

Abstract
Store-operated Ca2+ release-activated Ca2+ (CRAC) channels are a widespread mechanism for generating cellular Ca2+ signals and regulate many Ca2+-dependent functions, including transcription, motility and proliferation. The opening of CRAC channels in response to depletion of intracellular Ca2+ stores involves a cascade of cellular events that culminate in direct interactions between STIM1, the endoplasmic reticulum Ca2+ sensor, and the channels composed of Orai proteins. Evidence gathered over the last two decades indicates that CRAC channels display a unique functional pore fingerprint characterized by exquisite Ca2+ selectivity, low unitary conductance, and low permeability to large cations. Here, we review the key pore properties of CRAC channels and discuss recent progress in addressing the molecular foundations of these properties. Structure–function and cysteine-scanning studies have revealed the identity and organization of pore-lining residues, including those that form the selectivity filter, providing a structural framework for understanding CRAC channel pore properties. Recent studies in pore mutants that produce STIM1-independent constitutive channel activation indicate that exquisite Ca2+ selectivity in CRAC channels is not hardwired into Orai proteins, but is instead manifested only following the binding of STIM1 to the intrinsically poorly Ca2+-selective Orai channels. These findings reveal new functional aspects of CRAC channels and suggest that the selectivity filter of the CRAC channel is a dynamic structure whose conformation and functional properties are powerfully regulated by the channel activation stimulus.

Murali Prakriya is an Associate Professor in the Department of Molecular Pharmacology and Biological Chemistry at Northwestern University, Feinberg School of Medicine. He received a PhD in Neuroscience from Washington University, St Louis, and obtained his postdoctoral training at Stanford University in the laboratory of Dr Richard Lewis. Murali's laboratory at Northwestern University examines the molecular and cellular mechanisms of intracellular Ca2+ signalling, with a particular focus on the molecular physiology of store-operated CRAC channels. Beth McNally is a Postdoctoral Fellow in the Prakriya lab at Northwestern University. She obtained a BS in biology and chemistry from Hillsdale College and a PhD in biochemistry from the University of Notre Dame. Her research is focused on examining the molecular mechanisms of CRAC channels.
Introduction
Store-operated calcium channels (SOCs) are a family of plasma membrane ion channels that are activated by the depletion of endoplasmic reticulum calcium stores in response to the generation of 1,4,5-inositol trisphosphate. Ca2+ entry through SOCs regulates a wide variety of cellular functions including gene expression, cell motility, proliferation and the exocytosis of inflammatory mediators (Hogan et al. 2010; Lewis, 2011). SOC currents have been identified using patch-clamp techniques in several cell types including T-cells, mast cells, hepatocytes, pancreatic acinar cells, endothelial cells and smooth muscle cells (Parekh & Putney, 2005). Imaging studies indicate that a store-operated entry mechanism exists in an even wider range of tissues and cell types that includes skeletal muscle and even some types of neurons (Dirksen, 2009; Gemes et al. 2011). Nevertheless, the SOC subtype for which the most detailed information is available is the Ca2+ release-activated Ca2+ (CRAC) channel, a highly Ca2+ selective SOC that is expressed in T-cells, mast cells and related haematopoetic cells (Lewis, 2011). CRAC channels were investigated for more than two decades before their molecular composition was identified in 2005–2006, first with the discovery of STIM1, a protein in the endoplasmic reticulum (ER) membrane responsible for sensing the drop in ER calcium (Liou et al. 2005; Zhang et al. 2005), followed by the discovery of Orai1, a prototypic pore-forming CRAC channel protein (Feske et al. 2006; Vig et al. 2006b; Zhang et al. 2006). It is now known that STIM1 binds to and directly activates Orai1 and these two molecules appear both necessary and sufficient to reconstitute store-operated calcium entry (SOCE) (Mercer et al. 2006; Peinelt et al. 2006; Muik et al. 2008; Park et al. 2009; Zhou et al. 2010a). Much of the attention on CRAC channels has surrounded the mechanism of channel activation in response to store depletion, and significant progress has occurred in this area (Lewis, 2011). By comparison, the mechanisms of ion conduction and channel gating are less well understood, although this is rapidly changing in recent years. This review will focus on our current understanding of the ion conduction properties of CRAC channels and the molecular determinants of these properties.
