

Abstract
Store-operated Ca2+ entry (SOCE) in cells of the immune system is mediated by Ca2+ release-activated Ca2+ (CRAC) channels that are formed by ORAI1 and its homologues ORAI2 and ORAI3. They are activated by stromal interaction molecules (STIM) 1 and 2 in response to depletion of endoplasmic reticulum Ca2+ stores. Loss-of-function mutations in the human ORAI1 and STIM1 genes abolish CRAC channel function and SOCE in a variety of non-excitable cells including lymphocytes and other immune cells, resulting in a unique clinical syndrome termed CRAC channelopathy. It is dominated by severe immunodeficiency and autoimmunity due to impaired SOCE and defects in the function of several lymphocyte subsets. These include CD8+ T cells, CD4+ effector and regulatory T cells, natural killer (NK) cells and B cells. This review provides a concise discussion of the role of CRAC channels in these lymphocyte populations and the regulation of adaptive immune responses to infection, in autoimmunity and inflammation.

Stefan Feske (left) received his medical degree from the University of Freiburg, Germany, with a thesis on signalling defects in lymphocytes of immunodeficient patients. Following a residency in rheumatology with H. H. Peter, he joined the lab of A. Rao at Harvard Medical School for postdoctoral training. There he and colleagues discovered ORAI1 as the pore-forming subunit of the CRAC channel. The focus of his lab at New York University School of Medicine is on the mechanisms of CRAC channel function in cells of the immune system. Patrick Shaw (right) received his doctorate from Michigan State University in Pharmacology & Toxicology. His thesis examined the interactions between idiosyncratic hepatotoxicants and inflammation. Following graduation, he accepted a postdoctoral research fellowship at St Jude Children's Research Hospital working in the lab of T. Kanneganti. His projects there focused on the role of NLR proteins in adaptive immunity. Subsequently, he accepted a postdoctoral research position in the lab of S. Feske. His current projects focus on the role of CRAC channels in lymphocytes during autoimmunity and antiviral immune responses.
Introduction
Ca2+ signals in lymphocytes are critical regulators of immunity to infection, inflammation and autoimmunity. Elevations in intracellular Ca2+ concentrations originate from two sources: the release of Ca2+ from intracellular stores and influx from the extracellular space through plasma membrane Ca2+ channels. The best described Ca2+ influx pathway in lymphocytes involves Ca2+ release-activated Ca2+ (CRAC) channels, which are composed of ORAI proteins (Fig. 1). ORAI1 (and its homologues ORAI2 and ORAI3) are small tetraspanning plasma membrane proteins that form the pore of the CRAC channel. They are activated by STIM1 and STIM2, which are localized in the membrane of the endoplasmic reticulum (ER). STIM proteins sense the depletion of ER Ca2+ stores and directly bind to ORAI proteins in the plasma membrane. Since the activation of CRAC channels and the subsequent Ca2+ influx are controlled by the filling state of the ER, this Ca2+ signalling pathway is called store-operated Ca2+ entry (SOCE) or – using an older term – capacitative Ca2+ entry (Putney, 1986, 1990).
Figure 1. ORAI1, STIM1 and STIM2 mediate SOCE and control gene expression in distinct T cell subsets.
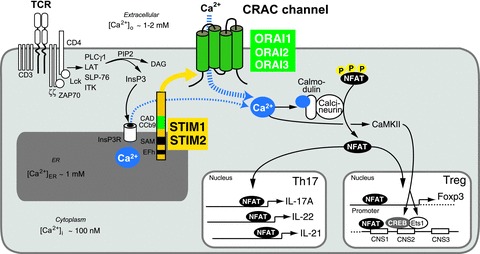
A, SOCE is activated following T cell receptor (TCR) or B cell receptor (BCR, not shown) ligation in T and B cells. In T cells, the protein tyrosine kinases Lck and ZAP-70 initiate a signalling cascade that results in the activation of PLCγ1 and production of InsP3, a second messenger whose binding to the InsP3 receptor leads to the release of Ca2+ from the ER. The subsequent reduction of [Ca2+]ER causes the dissociation of Ca2+ from EF hand (EFh) domains in the N terminus of STIM1 and STIM2, unfolding of the adjacent EFh-SAM domains and oligomerization of STIM molecules. Oligomerized STIM assembles in puncta localized at junctions formed by the ER and plasma membrane to which ORAI channels are recruited. STIM1 binds to and activates ORAI1 via a CRAC activation domain (CAD or CCb9) in its C terminus. Opening of ORAI CRAC channels in the plasma membrane results in sustained Ca2+ influx and activation of several Ca2+ regulated enzymes and transcription factors. Of particular importance in lymphocytes is the serine/threonine phosphatase calcineurin, which dephosphorylates NFAT (nuclear factor of activated T cells) and thereby enables it to translocate to the nucleus and bind to promoters and conserved non-coding DNA sequences (CNS) of many target genes. The expression of several cytokines (IL-17A, IL-22, IL-21) in Th17 cells and the lineage-specific transcription factor Foxp3 in Treg cells are regulated by NFAT. (Note: only NFAT and other Ca2+ dependent transcription factors regulating cytokine and Foxp3 expression are shown in this figure.)
Ca2+ signals in lymphocytes are initiated by a variety of antigen receptors on the cell surface including the T cell receptor (TCR), B cell receptor (BCR) and a variety of NK cell receptors such as FcγRIIIa/b, NKp46 and 2B4 (Bryceson et al. 2006). Engagement of antigen receptors results in the activation of phospholipase C (PLC) γ and the resulting hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2) into 1,4,5-inositol trisphosphate (InsP3) and diacylglycerol (DAG). InsP3 binding to InsP3 receptors in the ER causes the release of Ca2+ from ER stores and thereby SOCE (Fig. 1). Ca2+ signals arising from CRAC channels regulate a variety of Ca2+-dependent kinases, phosphatases and transcription factors such as the nuclear factor of activated T cells (NFAT), NF-κB and myocyte enhancer factor (MEF) 2 (Macian, 2005; Savignac et al. 2007) through which they control lymphocyte functions such as cytokine production, proliferation, differentiation and cytotoxicity (Pores-Fernando & Zweifach, 2009; Shaw & Feske, 2011). In this review, we will provide an overview of the role of CRAC channels in lymphocyte function. We will discuss how the deletion of ORAI1, STIM1 and STIM2 genes in human patients and mice results in immune dysregulation, and examine the consequences of impaired SOCE for immunity to infection and autoimmunity.
