSMCs comes from studies of chicken gizzard (Lehman et al. 1987; North et al. 1994b). The contractile actin filament length averages 4.5 μm, which is about 3 times longer than the myosin filament length average of 1.6 μm (Small et al. 1990; North et al. 1994b), but the architecture of the contractile actin filaments remains controversial and is discussed in more detail below. The actin that interacts with myosin generates tension and cell shortening and will be referred to here as contractile actin. Additional actin not associated with myosin is also present in dVSMCs and will be referred to as cytoplasmic actin or non-muscle actin.
Connection to the extracellular matrix/neighbouring cells
dVSMCs are connected to each other either indirectly through attachments to the extracellular matrix or directly via cell-to-cell contacts. The extracellular matrix of the blood vessel wall in essence serves as an intramuscular tendon, transmitting and summing the forces generated by individual contractile elements of the cells. These forces add in parallel, which is one of the reasons that dVSMCs, even though they only contain about 1/5th the amount of myosin present in skeletal muscle (Warshaw et al. 1988), can generate as much or more force/cross-sectional area than does skeletal muscle where the sarcomeres are arranged both in series and in parallel between two tendons.
Connection of the contractile units of dVSM cells with the extracellular matrix occurs through complicated multi-protein structures that attach to the actin cytoskeleton inside the cell and to the matrix through transmembrane proteins. These structures are discussed in detail below and are referred to by multiple names. A term that is unique to the smooth muscle field is the dense plaque (DP), reflecting the early description of these structures by EM or phase microscopy where they were observed as large (∼1 μm) dense structures. The dense plaques are also called adhesion plaques, or focal adhesions (FAs) (Somlyo, 1980; Zhang & Gunst, 2008). The latter term is generally used to describe such structures in non-muscle cells or proliferating VSMCs but can also be used globally for all adherent cell types and is the term that will be used here. There are also cytoplasmic dense bodies (DBs) that are points of insertion of the central contractile filaments on the way to transmitting force at a FA.
Actin isoforms
There are six vertebrate isoforms of actin. Excluding skeletal and cardiac α actins, there are 4 isoforms relevant to smooth muscle: α smooth muscle actin, β non-muscle actin, gamma smooth muscle actin, and γ cytoplasmic actin. They are all separate gene products. α Smooth muscle actin is the predominant isoform in vascular smooth muscle whereas γ smooth muscle actin is largely restricted to gastrointestinal muscles. Both α smooth and γ smooth muscle actin are known to be concentrated in the contractile filaments and will be referred to here as contractile actin. The remaining two isoforms, non-muscle β and non-muscle γ actins have far less clear function in dVSM. Since non-muscle γ and smooth muscle γ actins co-migrate on the 2-D gels generally used to quantitate protein content, to the best of our knowledge it is not known how much smooth muscle γ actin may be present in dVSM. In general, large arteries contain about 60%α smooth muscle actin, 20%β actin and, together, about 20%γ smooth muscle and γ non-muscle actin (Fatigati & Murphy, 1984).
Initially, the dVSM cells embedded in the vascular wall and known to be non-proliferative and non-migratory were viewed as having a rather static cytoskeleton, in a manner similar to healthy adult cardiac and striated muscle cells. However, studies from our group (Kim et al. 2008b, 2010a) and other vascular groups (Cipolla et al. 2002; McGregor et al. 2004; Flavahan et al. 2005) as well as earlier work in airway smooth muscle (Gerthoffer & Gunst, 2001) have made it clear that a small but important fraction of the actin cytoskeleton is remodelled during agonist activation of the tissue. The vasoconstrictors phenylephrine (PE), noradrenaline (NA) and endothelin (ET) (Ohanian et al. 2005; Kim et al. 2008b) have been shown to trigger increases in actin polymerization. Presumably, depolymerization occurs during relaxation to reverse the effect but this has been little studied.
It was initially thought that all filaments contained a mix of actin isoforms (Drew et al. 1991); however, with the availability of specific β and γ non-muscle antibodies and improved microscopic techniques, several more recent publications have demonstrated segregation of actin isoforms into different populations of actin filaments in dVSM (North et al. 1994b; Parker et al. 1994, 1998; Gallant et al. 2011). Our group has shown that γ non-muscle actin is restricted to the cell cortex in marked contrast to the α smooth muscle actin filaments that snake longitudinally down the length of the cell and that punctae of β actin are associated with DBs and FAs (see Fig. 1). Work from our group has also shown that agonist-induced actin incorporation into cortical filaments could be directly observed (Kim et al. 2010a,b). Furthermore, differential centrifugation has shown that the non-muscle actin isoforms are primarily responsible for agonist-induced actin polymerization (Kim et al. 2008b). These findings have led to the conclusion that cortical non-muscle actin is probably the more dynamic subpopulation of actin in dVSM during the action of vasoconstrictors.
Figure 1. Actin isoforms in dVSMCs.

Freshly isolated aortic smooth muscle cells were fixed and stained with anti-α, β and cytoplasmic γ actin antibodies. Shown is a deconvolution microscopy centre optical section of: A, γ actin filaments (red), primarily located in the cell cortex; B, β actin filaments (green), primarily located in the cell interior as well as punctate staining in the cell cortex; and C, α smooth muscle actin (green) in filamentous structures twisting through the length of the cell. Nuclei visualized by DAPI staining (blue). Calibration bar, 10 μm in all panels. Reproduced with permission from Gallant et al. 2011.