Functional fingerprint of the CRAC channel pore
Perhaps the most prominent hallmark of the CRAC channel's biophysical fingerprint is its extraordinarily high Ca2+ selectivity (PCa/PNa > 1000), which places it, together with voltage-gated Ca2+ (Cav) channels, in a unique category of highly Ca2+ selective channels (Hoth & Penner, 1993). Interestingly, the CRAC channel's strong preference for Ca2+ ions is seen only in solutions with mixtures of Ca2+ and monovalent ions. In the absence of extracellular Ca2+ and other divalent ions, CRAC channels readily conduct a variety of small monovalent ions including Na+, Li+ and K+ (Lepple-Wienhues & Cahalan, 1996; Bakowski & Parekh, 2002; Prakriya & Lewis, 2002). This feature suggests that exclusive permeation of Ca2+ is not hardwired into the CRAC channel pore but arises from competition between Ca2+ and monovalent ions for binding sites within the ion conduction pathway. Consistent with this interpretation, extracellular Ca2+ potently blocks Na+ currents (Ki∼20 μm at −100 mV; Hoth & Penner, 1993; Bakowski & Parekh, 2002; Su et al. 2004; Prakriya & Lewis, 2006), and, as expected for a binding site within the pore, Ca2+ block is voltage dependent (Prakriya & Lewis, 2006; Yamashita et al. 2007). Qualitatively, these characteristics are reminiscent of the properties of voltage-gated Ca2+ (Cav) channels, in which Ca2+ ions similarly bind tightly with a high-affinity site within the pore to occlude Na+ flux (Lansman et al. 1986; Sather & McCleskey, 2003). Unlike Cav channels, however, CRAC channels display a very low permeability to large monovalent cations such as Cs+. As described below, the feature probably arises from a narrow pore that limits the electrodiffusion of large cations within the restricted confines of the pore (Prakriya & Lewis, 2006; Yamashita et al. 2007; McNally et al. 2009; Zhou et al. 2010b). CRAC channels also display a very low unitary conductance that is estimated from noise analysis to be only 18–24 fS in 110 mm Ca2+ and ∼1 pS for Na+ (Zweifach & Lewis, 1993; Prakriya & Lewis, 2006). At physiological Ca2+ concentrations (2 mm), the unitary conductance has been estimated to be even lower, ∼9 fS (Zweifach & Lewis, 1993).
Pharmacologically, CRAC channels are distinguished by high sensitivity to blockade by trivalent lanthanide ions (Ki ≪ 1 μm) (Mason et al. 1991; Aussel et al. 1996; Yeromin et al. 2004, 2006), a feature that differentiates these channels from many less Ca2+ selective cation channels. Many other inhibitors of CRAC channels have been described over the years. These include the compounds 2-aminoethyldiphenyl borate (2-APB), which inhibits CRAC channels at concentrations >10 μm, and YM-58483, also known as BTP2, which inhibits CRAC channels and SOCE with a Ki of ∼10 nm (Zitt et al. 2004). Both compounds inhibit many other classes of ion channels including TRPC channels (Bootman et al. 2002; Takezawa et al. 2006) and therefore only have limited utility as specific probes for elucidating the cellular functions of CRAC channels. However, 2-APB elicits a spectrum of unusual CRAC channel behaviours that have become a popular focus of investigation. These effects include a strong enhancement of ICRAC at low drug concentrations, transient facilitation followed by persistent inhibition at high doses, and the removal of fast Ca2+-dependent inactivation (CDI) (Prakriya & Lewis, 2001; Ma et al. 2002; Yamashita et al. 2011). Adding to this complexity, recombinant Orai3 (and to a smaller extent Orai1) channels are also directly activated by 2-APB in a store-independent manner (DeHaven et al. 2008; Peinelt et al. 2008; Schindl et al. 2008; Zhang et al. 2008; Yamashita et al. 2011). In addition to these compounds, newer, more selective CRAC channel inhibitors developed for therapeutic applications include the drugs Synta-66 (Ashmole et al. 2012) and CM2489 (CalciMedica, http://www.calcimedica.com). With the exception of lanthanides, however, which block CRAC channels by binding to the acidic residues in the TM1–TM2 loop region (Yeromin et al. 2006; McNally et al. 2009), the mechanisms by which the different compounds inhibit CRAC channel activity are unknown. Nevertheless, when used in conjunction with the biophysical fingerprint described above, these pharmacological agents have proven useful for identifying and characterizing CRAC channels in many native tissues.