SOCE in lymphocytes is mediated by ORAI and STIM proteins
ORAI1 proteins form the highly Ca2+ selective CRAC channel in lymphocytes. The biophysical properties and gating mechanism of ORAI1 channels are discussed elsewhere in this issue (McNally & Prakriya, 2012). ORAI1 and its homologue ORAI3 are almost ubiquitously expressed in human and murine tissues and cell types, whereas ORAI2 has a more restricted expression pattern (Gwack et al. 2007; McCarl et al. 2009; Feske, 2010). ORAI1 protein expression is found in all murine lymphocyte subtypes (McCarl et al. 2010). mRNA for all three ORAI homologues can be detected in lymphoid and myeloid cells of the adaptive and innate immune system, but relative levels vary by database and gene expression array used for analysis (BioGPS; ImmunologicalGenomeProject). ORAI1 is the best-characterized and dominant CRAC channel homologue in lymphocytes and myeloid cells. Defects in ORAI1 expression or function caused by inherited mutations in the ORAI1 gene of immunodeficient patients abolishes CRAC currents and SOCE in T cells, NK cells, B cells and neutrophils (Partiseti et al. 1994; Le Deist et al. 1995; Feske et al. 2001, 2005, 2006; McCarl et al. 2009; Maul-Pavicic et al. 2011; and reviewed in Feske, 2010, 2011). In knockout mice lacking ORAI1 or knock-in mice expressing the non-functional ORAI1-R93W mutant protein, SOCE is partially impaired in naive CD4+ and CD8+ T cells, whereas in vitro differentiated T cells from these mice lack SOCE and CRAC channel function almost completely (Fig. 2; Vig et al. 2008; McCarl et al. 2010). These findings indicate that ORAI1 is essential for SOCE in murine lymphocytes, but that residual Ca2+ influx in naive T cells may be provided – unlike in human T cells – by ORAI2 and ORAI3 (Vig et al. 2008; McCarl et al. 2010). The roles of ORAI2 and ORAI3 in SOCE in T cells and other immune cells has, however, not been established and awaits the investigation of ORAI2- and ORAI3-deficient mice and human patients.
Figure 2. ORAI1, STIM1 and STIM2 control SOCE and cytokine production in CD4+ T cells.
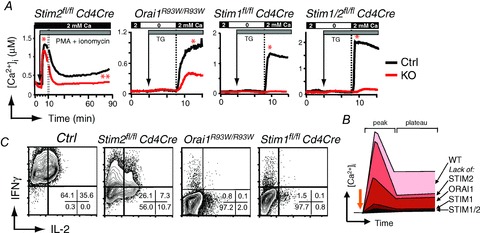
A, T cell-specific deletion of STIM2 in CD4+ T cells from Stim2fl/fl Cd4Cre mice does not interfere with the peak of Ca2+ influx (*) but impairs sustained SOCE (**) compared to wild-type control (Ctrl) T cells. Gradually more severe defects in SOCE are observed (in this order) in ORAI1-, STIM1- and STIM1/2-deficient CD4+ T cells (from Oh-Hora et al. 2008; McCarl et al. 2010 and authors unpublished observations). T cells were stimulated with PMA (10 nm), ionomycin (0.5 μm) and thapsigargin (TG, 1 μm) in Ringer solution containing 0 or 2 mm Ca2+ as indicated. In Orai1R93W knock-in mice, the endogenous Orai1 gene was replaced with a mutant (Arg → Trp) allele that encodes a non-functional ORAI1 protein. B, summary of SOCE levels in ORAI/STIM-deficient murine CD4+ T cells. C, cytokine production by CD4+ T cells from the indicated mice that were stimulated for 6 h with PMA and ionomycin and analysed by intracellular cytokine staining in flow cytometry (from Oh-Hora et al. 2008; McCarl et al. 2010; Copyright 2010. The American Association of Immunologists, Inc.). Note that >99% of wild-type Ctrl T cells express IFNγ or IFNγ and IL-2, whereas this percentage is gradually reduced in STIM2-, ORAI1- and STIM1-deficient CD4+ T cells.
STIM1 and STIM2 are single-pass membrane proteins predominantly localized in the ER that sense the depletion of ER Ca2+ stores and, through a series of multimerization and translocation events, bind to and activate ORAI channels. Their function in SOCE has been reviewed in great detail elsewhere (Cahalan, 2009; Hogan et al. 2010). STIM1 and STIM2 are expressed in all lymphoid lineages where they control SOCE (Oh-Hora et al. 2008; Beyersdorf et al. 2009; Matsumoto et al. 2011). While STIM1 has been widely studied and is considered to be the main activator of CRAC channels, STIM2 plays a lesser albeit important role for SOCE in lymphocytes (Fig. 2). Accounting for the distinct contributions of STIM1 and STIM2 to SOCE in murine lymphocytes are their different expression levels: STIM1 is constitutively expressed in naive and T helper (Th) cells whereas STIM2 is undetectable in naive CD4+ T cells and is upregulated upon differentiation into Th cells in vitro (Oh-Hora et al. 2008). Furthermore the kinetics and [Ca2+]ER levels that trigger STIM1 and STIM2 activation are distinct. STIM1 oligomerization occurs when [Ca2+]ER is decreased to <∼0.4 μm, whereas STIM2 responds to smaller reductions in resting [Ca2+]ER (<∼0.8 μm) (Brandman et al. 2007). In addition, the unfolding of the EFh-SAM domain of STIM1 occurs ∼3.5–4 times faster than that of STIM2, resulting in >70 times shorter oligomerization kinetics of STIM1 compared to STIM2 (Stathopulos et al. 2009). Collectively, these findings are consistent with a role of STIM1 as the main mediator of immediate SOCE in response to strong antigen receptor-induced signals that result in significant depletion of ER Ca2+ stores. By contrast, STIM2 responds to weaker stimuli that only partially deplete ER Ca2+ stores and is required to sustain prolonged SOCE.
Lack of STIM1 expression in human patients with loss-of-function mutations in STIM1 or mice with genetic deletion of the Stim1 gene results in almost completely abolished SOCE in T cells, B cells and a variety of myeloid cells (Fig. 2; Baba et al. 2008; Oh-Hora et al. 2008; Picard et al. 2009; Matsumoto et al. 2011; Fuchs et al. 2012; and authors' unpublished observations). By contrast, STIM2 deletion in murine T cells does not have a significant effect on peak Ca2+ influx occurring in the first minutes after antigen receptor stimulation or passive store depletion but abolishes sustained Ca2+ influx (Fig. 2; Oh-Hora et al. 2008). The latter is required for maintaining activation of the Ca2+ dependent transcription factor NFAT and cytokine gene expression (Dolmetsch et al. 1997; Oh-Hora et al. 2008). Consequently, STIM2-deficient CD4+ T cells fail to sustain nuclear translocation of NFAT and show a significant defect in IL-2 and IFNγ production (Oh-Hora et al. 2008). In B cells, deletion of STIM2 alone causes only a moderate reduction in SOCE (Matsumoto et al. 2011). Nevertheless, STIM2 has a significant role in murine lymphocyte function as only combined deletion of STIM1 and STIM2 abolishes SOCE in T and B cells compared to deletion of STIM1 alone (Oh-Hora et al. 2008; Matsumoto et al. 2011) and severe immune dysregulation is often observed only in STIM1/STIM2- but not STIM1- or ORAI1-deficient mice as will be discussed further below.