A host of actin-binding proteins are known, in other cell types, to regulate branching, elongation and crosslinking of actin as well as to regulate the interaction of the filaments with myosin. The best-studied actin-binding proteins of the dVSM contractile filaments are tropomyosin (Tm), caldesmon and calponin. Further details on the functions of these proteins can be found in other recent reviews (Kim et al. 2008a; Wang, 2008; Schevzov et al. 2011; Lehman & Morgan, 2012), but Tm is of special interest since it has recently been found that dVSMC contains at least five different isoforms of Tm. Furthermore, by cellular imaging it was shown that α smooth muscle Tm (Tm6) associates with α smooth muscle actin in the contractile filaments but β non-muscle Tm (Tm1) associates with γ non-muscle actin in the cortex of the cell, further indicating the existence of different cytoskeletal subdomains in the dVSMC (Fig. 2) (Gallant et al. 2011).
Figure 2. Tropomyosin isoforms Tm1 and Tm6 in isolated dVSMCs.

Freshly isolated aortic dVSMCs were fixed and stained to identify Tm1 (green, top panel) and Tm6 (green, bottom panel). Co-staining with phalloidin for identifies total actin (red, top panel). Co-staining for α actinin identifies focal adhesions and dense bodies (red, bottom panel). Reproduced with permission from Lehman & Morgan, 2012.
Actin polymerization mechanisms
Remodelling of actin cytoskeleton and dVSM plasticity
The smooth muscle of blood vessels is subjected to the mechanical forces due to the pulsatile nature of blood flow and shear stress. Thus, dVSM tissues possess a necessary capability for a rapid adaptation to such external physical changes in order to autoregulate blood flow and blood pressure. Additionally, it is known that the dVSMC is capable of shortening to a far greater degree (down to 50% or less of initial length) than do striated muscle cells (Gabella, 1976; Vetterkind & Morgan, 2012). This effect is especially important in the capacitative smooth muscle organs such as the stomach, the uterus or the venous vascular system. Recent studies provide evidence for the importance of cytoskeletal dynamics in these functions of dVSM. Cipolla et al. have demonstrated an involvement of actin polymerization in myogenic contractions and, conversely, that depolymerization of F-actin with cytochalasin D causes dVSM relaxation (Cipolla et al. 2002). The phenomenon of history-dependent ‘force augmentation’ has been related to increased actin polymerization in the swine carotid artery (Tejani et al. 2011). It has also been reported that histamine and potassium depolarization increase actin polymerization (Meeks et al. 2005; Rembold et al. 2007) in swine carotid artery.
Increases in actin polymerization can take the form of: (1) the nucleation of new actin filaments (against large energetic barriers), (2) the addition of branches to existing filaments, or (3) the elongation of linear filaments. Although an important agonist-induced activation of neural-Wiskott–Aldrich syndrome protein (N-WASP)/Arp2/3 complex (a seven subunit protein that promotes actin polymerization), resulting in the growth of branched actin filaments, has clearly been shown for airway smooth muscle (Zhang et al. 2005; Zhang et al. 2007), this has not yet been demonstrated in dVSM. However, we have demonstrated a role for VASP, a member of the ENA/VASP family responsible for linear filament elongation in dVSMC contractility and actin polymerization (Kim et al. 2010a). In this study, VASP was shown to co-localize with sites of actin polymerization near the cell membrane. It seems likely that agonist-induced actin polymerization in the cortex may involve both elongation and branching of filaments but the actual architecture of the dVSM cortical cytoskeleton, how it changes during contraction–relaxation cycles, and how that links to contractile function are areas where considerable future work is needed.
Biochemical mechanisms
Relatively little is known about the signalling pathways used by dVSMCs to remodel the cytoskeleton but the integrins are the main transmembrane component of FAs. They are also sites of tension transmission between the contractile apparatus and the extracellular matrix (inside-out signalling) as well as sites of transmission of intraluminal pressure to the interior of the dVSMC (outside-in signalling) (Gerthoffer & Gunst, 2001; Sun et al. 2005; Heerkens et al. 2007). It is well known from proliferative cell studies that actin cytoskeletal remodelling in VSM can be stimulated by an integrin-associated mechano-transduction pathway (Martinez-Lemus et al. 2005).
For both the mechano-transduction pathway and agonist-induced changes in actin polymerization, multiple biochemical pathways have been suggested. For example, several groups have shown a role for the GTPase Rho (Albinsson et al. 2004; Tang & Anfinogenova, 2008; Tejani et al. 2011) but three different types of downstream pathways have been suggested.
One Rho-mediated effect involves the formin homology domain protein 1 (FHOD1). FHOD1 is a member of the Dia1 formin family, a Rho GTPase effector protein, which binds to the plus end of actin filaments and recruits actin monomers through binding to profilin (Halayko & Solway, 2001; Wang et al. 2004). However, we are not aware of this pathway being studied directly in dVSM.
Another pathway by which RhoA effects cytoskeletal remodelling is by inhibiting actin filament disassembly. Tejani et al. (2011) observed the regulation of S3 cofilin phosphorylation through Rho signalling that results in actin polymerization in the swine carotid artery. Thus, these results are directly applicable to dVSM. In this pathway RhoA initiates a signalling cascade that leads to phosphorylation and subsequent inactivation of the ADF/cofilin family of actin filament severing/depolymerizing proteins through the action of LIM kinases (Lim domain containing serine/threonine kinases) (Saarikangas et al. 2010). Phosphorylation of cofilin by LIM kinase inhibits its ability to depolymerize actin filaments. Rho kinase induces the phosphorylation of LIM kinase, which enhances LIM kinase activity towards cofilin and promotes cofilin phosphorylation and net actin filament assembly (Gunst & Zhang, 2008; Tang & Anfinogenova, 2008; Zhao et al. 2008). Interestingly, it is difficult to predict the outcome of pathways that regulate cofilin activity since just as inhibition of cofilin inhibits depolymerization, its activation is also known to play a role regulating actin polymerization during SM contraction by creating new polymerization-competent actin filament barbed ends through its severing activity (Kuhn et al. 2000). Thus, the net result in each case needs to be determined by direct F/G actin ratio measurements.