Molecular underpinnings of selectivity and permeation in CRAC channels
Permeation and selectivity in ion channels are typically controlled by the organization and chemistry of pore-lining residues. For CRAC channels, a mechanistic understanding of ion permeation is confronted with the challenge that the Orai proteins bear little sequence homology to other ion channels. Hence, lessons drawn from other channels cannot be readily extrapolated to CRAC channels. Thus, an important focus of recent efforts has been the identification of amino acids that line the CRAC channel ion conduction pathway. In early studies, site-directed mutagenesis was employed to explore the role of conserved acidic amino acids located in the transmembrane (TM) domains for high Ca2+ selectivity. These studies resulted in the identification of several acidic residues that influenced ion permeation, including E106 in TM1, E190 in TM3, and D110, D112 and D114 in the TM1–TM2 linker region of human Orai1 (Fig. 1A; Prakriya et al. 2006; Vig et al. 2006a; Yeromin et al. 2006). Mutations at these positions resulted in a wide range of effects including loss of Ca2+ selectivity, increase in Cs+ permeation, changes in the voltage dependence of Ca2+ block, widening of the pore, and (for the TM1–TM2 loop residues) diminished La3+ block (Prakriya et al. 2006; Vig et al. 2006a; Yeromin et al. 2006; Yamashita et al. 2007). Based on these findings, early models of the CRAC channel pore concluded that both TM1 and TM3 flank the ion conduction pathway, with the Glu residues at 106 (TM1) and 190 (TM3) and the Asp residues in the TM1–TM2 loop forming rings of coordinating sites for the conducting ions (Vig et al. 2006a; Cai, 2007).
Figure 1.
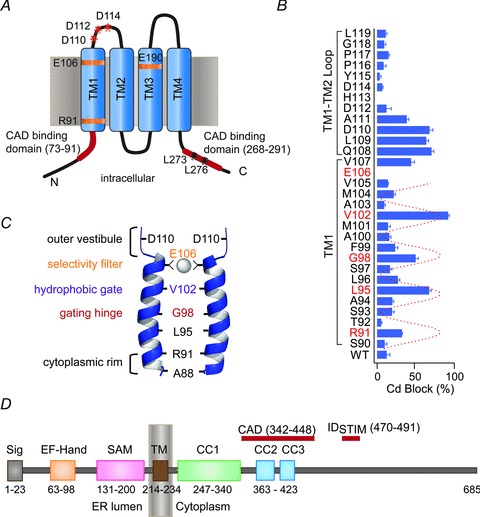
A, the predicted topology of Orai1. Key residues (mentioned in the text) are labelled. B, the pattern of Cd2+ blockade for residues in TM1 domain and TM1–TM2 loop regions. Residues V102, G98 and L95 in TM1 exhibit strong blockade, identifying them as pore-lining amino acids (adapted from McNally et al. 2009). C, a schematic representation of the Orai1 pore-lining residues in TM1, along with their identified structural or functional features. For clarity, only two of the four TM1 domains are depicted. D, a schematic representation of STIM1 and its key functional domains. These domains include: Sig, signal peptide; SAM, sterile α motif; TM, transmembrane domain; CC, coiled-coil domain; CAD, CRAC activation domain; IDSTIM1, inactivation domain of STIM1.
But are these residues directly exposed to the ion conduction pathway? Or do the mutations simply destabilize the energetic stability of the pore through long-range effects? To address this issue, we used the substituted cysteine accessibility method to identify and illuminate the organization of pore-lining residues (McNally et al. 2009). In this method, successive amino acids within specific target areas of Orai1 are substituted one at a time by Cys, and the sensitivity of the mutated channels to block by aqueous thiol-labelling reagents is examined (Karlin & Akabas, 1998). The highly polar nature of the thiol reagents (including methanethiosulphonate (MTS) reagents and Cd2+) causes them to react rapidly with water-exposed sulfhydryl groups resulting in the attachment of the reagent to single sulfhydryl groups (in the case of MTS reagents) or coordination by multiple SH-groups (in the case of Cd2+). If the cysteine residue is located in the pore of the channel, the attachment of the reagents to pore-lining residues results in rapid blockade of ion conduction, thereby revealing the identity of pore lining residues (Karlin & Akabas, 1998). Cysteine accessibility methods have been successfully applied to numerous ion channels to identify pore-lining amino acids and deduce their relative organization within the ion conduction trajectory (Karlin & Akabas, 1998; Yellen, 1998). In a complementary approach, another study examined the ability of Cys substitutions in the predicted TM1 segment to form intersubunit disulfide bonds across the pore (Zhou et al. 2010b).