CRAC channels are essential for immunity to infection
CRAC channelopathy due to mutations in ORAI1 and STIM1 genes causes immunodeficiency in patients
The importance of CRAC channel function for immunity is most evident in patients that are homozygous for autosomal recessive mutations in STIM1 or ORAI1 genes which either abolish mRNA and protein expression or result in non-functional proteins (Feske et al. 2006; McCarl et al. 2009; Picard et al. 2009; Fuchs et al. 2012). All mutations reported in patients to date severely impair CRAC channel function and SOCE resulting in a unique clinical disease syndrome we termed CRAC channelopathy (Feske, 2010). It is defined by severe immunodeficiency, autoimmunity, congenital myopathy with atrophy of type II muscle fibres and ectodermal dysplasia with anhydrosis and a dental enamel calcification defect (Feske, 2010, 2011). The leading symptom responsible for the high mortality of CRAC-deficient patients in their first year of life is immunodeficiency, which results in life-threatening infections with bacterial, fungal and viral pathogens. Immunodeficiency is largely due to impaired T cell function, but SOCE defects in other immune cell types are likely to contribute to the increased susceptibility to infections as well since SOCE was also found to be severely impaired in B cells, NK cells and neutrophils of ORAI1 and STIM1-deficient patients (Le Deist et al. 1995; Feske et al. 2001, 2005; Maul-Pavicic et al. 2011). The types of mutations in ORAI1 and STIM1 genes and the resulting clinical phenotypes have been reviewed in detail elsewhere (Feske, 2009, 2010, 2011; Feske et al. 2010).
CRAC channels are required for CD8+ T cell function
CRAC channel-deficient patients lacking functional ORAI1 or STIM1 are particularly susceptible to chronic viral infections, especially herpes viruses such as cytomegalovirus (CMV) and Epstein-Barr virus (EBV). In the most severe case, one patient suffered from disseminated Kaposi sarcoma due to human herpes virus (HHV) 8 infection (Byun et al. 2010; Sahin et al. 2010). CD8+ T cells, along with several other cell types in the adaptive and innate immune system, are critical to control viral infections and Ca2+ influx through CRAC channels is important for their function (reviewed in Pores-Fernando & Zweifach, 2009). Naive CD8+ T cells and differentiated cytotoxic T cells (CTL) from Orai1−/− and Orai1R93W/R93W knock-in mice had reduced SOCE and CRAC channel function indicating that ORAI1 is an essential CRAC channel homologue in CD8+ T cells (Gwack et al. 2008; McCarl et al. 2010). Lack of ORAI1 in naive CD8+ T cells partially reduced Ca2+ influx whereas SOCE was abolished in ORAI1-deficient CTL that had been cultured in vitro (Gwack et al. 2008; McCarl et al. 2010). Human CD8+ T cells from patients with non-functional ORAI1 or STIM1 completely lacked SOCE resulting in severely dysregulated NFAT activation and gene expression (Feske et al. 2001, 2005; Fuchs et al. 2012). Collectively, these findings indicate that ORAI1 is the main CRAC channel homologue in human CD8+ T cells and mouse CTL, whereas ORAI2 or ORAI3 may contribute to SOCE in naive murine CD8+ T cells.
In addition to transcriptional regulation, SOCE is critical for cytolytic effector functions of CD8+ T cells. Ca2+ signals are involved in many key steps of CTL-mediated lysis of target cells including the release of perforin- and granzyme-containing lytic granules at the immune synapse (IS) (Pores-Fernando & Zweifach, 2009). The IS is formed between CTL and their target cells, for instance virus-infected or tumour cells (Pipkin & Lieberman, 2007). It is stabilized by interactions between the TCR and peptide-MHC (major histocompatibility complex) complexes as well as the binding of integrin and co-stimulatory molecules on T cells and their target cells. Formation of the IS ensures the directed and spatially confined release of cytotoxic granules to kill target cells in an antigen-specific manner while limiting the killing of bystander cells. Granule release and cytotoxicity were shown to depend on Ca2+ influx via CRAC channels whereas Ca2+ release from intracellular stores was not sufficient to promote degranulation (Takayama & Sitkovsky, 1987; Pores-Fernando et al. 2005). A recent report furthermore showed that rapid and robust Ca2+ influx leads to the fast recruitment of lytic granules to the centre of the IS (Beal et al. 2009). This raises the question whether localized Ca2+ influx at the IS is required for subsequent granule exocytosis and whether CRAC channels are recruited to the IS to facilitate release. Localized Ca2+ influx at the IS, however, was not observed in studies using the cytotoxic T-cell leukemic line TALL-104 and Raji B cell lymphoma cells as targets, suggesting that CRAC channels in CTL may not be polarized during killing (Lyubchenko et al. 2001). By contrast, a more recent report showed that ORAI1 Ca2+ channels accumulate at the IS formed by Jurkat T cells and primary human T cells (Barr et al. 2008; Lioudyno et al. 2008; Quintana et al. 2011), consistent with a potential requirement for SOCE in the polarization of lytic granules to the IS and their directed secretion.
The significance of SOCE in CTL is emphasized by the impaired cytotoxic function of CD8+ T cells and NK cells (see below) from patients and mice lacking functional ORAI and STIM genes. CD8+ T cells from STIM1/STIM2-deficient mice lacked SOCE completely and showed impaired exocytosis of lytic granules and killing of tumour target cells (S. Feske, unpublished observations). By contrast, the cytotoxic function of human CD8+ T cells isolated from STIM1-deficient patients was normal (Maul-Pavicic et al. 2011). The cause for this discrepancy between human and mouse CD8+ T cell function is unknown, but may be related to species differences or the presence of residual SOCE in human CD8+ T cells lacking only STIM1. The latter would suggest that modest SOCE is sufficient for the cytolytic function of CD8+ T cells. Future experiments will have to assess the role of SOCE in cytotoxic CD8+ T cell function in vivo in the context of immunity to tumours and antiviral immune responses.