A third pathway involves Cdc42, another small GTPase activated by RhoA through an ERK and PAK1/p38 pathway, which has been found to activate the neural-Wiskott–Aldrich syndrome protein (N-WASP). Activated phosphorylated N-WASP regulates actin branching at FAs in airway dSMCs and cultured aortic SMCs (Li et al. 2003; Albiges-Rizo et al. 2009). However, the relevance of these airway studies to dVSM is yet to be determined.
Finally, the vasodilator-stimulated phosphoprotein VASP, is expressed in dVSMCs, and is targeted to the FAs and DBs in non-muscle as well as dVSM (Benz et al. 2009; Kim et al. 2010a). The details of this vasodilator signalling pathway are unknown but, interestingly, vasodilators that increase cAMP levels leading to PKA activation are known to phosphorylate VASP (hence its name) in a way that inhibits its actin polymerization activities. It is known that VASP also contains predicted PKC phosphorylation sites that could be linked to its vasoconstrictor and pro-actin polymerization activities (Kim et al. 2010a).
Rheology
Recently rheology studies of cytoskeleton dynamics in airway smooth muscle cells imply special constraints on protein–protein interactions that dominate cytoskeleton mechanical properties and have led to a model of the smooth muscle cytoskeleton as a ‘soft-glass’ (Deng et al. 2006; Rembold et al. 2007). However, the relationship between this biophysical model and the biochemical and physiological studies in the dVSM literature is not yet clear.
Cellular organization of the actin cytoskeleton and connection to the focal adhesions
Filament organization in dVSMCs
In 1994, two papers by North et al. (North et al. 1994a,b) described a model of cytoskeletal organization in the gizzard smooth muscle cell. This model was the first attempt at a complete picture of smooth muscle cellular cytoskeletal organization and continues to be a landmark study in the field. This model described contractile actin filaments running diagonally across the width of the cell alternating with myosin filaments, as had been seen by others in toad stomach cells (Fay & Delise, 1973). Another population of actin identified in the North et al. studies, specifically by a newly produced β actin antibody, was observed around DBs, and in longitudinal channels lacking myosin but also containing the intermediate filament protein desmin. The authors envisioned separate contractile filament channels intersecting the β actin/desmin structural channels at the DBs.
The limitations of this model for studies on dVSMCs are, firstly, that the tissue used was avian gizzard and there are likely to be both species-dependent and organ-specific differences in cytoskeletal organization. Secondly, the assumption was made that the majority of non-muscle actin in the cells was β actin, in the absence of any measurement of γ non-muscle actin content. Similarly, the actin in the diagonal fibres was assumed to be gamma smooth muscle actin using an antibody that cross-reacts with all muscle actins. Additionally, the model describes contractile actin filaments directly connecting with DBs and the FAs are not specifically detailed.
More recent evidence, directly from work on dVSMCs, indicates the presence of β actin surrounding both DBs and FAs (Gallant et al. 2011), similar to the North model, but α smooth muscle filaments are associated with myosin in the contractile filaments that snake through the cell, sometimes parallel to the long axis of the cell, sometimes diagonal to the long axis (Fig. 1). Additionally, in the dVSMC there is also a cortical γ non-muscle actin cytoskeleton not described in the North et al. model. Necessary details at the level of EM are still lacking but it appears that the contractile actin filaments insert through/into this γ actin cortical layer to transmit force to the surface of the cell. Similarly, it is likely that α actin filaments insert through some sort of β actin network to either directly or indirectly transmit force to the DBs and FAs. Thus, both the β actin and the γ actin networks, where changes in polymerization state are known to occur (Kim et al. 2008b), are sites of potential regulation of the cytoskeleton both structurally and functionally. A possible arrangement is illustrated in the model shown in Fig. 3.
Figure 3. Schematic model of cytoskeletal organization in aortic dVSMCs.
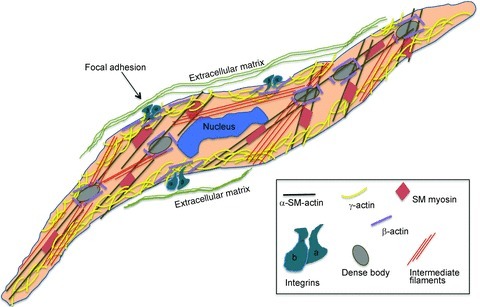
γ Actin mostly in the cell cortex and β actin surrounding the dense bodies and focal adhesions. Symbols and colours as indicated in box.
While the function of the contractile filaments – to generate tension and shorten the cell – is clear, the function of the non-muscle actin cytoskeleton is less clear. The non-muscle cytoskeleton could be purely structural in function as the North et al. model suggested, or it could play an active role in the regulation of cell shape, cell stiffness and force transmission (see below).
Focal adhesions in dVSMCs
FAs in non-muscle cells are known to contain more than 150 different proteins connected to the transmembrane protein integrin at one end and to the actin cytoskeleton at the other. dVSM FAs are probably just as complex, but have been far less studied (Romer et al. 2006). The vast majority of the work on contractile smooth muscle FAs has been performed in airway smooth muscle, where it has been shown that proteins such as paxillin, talin and vinculin undergo phosphorylation and subcellular translocations in response to cholinergic contractile stimuli (Gunst et al. 2003; Opazo Saez et al. 2004). Far less is known about the FA in contractile vascular smooth muscle and even in airway smooth muscle the outcome of phosphorylation-dependent regulation of FA proteins with respect to contractile function continues to be unclear.