The results of the two studies converged towards many similar conclusions. Both studies reaffirmed the hypothesis that the TM1 segment flanks the pore. However, residues in TM3 including E190 failed to exhibit any detectable reactivity, indicating that despite the effects of some E190 mutations on ion selectivity, TM3, in fact, does not flank the pore (McNally et al. 2009; Zhou et al. 2010b). Given that functional CRAC channels are tetramers of Orai subunits (Mignen et al. 2008; Penna et al. 2008), these results imply that the ion selective part of the pore is fashioned by four TM1 segments, with the selectivity filter formed by a quartet of E106 residues. The electrophysiological study also revealed that sequential residues of the TM1–TM2 loop segments interact tightly with both large (>8 Å) and small (<3 Å) as well as with positively charged and negatively charged probes, and form inter-subunit disulfide bonds (McNally et al. 2009). This pattern of solvent reactivity is consistent with the TM1–TM2 segments forming a flexible vestibule at the outer mouth of the pore.
Moving deeper into the pore, differences in the accessibility of thiol probes of different sizes showed that the pore narrows sharply at the base of the vestibule, probably above the Ca2+ binding site formed by E106 (McNally et al. 2009). Notably, several ‘hotspots’ of strong Cd2+ reactivity and disulfide bond formation were found in the TM1 segment (McNally et al. 2009; Zhou et al. 2010b). The positions of these highly reactive residues displayed a periodicity typical of a pore-facing α helix (Fig. 1B and C). Interestingly, because strong coordination of Cd2+ requires the presence of several (≥3) coordinating SH-groups in ion channel pores (Liu et al. 1997; Loussouarn et al. 2001), the ability of sequential pore-facing residues on the TM1 helix to coordinate Cd2+ indicates that the centrally located TM1 helices are located close to each other, effectively forming a long narrow pore. Even R91 at the cytoplasmic rim of TM1 displayed significant ability to coordinate Cd2+ (Fig. 1B and C), suggesting that introduction of hydrophobic bulky amino acids at this position, such as that found in the human severe combined immunodeficiency (SCID) mutation R91W, could interfere with ion conduction due to steric or hydrophobic occlusion of the inner pore. A88C, which is technically predicted to be in the cytoplasmic N-terminus, also formed inter-subunit disulfide bonds (Zhou et al. 2010b), raising the prospect that the cytoplasmic rim of the pore may extend further into the N-terminus than had been previously considered.
A long narrow pore may be a feature unique to CRAC channels. In the equally Ca2+ selective Cav channels, by contrast, the inner pore is reported to form a wide vestibule large enough to accommodate very large thiol reagents and one or more hydrated ions (Zhen et al. 2005). This structural feature of the CRAC channel pore could produce steric hindrance for the electrodiffusion of ions within the narrow confines of the pore, possibly accounting for its low permeability to large ions (>3.8 Å) and low unitary conductance. It is interesting that the results of the two cysteine scanning studies were in broad agreement about the identity and organization of the TM1 pore-lining residues despite differences in the methodologies employed (the electrophysiological study examined accessibility of thiol reagents in open STIM1-gated channels, whereas the disulfide cross-linking study studied a non-functional mutant in the absence of STIM1; McNally et al. 2009; Zhou et al. 2010b). This correspondence suggests that channel activation by STIM1 may not involve large conformational alterations in the TM1 segment. More studies are needed to understand the structural dynamics of the CRAC channel pore during gating. However, findings from these studies provided the first step towards building a structural model of the pore, and offer testable hypotheses for further functional, pharmacological and structural investigations.