CRAC channels are required for NK cell function
Like CD8+ T cells, natural killer (NK) cells have cytolytic function and mediate antitumour and antiviral immune responses. They express a variety of activating and inhibitory receptors. Crosslinking of several activating receptors such as the low affinity Fcγ receptors for IgG (FcγRIIIa/b or CD16a/b), NKp46 and 2B4 induces SOCE (Bryceson et al. 2006). A recent study using NK cells from patients with mutations in ORAI1 and STIM1 genes has shown that SOCE is required for the production of cytokines and the release of cytotoxic granules in NK cells (Maul-Pavicic et al. 2011; Fuchs et al. 2012). NK cells from these patients lacked SOCE following CD16 stimulation or passive store depletion with thapsigargin and showed impaired production of IFNγ and TNFα when co-incubated with tumour cells. Furthermore, SOCE-deficient NK cells failed to exocytose their lytic granules and accordingly were severely impaired in their ability to kill tumour cells in vitro (Maul-Pavicic et al. 2011). A partial defect in NK cell degranulation was also observed in NK cells from Orai1R93W/R93W mice (authors' unpublished observations).
While NK cell function requires SOCE, NK cell development does not and is normal in ORAI1- and STIM1-deficient patients and mice. By contrast, the development of NKT cells, a subgroup of CD1d-restricted T cells expressing an invariant T cell receptor that recognizes lipid and glycolipid antigens, appears to require SOCE. Virtually no CD3+ Va24+ Vb11+ NKT cells were found in the blood of a STIM1-deficient patient (Fuchs et al. 2012). This finding is consistent with the lack of NKT cells in calcineurin-deficient mice as well as mice lacking the NFAT-dependent transcription factor Egr2 (Lazarevic et al. 2009). The immunological significance of the functional defects observed in SOCE-deficient NK cells and the developmental defect of NKT cells is not yet understood. It also remains to be elucidated to what extent the impaired function and development of the individual cytotoxic lymphocyte subtypes (CD8+ T cells, NK cells, NKT cells) contribute to the increased susceptibility to viral infections and the increased incidence of virus-associated malignancies in patients (EBV-associated lymphoma, HHV8-associated Kaposi sarcoma) with loss-of-function mutations in ORAI1 and STIM1 genes (McCarl et al. 2009; Byun et al. 2010; Feske et al. 2010).
CRAC channels in inflammation and autoimmunity
SOCE controls effector CD4+ T cell functions
SOCE controls the function of CD4+ T cells, which are important mediators of inflammation and autoimmunity but also regulators of immunological tolerance. SOCE in CD4+ T cells is mediated predominantly by ORAI1 and STIM1 as CD4+ T cells from patients with mutations in either gene showed strongly reduced SOCE, NFAT activation and production of cytokines such as IL-2, IL-4, IFNγ and TNFα (Feske et al. 1996, 2001). The critical role of SOCE for CD4+ T cell function is confirmed by impaired cytokine production in several murine CD4+ T helper (Th) cell subsets derived from ORAI1- and STIM1-deficient mice. When naive CD4+ T cells from these mice were differentiated into Th1, Th2 and Th17 cells in vitro (Fig. 3), they produced drastically lower levels of IFNγ, TNFα, IL-10, IL-4 and IL-17, respectively, upon stimulation (Figs 2 and 3; Oh-Hora et al. 2008; Ma et al. 2010; McCarl et al. 2010). As discussed in an earlier section, STIM2 is required for sustained SOCE and NFAT activation and its genetic deletion in CD4+ T cells from Stim2fl/fl Cd4Cre mice impairs the expression of cytokines such as IFNγ and IL-17 (Fig. 2; Ma et al. 2010). SOCE controls the proliferation of human T cells, which is significantly reduced in patients with CRAC channelopathy (Le Deist et al. 1995; Feske et al. 1996). By contrast, the proliferation of murine CD4+ T cells is impaired only in T cells lacking both STIM1 and STIM2, but is normal in T cells lacking STIM1 or ORAI1 alone (Oh-Hora et al. 2008; Beyersdorf et al. 2009; McCarl et al. 2010). Kim et al. described enhanced proliferation of CD4+ T cells from Orai1−/− mice upon repeated TCR stimulation; however, this defect was due to increased resistance to apoptotic cell death (Kim et al. 2011). The cause of the more severe proliferation defect in ORAI1/STIM1-deficient human compared to mouse T cells is not known but may be related to a greater functional redundancy of ORAI/STIM proteins in mouse lymphocytes.
Figure 3. Role of SOCE in the differentiation and function of CD4+ T cells.
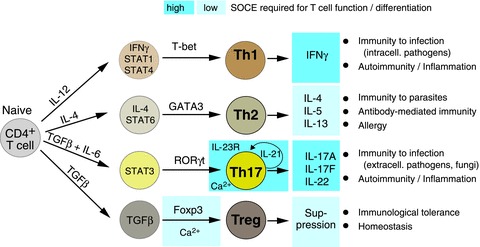
Naive CD4+ T cells differentiate into distinct T helper (Th) cell types and regulatory T (Treg) cells whose role in immunity is indicated on the right. Differentiation into Th1, Th2, Th17 or Treg cells is influenced by the cytokine milieu in which TCR stimulation occurs. Indicated are the cytokines and STAT (signal transducer and activator of transcription) molecules that initiate CD4+ T cell differentiation (left column) and promote expression of lineage specific transcription factors (indicated above arrows). In Treg cells, SOCE is required for Foxp3 expression and Treg differentiation. In Th17 cells, SOCE controls expression of IL-21 and the receptor for IL-23 (IL-23R), cytokines which have been implicated in the terminal differentiation and survival of Th17 cells. In addition to influencing CD4+ T cell differentiation, SOCE is essential for effector functions of Th1, Th2, Th17 and Treg cells. The levels of SOCE required for the differentiation and function of distinct CD4+ T cell types are indicated by blue boxes (light blue: low levels of SOCE sufficient; dark blue: robust, sustained SOCE required).