Knockdown of the FA proteins in airway smooth muscle has produced dramatic, almost complete ablation of contractile responses (Tang & Gunst, 2001; Opazo Saez et al. 2004), perhaps simply indicating that the contractile apparatus has been disconnected from the integrins and that the FAs have a simple structural function in the cell. At least two major functions of FAs are possible: (1) they could be necessary for force transmission to the extracellular matrix and/or (2) they could be necessary for rearrangement of the actin cytoskeleton during shortening of cells. If actin polymerization occurs at the barbed end of dVSM actin filaments in response to agonists, as has been reported, then the connection to the FAs would have to be, at least transiently, remodelled.
However, it is worth noting that in non-muscle cells and even in proliferative VSMCs, additional, more complex, roles have been proposed for FAs. Integrin-mediated adhesions have been shown to play a role not only in cell spreading and migration, which is not applicable to dVSMCs, but also in regulating the strength of the connection of the cell to the matrix, in remodelling the matrix, in sensing of the chemical and physical properties of the cell environment and as serving as signalling scaffolds (Geiger et al. 2001; Zaidel-Bar et al. 2007a; Wang & McNiven, 2012). Also, in non-muscle cells, early FAs are distinguished from more mature FAs and from very mature fibrillar adhesions, each having specialized functions (Zaidel-Bar et al. 2007b). Work by Davis, Meininger and colleagues has related Ca2+ changes to mechanical stimuli that are linked to focal adhesions. They have demonstrated an involvement of mechano-sensitive ion channels and spatiotemporal aspects of intracellular Ca2+ signalling associated with mechanotransduction (Hill et al. 2001; Sun et al. 2005).
An often overlooked fact is that not all FAs connect with integrins (Wilcox-Adelman et al. 2002; Bellin et al. 2009). Syndecans, transmembrane proteoglycans, also connect the extracellular matrix to the intracellular actin cytoskeleton. Syndecans are present in smooth muscle and at least in one case, myometrial smooth muscle, they are thought to play a regulatory role. Reduced syndecan3 levels have been shown to be associated with prolonged labour in humans (Cluff et al. 2006).
The actual connection of actin filaments to FA proteins remains a bit of a mystery even in thoroughly studied non-muscle cells. Hu et al. (2007) have shown that different FA molecules exhibit varying degrees of correlated motions with actin filaments and have proposed that regulation of protein–protein interactions leads to a ‘hierarchical molecular clutch’ between actin filaments, FA proteins and the extracellular matrix that presumably can be regulated under certain circumstances to ‘shift gears’. One can imagine an action of vasoconstrictors to engage the ‘clutch’ to move dVSM contractility into the ‘next gear’.
Consequences of cytoskeletal remodelling on contractility, stiffness and disease
As described above, dVSMCs are constantly subjected to a continuum of haemodynamic stimuli (e.g. shear stress, flow disturbances, mechanical stimuli from pulsatile blood flow) that alter cytoskeletal dynamics, organization and associated signalling pathways. Under physiological conditions, these alterations in the vessel wall are aimed to autoregulate and ultimately restore basal levels of tensile pressure and stress.
The disease of hypertension is multifactorial in its causes, but in most cases there is a primary or secondary increase in total contractility of the blood vessel wall; hence, many antihypertensives are vasodilators. Under conditions of prolonged increased blood pressure, the VSMCs also remodel to increase wall thickness and normalize the stress on the vessel wall (Folkow, 1993; Skov & Mulvany, 2004). In the long term, vascular hypertrophy and hyperplasia lead to narrowing of small vessels and an undesirable further increase in blood pressure. One could imagine that, in the very earliest stages, mechanotransduction in response to increased pressure would invoke considerable actin cytoskeletal remodelling. Such cytoskeletal remodelling could either be beneficial or could further promote increased remodelling. Additional investigation into the role of subcellular actin remodelling is greatly needed and may point to valuable potential target molecules for drug discovery programs to combat vascular disease, possibly with fewer side effects than direct vasodilators such as calcum channel blockers.
As a further adaption to the elevation in tensile stress, both nuclear factor kappa B (NFkB) and MAP kinase signalling pathways are activated, leading the VSMCs to synthesize key extracellular matrix proteins including increased collagen type I, III and IV in order to increase wall thickness and normalize this stress (Lehoux et al. 2006). However, increased collagen synthesis as well as breakdown and crosslinking of elastin, common also with ageing, are known to lead to increased stiffness of the vasculature. Much attention has recently been given to the fact that not only increased vascular contractility, but also increased vascular stiffness, can serve as biomarkers for cardiovascular disease. In fact, the increase in aortic stiffness associated with ageing, as measured in vivo as an increased pulse wave velocity, has been shown to precede and predict strongly and significantly for increased adverse cardiovascular events (Mitchell, 2008). Stiffer large arteries are less compliant and to a lesser degree dampen the upstream pressure of the heartbeat. Thus, the small vessels are exposed to higher pressures and, especially in the heart, brain and kidneys, pressure-induced damage can lead to the major cardiovascular end points of heart disease, stroke and kidney disease (Mitchell, 2008).
Very recently, changes in the VSMC actin cytoskeleton have been shown to contribute to ageing-induced increases in aortic stiffness in a primate model (Qiu et al. 2010), pointing again to the need for further knowledge of the mechanisms of function of the actin cytoskeleton in order to identify potential therapeutic target molecules (Folkow, 1993).
Summary
An extensive literature on proliferative cells has demonstrated that actin filament polymerization and depolymerization and FA turnover are essential for cell movement, adhesion and division. That literature has been essential in the development of new therapeutic approaches in the fields such as cancer biology. In contrast, very little is now known on the role and mechanism of actin cytoskeletal dynamics and FA function in contractile dVSMCs. We have reviewed here the current understanding of actin cytoskeletal dynamics and FA function in dVSMCs. Recent studies have shown that the cytoskeletal/adhesion connections are far more plastic than once thought. Since the dVSMCs are constantly exposed to mechanical stimuli, understanding the role of actin cytoskeleton dynamics in the dVSMCs is of great importance and may pave the way to identifying potential therapeutic targets to prevent cardiovascular diseases.