STIM1 binds to Orai1
As noted above, CRAC channels are operationally defined as store-operated channels. The molecular basis of this fundamental property originates in the direct binding of the ER Ca2+ sensor, STIM1, to the pore-forming Orai proteins. Under basal conditions in cells with full ER Ca2+ stores, STIM1 is diffusely distributed through the ER membrane. Store depletion initiates a highly stereotyped set of events that culminates in the direct binding of STIM1 to Orai1. The principal events of this process include the release of Ca2+ from the luminal EF-SAM domain of STIM1, the oligomerization of STIM1, and redistribution of the STIM1 oligomers into distinct puncta at junctional ER sites located in close proximity to the plasma membrane (Lewis, 2007, 2011). Thus, STIM1 fulfils two critical roles for CRAC channel activation: first, it senses the depletion of ER Ca2+ stores, and second, it communicates store depletion to the CRAC channels located in the plasma membrane. STIM1 oligomerization is an early and essential step in this signalling pathway and serves as a critical switch that can unfold all the subsequent steps of the channel activation process (Stathopulos et al. 2006, 2008; Liou et al. 2007; Luik et al. 2008). Interestingly, recent studies indicate that activation of STIM1 can occur even in the absence of ER Ca2+ store depletion under certain conditions, for example following S-glutathionylation of the cysteine residue located in the EF-SAM domain (Hawkins et al. 2010), and in elevated temperatures (35–37°C; Xiao et al. 2011). The mechanisms of these latter effects remain to be firmly established, but are thought to be related to a reduction in the Ca2+ binding capacity of the STIM1 EF-SAM domain, thereby triggering STIM1 oligomerization (Hawkins et al. 2010; Xiao et al. 2011).
Association of STIM1 with Orai1 has been shown under a variety of conditions and chemical environments including in live cells, cell lysates, and even in a system of only purified components in solution (Yeromin et al. 2006; Muik et al. 2008; Park et al. 2009; Zhou et al. 2010a). Harnessing this finding, several groups employed deletions and serial truncations of STIM1 to identify a domain in the cytoplasmic region of STIM1 (amino acids 342–448) as a necessary and sufficient element for binding and activation of Orai1 channels. The structure of this minimal activation domain, termed the CRAC activation domain (CAD), (Fig. 1D) has recently been solved (Yang et al., 2012). The structure reveals a V-shaped dimeric module with the main functional domains of CAD, two coiled-coil regions (CC2 and CC3), forming a hair-pin motif (Yang et al., 2012). How the dimeric CAD structure is altered upon binding to Orai1 is not known yet, nevertheless, this high-resolution structure provides several testable hypotheses for dissecting the structural underpinnings of STIM1 function.
In Orai1, structure–function studies have identified two regions located in the C- and N-termini that are essential for STIM1 binding and channel activation (Li et al. 2007; Muik et al. 2008; Navarro-Borelly et al. 2008; Park et al. 2009). As in STIM1, the critical binding motif in the Orai1 C-terminus is a coiled-coil domain encompassing amino acids 268–291. Deletion of this motif (Li et al. 2007) or mutations of key hydrophobic residues (L273S and L276D) (Muik et al. 2008; Navarro-Borelly et al. 2008) strongly affect STIM1–Orai1 coupling and Orai1 activation, probably due to disruption of the coiled-coil helix. In the Orai1 N-terminus, a weak interaction of CAD with a region corresponding to amino acids 68–91 was found in co-immunoprecipitation and pull-down assays (Park et al. 2009; Zhou et al. 2010a). Interestingly, mutation of a critical lysine residue in this region (K85E) attenuates Orai1 activation by STIM1 without detectably reducing the ability of the Orai1 N-terminus (amino acids 48–91) to co-immunoprecipitate CAD (Lis et al. 2010), suggesting that this residue has a critical role in channel gating independent of STIM1 binding.
Collectively, these studies have radically altered our understanding of how CRAC channels are activated. They reaffirmed an old theory (Irvine, 1990) that the physical basis of CRAC channel activation involves local and direct interactions between an ER molecule and the channel. Mechanistically, we now have a firm understanding of the minimal structural requirements of CRAC channel activation by STIM1. Further progress in this area will depend on illumination of some key questions surrounding the binding of STIM1 and Orai1. For example, the specific contributions of the two distinct STIM1 binding sites in Orai1 for channel function are unknown. In addition, the thermodynamic features of STIM1–Orai1 binding have not been quantified and evidence exists for both strong as well as weak interactions. These questions are likely to be addressed in time with the use of more in-depth biophysical and protein biochemistry approaches.