While SOCE is not required for the proliferation of murine CD4+ T cells in general, the growth and differentiation of Th17 cells appears to require SOCE. Naive CD4+ T cells differentiate into proinflammatory Th17 cells under the influence of the transcription factor RORγt and cytokines such as TGFβ, IL-6, IL-21 and IL-23 (Fig. 3; Zhou & Littman, 2009). STIM1-deficient CD4+ T cells grown under Th17 polarizing conditions in vitro (in the presence of IL-6 and TGFβ) failed to proliferate, a defect that is likely to be related to their impaired expression of IL-21 and the receptor for IL-23 (Fig. 3; Ma et al. 2010). Consistent with an important role of SOCE in the function of Th17 cells is the profound defect in the production of effector cytokines IL-17A, IL-17F and IL-22 by STIM1- or STIM2-deficient T cells (Ma et al. 2010; Schuhmann et al. 2010). Importantly, SOCE-deficient T cells failed to induce autoimmunity in two animal models of disease in which Th17 plays an important pathophysiological role: inflammatory bowel disease (IBD), a mouse model for ulcerative colitis and Crohn's disease, and experimental autoimmune encephalomyelitis (EAE), a mouse model for multiple sclerosis. When STIM1-deficient mice were immunized with myelin-oligodendrocyte glycoprotein peptide to induce EAE, they were protected from autoimmune CNS inflammation (Ma et al. 2010; Schuhmann et al. 2010). T cells isolated from the CNS and draining lymph nodes of mice failed to produce IL-17A (Ma et al. 2010). Interestingly, STIM2-deficient mice were partially protected from EAE suggesting that STIM2 is required for SOCE and Th17 cell function in vivo (Ma et al. 2010; Schuhmann et al. 2010). In a murine model of colitis, adoptive transfer of naive CD4+ T cells from ORAI1- or STIM1-deficient mice failed to induce IBD in contrast to the transfer of wild-type T cells. Protection was associated with a complete absence of mucosal inflammation in the colon of recipient mice and strongly attenuated production of proinflammatory cytokines, especially IFNγ and IL-17, in the gut (McCarl et al. 2010). These findings are consistent with reduced SOCE and expression of IFNγ observed in lamina propria (LP) mononuclear cells that were isolated from biopsies of patients with IBD and treated with a CRAC channel inhibitor in vitro (Di Sabatino et al. 2009). Interestingly, CD4+ and CD8+ T cells isolated from the LP of healthy individuals have significantly lower SOCE compared to T cells in the peripheral blood (Schwarz et al. 2004) whereas LP T cells isolated from patients with IBD have robust SOCE similar to that in blood T cells. While the study by Schwarz et al. did not compare the activation status or cytokine production by LP T cells isolated from IBD patients and healthy controls, these findings suggest that T cells present in the inflamed LP of IBD patients are effector T cells (most likely Th1 and Th17 cells) that have upregulated expression of SOCE genes on which they depend for their proinflammatory function. Collectively, the studies described above show that CRAC channels are important regulators of T cell-mediated inflammation and autoimmunity. How they control the function of T cells and the balance of different Th subsets in inflammation remains to be elucidated.
SOCE controls regulatory T cell function and the maintenance of immunological tolerance
In addition to immunodeficiency, SOCE-deficient patients suffer from autoimmunity characterized by haemolytic anaemia and thrombocytopenia caused by autoantibodies against red blood cells and platelets (Picard et al. 2009; Fuchs et al. 2012). The autoimmunity and inflammatory disease observed in patients may have several, not mutually exclusive causes including a defect in the negative selection of autoreactive T cells in the thymus (also referred to as central tolerance) or impaired peripheral T cell tolerance (Qu et al. 2011). The role of SOCE in negative selection in the thymus is not clear, but STIM1-deficient patients and mice have a normal TCR repertoire (Picard et al. 2009; Fuchs et al. 2012), suggesting that SOCE is not critical for eliminating potentially autoreactive T cells. Two of the most important peripheral tolerance mechanisms involve T cell hyporesponsiveness (or T cell anergy) and regulatory T (Treg) cells. T cells can become anergic following weak TCR stimulation or TCR engagement in the absence of co-stimulatory signals. Anergy is characterized by the inability of T cells to mount a productive immune response, for instance against pathogens (Chappert & Schwartz, 2010). Induction of anergy in T cells was shown to require Ca2+ influx and activation of the transcription factor NFAT (reviewed in Baine et al. 2009). Conversely, however, the lack of SOCE and NFAT activation observed in T cells from CRAC-deficient patients, does not result in enhanced T cell activation or hyperproliferation. Autoimmunity in CRAC-deficient patients therefore is unlikely to be caused by impaired T cell anergy.
By contrast, the numbers of Treg cells, defined by co-expression of the Treg lineage-specific transcription factor Foxp3+ and the surface receptors CD4 and CD25 are reduced in STIM1-deficient patients (Picard et al. 2009; Fuchs et al. 2012). Treg cells are essential for maintaining immunological tolerance to self-antigens as well as regulating, for instance, immunity to infection and tumours (Nishikawa & Sakaguchi, 2010; Wing & Sakaguchi, 2010). Lack of Treg cells in human patients or mice with mutations in the Foxp3 gene results in severe autoimmunity (Mercer & Unutmaz, 2009). SOCE is required for the development and function of Foxp3+ Treg cells (Fig. 3). This is most clearly shown in SOCE-deficient mice with T cell-specific deletion of both Stim1 and Stim2 genes, which have ∼80–90% reduced numbers of Foxp3+ Treg cells and abolished suppressive Treg function (Oh-Hora et al. 2008). Accordingly, these mice showed a severe inflammatory phenotype with lymphadenopathy, splenomegaly and progressive infiltration of the lung, colon and other organs with lymphoid and myeloid cells (Oh-Hora et al. 2008). A similar lymphadenopathy and splenomegaly is present in STIM1-deficient patients (Feske, 2009; Picard et al. 2009). Treg development and function are not or only moderately impaired in Treg cells from STIM1- or ORAI1-deficient mice (Beyersdorf et al. 2009; McCarl et al. 2010), suggesting that moderate SOCE levels in T cells are sufficient for Treg development and function (Fig. 3). The mechanisms by which SOCE controls Treg cell physiology remain to be investigated, but they are likely to involve the ability of SOCE to regulate NFAT activation and its binding to gene regulatory elements in the Foxp3 promoter and enhancer (Fig. 1; Tone et al. 2008) and to regulate the NFAT-dependent expression of IL-2.
CRAC channels in B cell function and (auto) antibody production
The cause of autoantibody production by B cells in CRAC-deficient patients is unknown. Besides impaired Treg development and/or function resulting in failed suppression of autoreactive T and B cells, an alternative explanation may be that SOCE is required for the negative selection of autoreactive B cells during their development. Developing B cells that recognize self-antigens are eliminated in a process called negative selection to prevent the production of autoantibodies that can cause autoimmune disease. SOCE in B cells has been implicated in diverse B cell functions such as clonal expansion, differentiation and apoptotic cell death (Engelke et al. 2007; King & Freedman, 2009) but its role in B cell development and selection has remained unclear. STIM1 expression levels and SOCE amplitudes were higher in immature and transitional B cells than in mature B cells, potentially increasing antigen sensitivity during B cell development and promoting deletion of autoreactive B cells (Yarkoni & Cambier, 2011). Consistent with this idea, Linmander et al. reported that STIM1 regulates ERK activation in B cells during negative selection (Limnander et al. 2011). SOCE mediated by STIM1 induced the apoptosis of B cells recognizing self-antigen, and overexpression of STIM1 in bone marrow-derived B cells conferred a competitive disadvantage to developing B cells. By contrast, no defect in B cell development was observed in mice with B cell-specific deletion of both Stim1 and Stim2 genes despite severely impaired SOCE in STIM1/2-deficient B cells (Matsumoto et al. 2011). These results call into question the role of SOCE in B cell development and selection.