Acknowledgments
The preparation of this review was supported by an NIH grant to K.G.M. (NIH HL086655).
Glossary
- DB
dense body
- DP
dense plaque
- dVSMC
differentiated vascular smooth muscle cell
- FA
focal adhesion
- MLC
myosin light chain
- N-WASP
neural-Wiskott–Aldrich syndrome protein
- PE
phenylephrine
- VASP
vasodilator-stimulated phosphoprotein
References
- Albiges-Rizo C, Destaing O, Fourcade B, Planus E, Block MR. Actin machinery and mechanosensitivity in invadopodia, podosomes and focal adhesions. J Cell Sci. 2009;122:3037–3049. doi: 10.1242/jcs.052704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albinsson S, Nordstrom I, Hellstrand P. Stretch of the vascular wall induces smooth muscle differentiation by promoting actin polymerization. J Biol Chem. 2004;279:34849–34855. doi: 10.1074/jbc.M403370200. [DOI] [PubMed] [Google Scholar]
- Beamish JA, He P, Kottke-Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng Part B Rev. 2010;16:467–491. doi: 10.1089/ten.teb.2009.0630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellin RM, Kubicek JD, Frigault MJ, Kamien AJ, Steward RL, Jr, et al. Defining the role of syndecan-4 in mechanotransduction using surface-modification approaches. Proc Natl Acad Sci U S A. 2009;106:22102–22107. doi: 10.1073/pnas.0902639106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz PM, Blume C, Seifert S, Wilhelm S, Waschke J, Schuh K, et al. Differential VASP phosphorylation controls remodeling of the actin cytoskeleton. J Cell Sci. 2009;122:3954–3965. doi: 10.1242/jcs.044537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berner PF, Somlyo AV, Somlyo AP. Hypertrophy-induced increase of intermediate filaments in vascular smooth muscle. J Cell Biol. 1981;88:96–100. doi: 10.1083/jcb.88.1.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum Cde A, Duarte ID, Webb RC, Leite R. Disruption of microtubular network attenuates histamine-induced dilation in rat mesenteric vessels. Am J Physiol Cell Physiol. 2005;288:C443–C449. doi: 10.1152/ajpcell.00130.2004. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. 2002;16:72–76. doi: 10.1096/cj.01-0104hyp. [DOI] [PubMed] [Google Scholar]
- Cluff AH, Bystrom B, Klimaviciute A, Dahlqvist C, Cebers G, Malmstrom A, Ekman-Ordeberg G. Prolonged labour associated with lower expression of syndecan 3 and connexin 43 in human uterine tissue. Reprod Biol Endocrinol. 2006;4:24. doi: 10.1186/1477-7827-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L, Trepat X, Butler JP, Millet E, Morgan KG, Weitz DA, Fredberg JJ. Fast and slow dynamics of the cytoskeleton. Nat Mater. 2006;5:636–640. doi: 10.1038/nmat1685. [DOI] [PubMed] [Google Scholar]
- Devine CE, Somlyo AP. Thick filaments in vascular smooth muscle. J Cell Biol. 1971;49:636–649. doi: 10.1083/jcb.49.3.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew JS, Moos C, Murphy RA. Localization of isoactins in isolated smooth muscle thin filaments by double gold immunolabeling. Am J Physiol Cell Physiol. 1991;260:C1332–C1340. doi: 10.1152/ajpcell.1991.260.6.C1332. [DOI] [PubMed] [Google Scholar]
- Fatigati V, Murphy RA. Actin and tropomyosin variants in smooth muscle. Dependence on tissue type. J Biol Chem. 1984;10:14383–14388. [PubMed] [Google Scholar]
- Fay FS, Delise CM. Contraction of isolated smooth-muscle cells–structural changes. Proc Natl Acad Sci U S A. 1973;70:641–645. doi: 10.1073/pnas.70.3.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan NA, Bailey SR, Flavahan WA, Mitra S, Flavahan S. Imaging remodeling of the actin cytoskeleton in vascular smooth muscle cells after mechanosensitive arteriolar constriction. Am J Physiol Heart Circ Physiol. 2005;288:H660–H669. doi: 10.1152/ajpheart.00608.2004. [DOI] [PubMed] [Google Scholar]
- Folkow B. Early structural changes in hypertension: pathophysiology and clinical consequences. J Cardiovasc Pharmacol. 1993;22:S1–S6. [PubMed] [Google Scholar]
- Frank ED, Warren L. Aortic smooth muscle cells contain vimentin instead of desmin. Proc Natl Acad Sci U S A. 1981;78:3020–3024. doi: 10.1073/pnas.78.5.3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G, Schmid E, Winter S, Chaponnier C, de Ckhastonay C, Vandekerckhove J, Weber K, Franke WW. Vascular smooth muscle cells differ from other smooth muscle cells: predominance of vimentin filaments and a specific alpha-type actin. Proc Natl Acad Sci U S A. 1981;78:298–302. doi: 10.1073/pnas.78.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabella G. Structural changes in smooth muscle cells during isotonic contraction. Cell Tissue Res. 1976;170:187–201. doi: 10.1007/BF00224298. [DOI] [PubMed] [Google Scholar]