Upon antigen encounter, B cells undergo clonal expansion as well as class switch recombination and somatic hypermutation of their B cell receptor to produce high affinity IgG, IgA and IgE antibodies. SOCE appears to be dispensable for the ability of B cells to produce antigen-specific antibodies as T-dependent and T-independent IgG antibody responses were normal in STIM1-deficient (Beyersdorf et al. 2009) and STIM1/STIM2-deficient mice (Matsumoto et al. 2011). These findings are consistent with normal B cell numbers and normal to elevated serum immunoglobulin levels for IgA, IgM and IgG in CRAC-deficient patients with mutations in ORAI1 or STIM1 genes (Le Deist et al. 1995; McCarl et al. 2009; Picard et al. 2009; Feske et al. 2010). Some SOCE-deficient patients, however, showed impaired seroconversion with a lack of specific antibodies against vaccination and recall antigens (McCarl et al. 2009). While this defect is most likely due to impaired CD4+ T cell help rather than a B cell-intrinsic defect, further studies are necessary to clarify whether SOCE in T cells, B cells or dendritic cells has a role in controlling the generation of antigen-specific antibodies and/or autoantibodies. Interestingly, mice with B cell-specific deletion of Stim1 and Stim2 genes showed impaired expression of IL-10 by a subset of regulatory B cells (CD1dhiCD5+). Accordingly, these mice were increasingly susceptible to experimental autoimmune encephalomyelitis (Matsumoto et al. 2011). It remains to be studied whether impaired SOCE in CD1dhiCD5+ regulatory B cells predisposes mice to develop autoantibodies and spontaneous autoimmunity, which might explain the occurrence of autoantibodies against red blood cells and platelets in CRAC-deficient patients.
Conclusions
The discovery of CRAC-deficient patients with mutations in ORAI1 and STIM1 genes and the generation of genetically engineered mice lacking functional STIM1, STIM2 and ORAI1 has greatly enhanced our knowledge of the physiological role of CRAC channels and SOCE in the immune system and other organs. Additional insights are likely to be gained from ORAI2- and ORAI3-deficient mice or human patients, but these are currently not available for study. Residual Ca2+ influx in naive CD4+ and CD8+ T cells from Orai1−/− and Orai1R93W/R93W mice suggests that ORAI2 and ORAI3 may play a role in SOCE in murine T cells and that functional redundancy between ORAI channels may exist in mouse T cells. Whether this is also the case in human T cells is questionable since T cells isolated from ORAI1-deficient patients have no detectable CRAC channel currents or SOCE. Further genetic and functional studies will be necessary to clarify if functional redundancy between ORAI proteins exists in human and murine lymphocytes.
Recent studies in gene-targeted mice lacking functional ORAI1, STIM1 and STIM2 have demonstrated an important role for SOCE in lymphocyte function in vivo in a variety of processes including allograft rejection (McCarl et al. 2010), graft-vs.-host disease (Beyersdorf et al. 2009), hypersensitivity responses (McCarl et al. 2010) and autoimmunity such as inflammatory bowel disease (McCarl et al. 2010) and multiple sclerosis (McCarl et al. 2010; Schuhmann et al. 2010). Other immune responses are likely to be controlled by SOCE as well and further studies in primary human immune cells as well as animal models will be required to define the role of ORAI/STIM proteins in adaptive and innate immunity. Interfering with SOCE by blocking CRAC channel function is a promising approach to therapeutic immune regulation in autoimmune and allergic diseases, inflammation and for the prevention of transplant rejection. An important consideration for the development of such drugs will be to understand the quantitative requirements for SOCE in different types of immune responses. Experience with ORAI1- and STIM1-deficient patients tells us that complete deletion of CRAC channel function results in deleterious immune dysregulation with increased susceptibility to infection and autoimmunity due to Treg cell dysfunction. It remains to be investigated whether there is a level of CRAC channel inhibition that does not interfere with protective immunity to infection and Treg-mediated self-tolerance, but allows for inhibiting proinflammatory functions of T cells and other immune cell types. In other words, it needs to be established if a therapeutic window for CRAC channel inhibition exist that can be exploited for immunosuppression in patients.
Acknowledgments
The authors thank Dr. Bin Qu for helpful suggestions. This work was funded by NIH grant AI066128 to S.F. and a fellowship by the National Multiple Sclerosis Society to P.J.S.
Conflict of interest disclosure
S.F. is scientific co-founder of Calcimedica, a company that seeks to develop CRAC inhibitors.
References
- Baba Y, Nishida K, Fujii Y, Hirano T, Hikida M, Kurosaki T. Essential function for the calcium sensor STIM1 in mast cell activation and anaphylactic responses. Nat Immunol. 2008;9:81–88. doi: 10.1038/ni1546. [DOI] [PubMed] [Google Scholar]
- Baine I, Abe BT, Macian F. Regulation of T-cell tolerance by calcium/NFATsignaling. Immunol Rev. 2009;231:225–240. doi: 10.1111/j.1600-065X.2009.00817.x. [DOI] [PubMed] [Google Scholar]
- Barr VA, Bernot KM, Srikanth S, Gwack Y, Balagopalan L, Regan CK, Helman DJ, Sommers CL, Oh-Hora M, Rao A, Samelson LE. Dynamic movement of the calcium sensor STIM1 and the calcium channel Orai1 in activated T-cells: puncta and distal caps. Mol Biol Cell. 2008;19:2802–2817. doi: 10.1091/mbc.E08-02-0146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal AM, Anikeeva N, Varma R, Cameron TO, Vasiliver-Shamis G, Norris PJ, Dustin ML, Sykulev Y. Kinetics of early T cell receptorsignaling regulate the pathway of lytic granule delivery to the secretory domain. Immunity. 2009;31:632–642. doi: 10.1016/j.immuni.2009.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyersdorf N, Braun A, Vogtle T, Varga-Szabo D, Galdos RR, Kissler S, Kerkau T, Nieswandt B. STIM1-independent T cell development and effector function in vivo. J Immunol. 2009;182:3390–3397. doi: 10.4049/jimmunol.0802888. [DOI] [PubMed] [Google Scholar]
- BioGPS. ( http://biogps.gnf.org)