- Gallant C, Appel S, Graceffa P, Leavis P, Lin JJ, Gunning PW, et al. Tropomyosin variants describe distinct functional subcellular domains in differentiated vascular smooth muscle cells. Am J Physiol Cell Physiol. 2011;300:C1356–C1365. doi: 10.1152/ajpcell.00450.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger B, Bershadsky A, Pankov R, Yamada KM. Transmembrane crosstalk between the extracellular matrix–cytoskeleton crosstalk. Nat Rev Mol Cell Biol. 2001;2:793–805. doi: 10.1038/35099066. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT, Gunst SJ. Invited review: focal adhesion and small heat shock proteins in the regulation of actin remodeling and contractility in smooth muscle. J Appl Physiol. 2001;91:963–972. doi: 10.1152/jappl.2001.91.2.963. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Tang DD, Opazo Saez A. Cytoskeletal remodeling of the airway smooth muscle cell: a mechanism for adaptation to mechanical forces in the lung. Respir Physiol Neurobiol. 2003;137:151–168. doi: 10.1016/s1569-9048(03)00144-7. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Zhang W. Actin cytoskeletal dynamics in smooth muscle: a new paradigm for the regulation of smooth muscle contraction. Am J Physiol Cell Physiol. 2008;295:C576–C587. doi: 10.1152/ajpcell.00253.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halayko AJ, Solway J. Molecular mechanisms of phenotypic plasticity in smooth muscle cells. J Appl Physiol. 2001;90:358–368. doi: 10.1152/jappl.2001.90.1.358. [DOI] [PubMed] [Google Scholar]
- Heerkens EH, Izzard AS, Heagerty AM. Integrins, vascular remodeling and hypertension. Hypertension. 2007;49:1–4. doi: 10.1161/01.HYP.0000252753.63224.3b. [DOI] [PubMed] [Google Scholar]
- Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol. 2001;91:973–983. doi: 10.1152/jappl.2001.91.2.973. [DOI] [PubMed] [Google Scholar]
- House SJ, Potier M, Bisaillon J, Singer HA, Trebak M. The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. 2008;456:769–785. doi: 10.1007/s00424-008-0491-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu K, Ji L, Applegate K, Danuser G, Waterman-Storer C. Differential transmission of actin motion within focal adhesions. Science. 2007;315:111–115. doi: 10.1126/science.1135085. [DOI] [PubMed] [Google Scholar]
- Johansson B, Eriksson A, Virtanen I, Thornell LE. Intermediate filament proteins in adult human arteries. Anat Rec. 1997;247:439–448. doi: 10.1002/(SICI)1097-0185(199704)247:4<439::AID-AR1>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Kim HR, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med. 2008a;12:2165–2180. doi: 10.1111/j.1582-4934.2008.00552.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Gallant C, Leavis PC, Gunst SJ, Morgan KG. Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. 2008b;295:C768–C778. doi: 10.1152/ajpcell.00174.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Graceffa P, Ferron F, Gallant C, Boczkowska M, Dominguez R, Morgan KG. Actin polymerization in differentiated vascular smooth muscle cells requires vasodilator-stimulated phosphoprotein. Am J Physiol Cell Physiol. 2010a;298:C559–C571. doi: 10.1152/ajpcell.00431.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Leavis PC, Graceffa P, Gallant C, Morgan KG. A new method for direct detection of the sites of actin polymerization in intact cells and its application to differentiated vascular smooth muscle. Am J Physiol Cell Physiol. 2010b;299:C988–C993. doi: 10.1152/ajpcell.00210.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn TB, Meberg PJ, Brown MD, Bernstein BW, Minamide LS, Jensen JR, et al. Regulating actin dynamics in neuronal growth cones by ADF/cofilin and rho family GTPases. J Neurobiol. 2000;44:126–144. [PubMed] [Google Scholar]
- Lehman W, Morgan KG. Structure and dynamics of the actin-based smooth muscle contractile and cytoskeletal apparatus. J Muscle Res Cell Motil. 2012 doi: 10.1007/s10974-012-9283-z. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehman W, Sheldon A, Madonia W. Diversity in smooth muscle thin filament composition. Biochim Biophys Acta. 1987;914:35–39. doi: 10.1016/0167-4838(87)90158-0. [DOI] [PubMed] [Google Scholar]
- Lehoux S, Castier Y, Tedgui A. Molecular mechanisms of the vascular responses to haemodynamic forces. J Intern Med. 2006;259:381–392. doi: 10.1111/j.1365-2796.2006.01624.x. [DOI] [PubMed] [Google Scholar]
- Li S, Moon JJ, Miao H, Jin G, Chen BP, Yuan S, et al. Signal transduction in matrix contraction and the migration of vascular smooth muscle cells in three-dimensional matrix. J Vasc Res. 2003;40:378–388. doi: 10.1159/000072702. [DOI] [PubMed] [Google Scholar]
- Lofgren M, Ekblad E, Morano I, Arner A. Nonmuscle myosin motor of smooth muscle. J Gen Physiol. 2003;121:301–310. doi: 10.1085/jgp.200208720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X, Slivano OJ, Cowan SL, Georger MA, Lee TH, Miano JM. Smooth muscle calponin: an unconventional CArG-dependent gene that antagonizes neointimal formation. Arterioscler Thromb Vasc Biol. 2011;31:2172–2180. doi: 10.1161/ATVBAHA.111.232785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGregor E, Kempster L, Wait R, Gosling M, Dunn MJ, Powell JT. F-actin capping (CapZ) and other contractile saphenous vein smooth muscle proteins are altered by hemodynamic stress: a proteonomic approach. Mol Cell Proteomics. 2004;3:115–124. doi: 10.1074/mcp.M300046-MCP200. [DOI] [PubMed] [Google Scholar]
- Marganski WA, Gangopadhyay SS, Je HD, Gallant C, Morgan KG. Targeting of a novel Ca2+/calmodulin-dependent protein kinase II is essential for extracellular signal-regulated kinase-mediated signaling in differentiated smooth muscle cells. Circ Res. 2005;97:541–549. doi: 10.1161/01.RES.0000182630.29093.0d. [DOI] [PubMed] [Google Scholar]
- Martinez-Lemus LA, Sun Z, Trache A, Trzciakowski JP, Meininger GA. Integrins and regulation of the microcirculation: from arterioles to molecular studies using atomic force microscopy. Microcirculation. 2005;12:99–112. doi: 10.1080/10739680590896054. [DOI] [PubMed] [Google Scholar]
- Meeks MK, Ripley ML, Jin Z, Rembold CM. Heat shock protein 20-mediated force suppression in forskolin-relaxed swine carotid artery. Am J Physiol Cell Physiol. 2005;288:C633–C639. doi: 10.1152/ajpcell.00269.2004. [DOI] [PubMed] [Google Scholar]
- Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol. 2008;105:1652–1660. doi: 10.1152/japplphysiol.90549.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy RA, Herlihy JT, Megerman J. Force generating capacity and contractile protein contenet of arterial smooth muscle. J Gen Physiol. 1974;64:691–705. doi: 10.1085/jgp.64.6.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura J, Bi D, Kanaide H. Dependence of proliferating dedifferentiated vascular smooth muscle contraction on Rho-Rho kinase system. Trends Cardiovasc Med. 2006;16:124–128. doi: 10.1016/j.tcm.2006.02.004. [DOI] [PubMed] [Google Scholar]
- North AJ, Gimona M, Cross RA, Small JV. Calponin is localised in both the contractile apparatus and the cytoskeleton of smooth muscle cells. J Cell Sci. 1994a;107:437–444. doi: 10.1242/jcs.107.3.437. [DOI] [PubMed] [Google Scholar]
- North AJ, Gimona M, Lando Z, Small JV. Actin isoform compartments in chicken gizzard smooth muscle cells. J Cell Sci. 1994b;107:445–455. doi: 10.1242/jcs.107.3.445. [DOI] [PubMed] [Google Scholar]
- Ohanian V, Gatfield K, Ohanian J. Role of the actin cytoskeleton in G-protein-coupled receptor activation of PYK2 and paxillin in vascular smooth muscle. Hypertension. 2005;46:93–99. doi: 10.1161/01.HYP.0000167990.82235.3c. [DOI] [PubMed] [Google Scholar]
- Opazo Saez A, Zhang W, Wu Y, Turner CE, Tang DD, Gunst SJ. Tension development during contractile stimulation of smooth muscle requires recruitment of paxillin and vinculin to the membrane. Am J Physiol Cell Physiol. 2004;286:C433–C447. doi: 10.1152/ajpcell.00030.2003. [DOI] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tang JX, Tao T, Morgan KG. Cytoskeletal targeting of calponin in differentiated, contractile smooth muscle cells of the ferret. J Physiol. 1998;508:187–198. doi: 10.1111/j.1469-7793.1998.187br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker CA, Takahashi K, Tao T, Morgan KG. Agonist-induced redistribution of calponin in contractile vascular smooth muscle cells. Am J Physiol Cell Physiol. 1994;267:C1262–C1270. doi: 10.1152/ajpcell.1994.267.5.C1262. [DOI] [PubMed] [Google Scholar]
- Paul RJ, Bowman PS, Kolodney MS. Effects of microtubule disruption on force, velocity, stiffness and [Ca2+]i in porcine coronary arteries. Am J Physiol Heart Circ Physiol. 2000;279:H2493–H2501. doi: 10.1152/ajpheart.2000.279.5.H2493. [DOI] [PubMed] [Google Scholar]
- Pfitzer G. Invited review: regulation of myosin phosphorylation in smooth muscle. J Appl Physiol. 2001;91:497–503. doi: 10.1152/jappl.2001.91.1.497. [DOI] [PubMed] [Google Scholar]
- Qiu H, Zhu Y, Sun Z, Trzeciakowski JP, Gansner M, Depre C, et al. Short communication: vascular smooth muscle cell stiffness as a mechanism for increased aortic stiffness with aging. Circ Res. 2010;107:615–619. doi: 10.1161/CIRCRESAHA.110.221846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM, Tejani AD, Ripley ML, Han S. Paxillin phosphorylation, actin polymerization, noise temperature & the sustained phase of swine carotid artery contraction. Am J Physiol Cell Physiol. 2007;293:C993–C1002. doi: 10.1152/ajpcell.00090.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romer LH, Birukov KG, Garcia JG. Focal adhesions: paradigm for a signaling nexus. Circ Res. 2006;98:606–616. doi: 10.1161/01.RES.0000207408.31270.db. [DOI] [PubMed] [Google Scholar]
- Saarikangas J, Zhao H, Lappalainen P. Regulation of the actin cytoskeleton-plasma membrane interplay by phosphoinositides. Physiol Rev. 2010;90:259–289. doi: 10.1152/physrev.00036.2009. [DOI] [PubMed] [Google Scholar]
- Schevzov G, Whittaker SP, Fath T, Lin JJ, Gunning PW. Tropomyosin isoforms and reagents. Bioarchitecture. 2011;1:135–164. doi: 10.4161/bioa.1.4.17897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skov K, Mulvany MJ. Structure of renal afferent arterioles in the pathogenesis of hypertension. Acta Physiol Scand. 2004;181:397–405. doi: 10.1111/j.1365-201X.2004.01311.x. [DOI] [PubMed] [Google Scholar]
- Small JV, Herzog M, Barth M, Draeger A. Supercontracted state of vertebrate smooth muscle cell fragments reveals myofilament lengths. J Cell Biol. 1990;111:2451–2461. doi: 10.1083/jcb.111.6.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AV. Ultrastructure of vascular smooth muscle. In: Bohr DF, Somlyo AP, Sparks HV Jr, editors. Handbook of Physiology, section 2, The Cardiovascular System. II. American Physiological Society; 1980. pp. 33–67. [Google Scholar]