- Brandman O, Liou J, Park WS, Meyer T. STIM2 is a feedback regulator that stabilizes basal cytosolic and endoplasmic reticulum Ca2+ levels. Cell. 2007;131:1327–1339. doi: 10.1016/j.cell.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryceson YT, March ME, Ljunggren HG, Long EO. Synergy among receptors on resting NK cells for the activation of natural cytotoxicity and cytokine secretion. Blood. 2006;107:159–166. doi: 10.1182/blood-2005-04-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun M, Abhyankar A, Lelarge V, Plancoulaine S, Palanduz A, Telhan L, Boisson B, Picard C, Dewell S, Zhao C, Jouanguy E, Feske S, Abel L, Casanova JL. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan MD. STIMulating store-operated Ca2+ entry. Nat Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappert P, Schwartz RH. Induction of T cell anergy: integration of environmental cues and infectious tolerance. Curr Opin Immunol. 2010;22:552–559. doi: 10.1016/j.coi.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Sabatino A, Rovedatti L, Kaur R, Spencer JP, Brown JT, Morisset VD, Biancheri P, Leakey NA, Wilde JI, Scott L, Corazza GR, Lee K, Sengupta N, Knowles CH, Gunthorpe MJ, McLean PG, MacDonald TT, Kruidenier L. Targeting gut T cell Ca2+ release-activated Ca2+ channels inhibits T cell cytokine production and T-box transcription factor T-bet in inflammatory bowel disease. J Immunol. 2009;183:3454–3462. doi: 10.4049/jimmunol.0802887. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Engelke M, Engels N, Dittmann K, Stork B, Wienands J. Ca2+signaling in antigen receptor-activated B lymphocytes. Immunol Rev. 2007;218:235–246. doi: 10.1111/j.1600-065X.2007.00539.x. [DOI] [PubMed] [Google Scholar]
- Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunol Rev. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S. CRAC channelopathies. Pflugers Arch. 2010;460:417–435. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S. Immunodeficiency due to defects in store-operated calcium entry. Ann N Y Acad Sci. 2011;1238:74–90. doi: 10.1111/j.1749-6632.2011.06240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Giltnane J, Dolmetsch R, Staudt LM, Rao A. Gene regulation mediated by calcium signals in T lymphocytes. Nat Immunol. 2001;2:316–324. doi: 10.1038/86318. [DOI] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Feske S, Muller JM, Graf D, Kroczek RA, Drager R, Niemeyer C, Baeuerle PA, Peter HH, Schlesier M. Severe combined immunodeficiency due to defective binding of the nuclear factor of activated T cells in T lymphocytes of two male siblings. Eur J Immunol. 1996;26:2119–2126. doi: 10.1002/eji.1830260924. [DOI] [PubMed] [Google Scholar]
- Feske S, Picard C, Fischer A. Immunodeficiency due to mutations in ORAI1 and STIM1. Clin Immunol. 2010;135:169–182. doi: 10.1016/j.clim.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Prakriya M, Rao A, Lewis RS. A severe defect in CRAC Ca2+ channel activation and altered K+ channel gating in T cells from immunodeficient patients. J Exp Med. 2005;202:651–662. doi: 10.1084/jem.20050687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs S, Rensing-Ehl A, Speckmann C, Bengsch B, Schmitt-Graeff A, Bondzio I, Maul-Pavicic A, Bass T, Vraetz T, Strahm B, Ankermann T, Benson M, Caliebe A, Folster-Holst R, Kaiser P, Thimme R, Schamel WW, Schwarz K, Feske S, Ehl S. Antiviral and regulatory T cell immunity in a patient with stromal interaction molecule 1 deficiency. J Immunol. 2012;188:1523–1533. doi: 10.4049/jimmunol.1102507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Feske S, Cruz-Guilloty F, Oh-hora M, Neems DS, Hogan PG, Rao A. Biochemical and functional characterization of Orai proteins. J Biol Chem. 2007;282:16232–16243. doi: 10.1074/jbc.M609630200. [DOI] [PubMed] [Google Scholar]
- Gwack Y, Srikanth S, Oh-Hora M, Hogan PG, Lamperti ED, Yamashita M, Gelinas C, Neems DS, Sasaki Y, Feske S, Prakriya M, Rajewsky K, Rao A. Hair loss and defective T- and B-cell function in mice lacking ORAI1. Mol Cell Biol. 2008;28:5209–5222. doi: 10.1128/MCB.00360-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calciumsignaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ImmunologicalGenomeProject. ( http://www.immgen.org/index_content.html)
- Kim KD, Srikanth S, Yee MK, Mock DC, Lawson GW, Gwack Y. ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J Immunol. 2011;187:3620–3630. doi: 10.4049/jimmunol.1100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King LB, Freedman BD. B-lymphocyte calcium influx. Immunol Rev. 2009;231:265–277. doi: 10.1111/j.1600-065X.2009.00822.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarevic V, Zullo AJ, Schweitzer MN, Staton TL, Gallo EM, Crabtree GR, Glimcher LH. The gene encoding early growth response 2, a target of the transcription factor NFAT, is required for the development and maturation of natural killer T cells. Nat Immunol. 2009;10:306–313. doi: 10.1038/ni.1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Deist F, Hivroz C, Partiseti M, Thomas C, Buc HA, Oleastro M, Belohradsky B, Choquet D, Fischer A. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–1062. [PubMed] [Google Scholar]
- Limnander A, Depeille P, Freedman TS, Liou J, Leitges M, Kurosaki T, Roose JP, Weiss A. STIM1, PKC-δ and RasGRP set a threshold for proapoptotic Erksignaling during B cell development. Nat Immunol. 2011;12:425–433. doi: 10.1038/ni.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lioudyno MI, Kozak JA, Penna A, Safrina O, Zhang SL, Sen D, Roos J, Stauderman KA, Cahalan MD. Orai1 and STIM1 move to the immunological synapse and are up-regulated during T cell activation. Proc Natl Acad Sci U S A. 2008;105:2011–2016. doi: 10.1073/pnas.0706122105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyubchenko TA, Wurth GA, Zweifach A. Role of calcium influx in cytotoxic T lymphocyte lytic granule exocytosis during target cell killing. Immunity. 2001;15:847–859. doi: 10.1016/s1074-7613(01)00233-3. [DOI] [PubMed] [Google Scholar]