- Sun Z, Martinez-Lemus LA, Trache A, Trzeciakowski JP, Davis GE, Pohl U, Meininger GA. Mechanical properties of the interaction between fibronectin and α5β1-integrin on vascular smooth muscle cells studied using atomic force microscopy. Am J Physiol Heart Circ Physiol. 2005;289:H2526–H2535. doi: 10.1152/ajpheart.00658.2004. [DOI] [PubMed] [Google Scholar]
- Tang D, Gunst S. Depletion of focal adhesion kinase by antisense depresses contractile activation of smooth muscle. Am J Physiol Cell Physiol. 2001;280:C874–C883. doi: 10.1152/ajpcell.2001.280.4.C874. [DOI] [PubMed] [Google Scholar]
- Tang DD. Intermediate filaments in smooth muscle. Am J Physiol Cell Physiol. 2008;294:C869–C878. doi: 10.1152/ajpcell.00154.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DD, Anfinogenova Y. Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther. 2008;13:130–140. doi: 10.1177/1074248407313737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tejani AD, Walsh MP, Rembold CM. Tissue length modulates “stimulated actin polymerization,” force augmentation & the rate of swine carotid arterial contraction. Am J Physiol Cell Physiol. 2011;301:C1470–C1478. doi: 10.1152/ajpcell.00149.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetterkind S, Morgan KG. Regulation of smooth muscle contraction. In: Hil JA, Olson EN, editors. Muscle; Fundamental Biology and Mechanism of Disease. London: Elsevier; 2012. [Google Scholar]
- Wang CLA. Caldesmon and the regulation of cytoskeletal functions. In: Gunning P, editor. Tropomyosin. Landes Bioscience and Springer Science + Business media; 2008. pp. 250–272. [Google Scholar]
- Wang M, Monticone RE, Lakatta EG. Arterial aging: a journey into subclinical arterial disease. Curr Opin Nephrol Hypertens. 2010;19:201–207. doi: 10.1097/MNH.0b013e3283361c0b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, El-Zaru MR, Surks HK, Mendelsohn ME. Formin homology domain protein (FHOD1) is a cyclic GMP-dependent protein kinase I-binding protein and substrate in vascular smooth muscle cells. J Biol Chem. 2004;279:24420–24426. doi: 10.1074/jbc.M313823200. [DOI] [PubMed] [Google Scholar]
- Wang Y, McNiven MA. Invasive matrix degradation at focal adhesions occurs via protease recruitment by a FAK-p130Cas complex. J Cell Biol. 2012;196:375–385. doi: 10.1083/jcb.201105153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshaw DM, Rees DD, Fay FS. Characterization of cross-bridge elasticity and kinetics of cross-bridge cycling during force development in single smooth muscle cells. J Gen Physiol. 1988;91:761–779. doi: 10.1085/jgp.91.6.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox-Adelman SA, Denhez F, Goetinck PF. Syndecan-4 modulates focal adhesion kinase phosphorylation. J Biol Chem. 2002;277:32970–32977. doi: 10.1074/jbc.M201283200. [DOI] [PubMed] [Google Scholar]
- Yuen S, Ogut O, Brozovich FV. Nonmuscle myosin is regulated during smooth muscle contraction. Am J Physiol Heart Circ Physiol. 2009;297:H191–H199. doi: 10.1152/ajpheart.00132.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Itzkovitz S, Ma’ayan A, Iyengar R, Geiger B. Functional atlas of the integrin adhesome. Nat Cell Biol. 2007a;9:858–867. doi: 10.1038/ncb0807-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidel-Bar R, Milo R, Kam Z, Geiger B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci. 2007b;120:137–148. doi: 10.1242/jcs.03314. [DOI] [PubMed] [Google Scholar]
- Zhang D, Jin N, Rhoades RA, Yancey KW, Swartz DR. Influence of microtubules on vascular smooth muscle contraction. J Muscle Res Cell Motil. 2000;21:293–300. doi: 10.1023/a:1005600118157. [DOI] [PubMed] [Google Scholar]
- Zhang D, Sherwood J, Li L, Swartz DR. Unloaded shortening velocity in single permeabilized vascular smooth muscle cells is independent of microtubule status. J Muscle Res Cell Motil. 2004;25:167–175. doi: 10.1023/b:jure.0000035898.10847.a1. [DOI] [PubMed] [Google Scholar]
- Zhang W, Gunst SJ. Interactions of airway smooth muscle cells with their tissue matrix: implications for contraction. Proc Am Thorac Soc. 2008;5:32–39. doi: 10.1513/pats.200704-048VS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wu Y, Du L, Tang DD, Gunst SJ. Activation of the Arp2/3 complex by N-WASp is required for actin polymerization and contraction in smooth muscle. Am J Physiol Cell Physiol. 2005;288:C1145–C1160. doi: 10.1152/ajpcell.00387.2004. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wu Y, Wu C, Gunst SJ. Integrin-linked kinase regulates N-WASp-mediated actin polymerization and tension development in tracheal smooth muscle. J Biol Chem. 2007;282:34568–34580. doi: 10.1074/jbc.M704966200. [DOI] [PubMed] [Google Scholar]
- Zhao R, Du L, Huang Y, Wu Y, Gunst SJ. Actin depolymerization factor/cofilin activation regulates actin polymerization and tension development in canine tracheal smooth muscle. J Biol Chem. 2008;283:36522–36531. doi: 10.1074/jbc.M805294200. [DOI] [PMC free article] [PubMed] [Google Scholar]