- Ma J, McCarl CA, Khalil S, Luthy K, Feske S. T-cell-specific deletion of STIM1 and STIM2 protects mice from EAE by impairing the effector functions of Th1 and Th17 cells. Eur J Immunol. 2010;40:3028–3042. doi: 10.1002/eji.201040614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarl CA, Khalil S, Ma J, Oh-hora M, Yamashita M, Roether J, Kawasaki T, Jairaman A, Sasaki Y, Prakriya M, Feske S. Store-operated Ca2+ entry through ORAI1 is critical for T cell-mediated autoimmunity and allograft rejection. J Immunol. 2010;185:5845–5858. doi: 10.4049/jimmunol.1001796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarl CA, Picard C, Khalil S, Kawasaki T, Rother J, Papolos A, Kutok J, Hivroz C, Ledeist F, Plogmann K, Ehl S, Notheis G, Albert MH, Belohradsky BH, Kirschner J, Rao A, Fischer A, Feske S. ORAI1 deficiency and lack of store-operated Ca2+ entry cause immunodeficiency, myopathy, and ectodermal dysplasia. J Allergy Clin Immunol. 2009;124:1311–1318 e1317. doi: 10.1016/j.jaci.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- McNally BA, Prakriya M. Permeation, selectivity and gating in store-operated CRAC channels. J Physiol. 2012;590:4179–4191. doi: 10.1113/jphysiol.2012.233098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto M, Fujii Y, Baba A, Hikida M, Kurosaki T, Baba Y. The calcium sensors STIM1 and STIM2 control B cell regulatory function through interleukin-10 production. Immunity. 2011;34:703–714. doi: 10.1016/j.immuni.2011.03.016. [DOI] [PubMed] [Google Scholar]
- Maul-Pavicic A, Chiang SC, Rensing-Ehl A, Jessen B, Fauriat C, Wood SM, Sjoqvist S, Hufnagel M, Schulze I, Bass T, Schamel WW, Fuchs S, Pircher H, McCarl CA, Mikoshiba K, Schwarz K, Feske S, Bryceson YT, Ehl S. ORAI1-mediated calcium influx is required for human cytotoxic lymphocyte degranulation and target cell lysis. Proc Natl Acad Sci U S A. 2011;108:3324–3329. doi: 10.1073/pnas.1013285108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer F, Unutmaz D. The biology of FoxP3: a key player in immune suppression during infections, autoimmune diseases and cancer. Adv Exp Med Biol. 2009;665:47–59. doi: 10.1007/978-1-4419-1599-3_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa H, Sakaguchi S. Regulatory T cells in tumor immunity. Int J Cancer. 2010;127:759–767. doi: 10.1002/ijc.25429. [DOI] [PubMed] [Google Scholar]
- Oh-Hora M, Yamashita M, Hogan PG, Sharma S, Lamperti E, Chung W, Prakriya M, Feske S, Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat Immunol. 2008;9:432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partiseti M, Le Deist F, Hivroz C, Fischer A, Korn H, Choquet D. The calcium current activated by T cell receptor and store depletion in human lymphocytes is absent in a primary immunodeficiency. J Biol Chem. 1994;269:32327–32335. [PubMed] [Google Scholar]
- Picard C, McCarl CA, Papolos A, Khalil S, Luthy K, Hivroz C, LeDeist F, Rieux-Laucat F, Rechavi G, Rao A, Fischer A, Feske S. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N Engl J Med. 2009;360:1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipkin ME, Lieberman J. Delivering the kiss of death: progress on understanding how perforin works. Curr Opin Immunol. 2007;19:301–308. doi: 10.1016/j.coi.2007.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pores-Fernando AT, Bauer RA, Wurth GA, Zweifach A. Exocytic responses of single leukaemic human cytotoxic T lymphocytes stimulated by agents that bypass the T cell receptor. J Physiol. 2005;567:891–903. doi: 10.1113/jphysiol.2005.089565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pores-Fernando AT, Zweifach A. Calcium influx andsignaling in cytotoxic T-lymphocyte lytic granule exocytosis. Immunol Rev. 2009;231:160–173. doi: 10.1111/j.1600-065X.2009.00809.x. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- Qu B, Al-Ansary D, Kummerow C, Hoth M, Schwarz EC. ORAI-mediated calcium influx in T cell proliferation, apoptosis and tolerance. Cell Calcium. 2011;50:261–269. doi: 10.1016/j.ceca.2011.05.015. [DOI] [PubMed] [Google Scholar]
- Quintana A, Pasche M, Junker C, Al-Ansary D, Rieger H, Kummerow C, Nunez L, Villalobos C, Meraner P, Becherer U, Rettig J, Niemeyer BA, Hoth M. Calcium microdomains at the immunological synapse: how ORAI channels, mitochondria and calcium pumps generate local calcium signals for efficient T-cell activation. EMBO J. 2011;30:3895–3912. doi: 10.1038/emboj.2011.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin G, Palanduz A, Aydogan G, Cassar O, Ertem AU, Telhan L, Canpolat N, Jouanguy E, Picard C, Gessain A, Abel L, Casanova JL, Plancoulaine S. Classic Kaposi sarcoma in 3 unrelated Turkish children born to consanguineous kindreds. Pediatrics. 2010;125:e704–708. doi: 10.1542/peds.2009-2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savignac M, Mellstrom B, Naranjo JR. Calcium-dependent transcription of cytokine genes in T lymphocytes. Pflugers Arch. 2007;454:523–533. doi: 10.1007/s00424-007-0238-y. [DOI] [PubMed] [Google Scholar]
- Schuhmann MK, Stegner D, Berna-Erro A, Bittner S, Braun A, Kleinschnitz C, Stoll G, Wiendl H, Meuth SG, Nieswandt B. Stromal interaction molecules 1 and 2 are key regulators of autoreactive T cell activation in murine autoimmune central nervous system inflammation. J Immunol. 2010;184:1536–1542. doi: 10.4049/jimmunol.0902161. [DOI] [PubMed] [Google Scholar]
- Schwarz A, Tutsch E, Ludwig B, Schwarz EC, Stallmach A, Hoth M. Ca2+signaling in identified T-lymphocytes from human intestinal mucosa. Relation to hyporeactivity, proliferation, and inflammatory bowel disease. J Biol Chem. 2004;279:5641–5647. doi: 10.1074/jbc.M309317200. [DOI] [PubMed] [Google Scholar]
- Shaw PJ, Feske S. Physiological and pathophysiological functions of SOCE in the immune system. Front Biosci (Elite Ed) 2011:2253–2268. doi: 10.2741/540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Zheng L, Ikura M. Stromal interaction molecule (STIM) 1 and STIM2 calcium sensing regions exhibit distinct unfolding and oligomerization kinetics. J Biol Chem. 2009;284:728–732. doi: 10.1074/jbc.C800178200. [DOI] [PubMed] [Google Scholar]
- Takayama H, Sitkovsky MV. Antigen receptor-regulated exocytosis in cytotoxic T lymphocytes. J Exp Med. 1987;166:725–743. doi: 10.1084/jem.166.3.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M. Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat Immunol. 2008;9:194–202. doi: 10.1038/ni1549. [DOI] [PubMed] [Google Scholar]
- Vig M, DeHaven WI, Bird GS, Billingsley JM, Wang H, Rao PE, Hutchings AB, Jouvin MH, Putney JW, Kinet JP. Defective mast cell effector functions in mice lacking the CRACM1 pore subunit of store-operated calcium release-activated calcium channels. Nat Immunol. 2008;9:89–96. doi: 10.1038/ni1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- Yarkoni Y, Cambier JC. Differential STIM1 expression in T and B cell subsets suggests a role in determining antigen receptor signal amplitude. Mol Immunol. 2011;48:1851–1858. doi: 10.1016/j.molimm.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Littman DR. Transcriptional regulatory networks in Th17 cell differentiation. Curr Opin Immunol. 2009;21:146–152. doi: 10.1016/j.coi.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]