STIM1 stoichiometry of CRAC channel activation
A fundamental aspect of the interaction between STIM1 and Orai1 crucial to our understanding of the CRAC channel activation process is the stoichiometric requirement of Orai1 activation by STIM1. How many STIM1 molecules does it take to activate CRAC channels? Two recent studies have addressed this question using very different approaches (Hoover & Lewis, 2011; Li et al. 2011). In one study, Li et al. (2011) used tandem constructs to determine the effect of the STIM:Orai1 ratio on CRAC channel activity. Tandem constructs with a varying number of Orai1 protomers (1–4) were fused to a STIM1 region containing two repeats of the minimal activation domain (amino acids 336–485, termed the ‘S’ domain). ICRAC was largest for constructs with a 2:1 S:Orai1 ratio, and decreased as the STIM1:Orai1 ratio decreased. Moreover, knocking out STIM1 binding sites sequentially in the C-terminus of Orai1 in a concatamer containing four Orai1 molecules revealed a graded relationship between STIM1 binding and Orai1 activation. Together, their data suggested that a 2:1 STIM1:Orai1 ratio gives optimal CRAC activity. Given that the active Orai1 complex is expected to contain four copies of Orai1 (Mignen et al. 2008; Ji et al. 2008; Penna et al. 2008), these results hence indicate that eight copies of STIM1 are required for optimal activation of single CRAC channels. Additionally, the data indicated that progressively decreasing the functional STIM1:Orai1 ratio produces a graded decline in ICRAC amplitude (Hoover & Lewis, 2011; Li et al. 2011).
In an alternative approach, Hoover & Lewis (2011) varied the relative expression of full-length STIM1 and Orai1 that were fused to fluorophores mCherry and GFP to study the functional requirements of Orai1 activation as well as trapping at the ER–plasma membrane junctions. Although their results indicated that activation was graded with increasing amounts of STIM1 bound to Orai1 channels, channel activation was highly non-linear (Hoover & Lewis, 2011). Maximal CRAC current activation required the binding of eight STIM1 molecules to each channel, and activation declined sharply with diminishing STIM1 such that the minimal stoichiometry for trapping Orai channels at puncta fails to evoke significant activation (Hoover & Lewis, 2011). Thus, their results suggested that once the optimal STIM threshold is reached, individual channels would presumably open abruptly to their fully active state in an all-or-none manner. This model seems harmonious with earlier findings in native CRAC channels in Jurkat T-cells indicating that the slow increase in ICRAC following store depletion occurs from the stepwise recruitment of closed channels to a very high open probability state (Prakriya & Lewis, 2006). The reasons for the apparent discrepancy in the degree of non-linearity of Orai1 activation by STIM1 between the studies of Li et al. (2011) and Hoover & Lewis (2011) are unknown, but it is worth noting that the Hoover and Lewis study employed full length STIM1 and monomeric Orai1, whereas Li et al. (2011) employed a tandem SS domain to activate Orai1 tetramers. This difference in methodology raises the possibility that the activation behaviour of tetrameric Orai1 constructs may be different from that of monomeric Orai1. In native tissues, the presence of additional modulatory proteins that regulate STIM–Orai binding (e.g. the CRACR2A/B proteins; Srikanth et al. 2010) may further accentuate the all-or-none activation behaviour by modulating the cooperativity of STIM1–Orai1 binding.
A surprising finding from the Hoover and Lewis study was that in addition to activation gating, STIM1 also regulates Ca2+-dependent inactivation (CDI) of CRAC channels. CDI is a prominent hallmark of CRAC channels seen as a decline in ICRAC amplitude over tens of milliseconds during hyperpolarizing voltage steps (Zweifach & Lewis, 1995). CDI is mediated by the high local [Ca2+]i around individual Ca2+ channels and is driven by Ca2+ binding to calmodulin (Litjens et al. 2004; Mullins et al. 2009), resulting in the binding of Ca2+-calmodulin to the membrane proximal N-terminal region of Orai1 (residues 68–91; Mullins et al. 2009). There is strong evidence that STIM1 regulates this process. CDI is absent in CRAC currents activated by the minimal activation domain of STIM1 (the CAD domain; Park et al. 2009), and the extent of CDI is correlated to the relative abundance of STIM1 (Scrimgeour et al. 2009), suggesting that a region within STIM1 distinct from CAD confers CDI. Subsequent work has revealed that an acidic region in the C-terminus of STIM1 (amino acids 470–491) is critical for this gating process although the precise mechanism by which this domain confers CDI remains unclear (Derler et al. 2009b; Lee et al. 2009; Mullins et al. 2009). The finding that CDI, a functional property closely associated with the CRAC channel pore, is regulated by STIM1 indicates that STIM1 is, in effect, a subunit of the channel that confers inactivation by direct and specific interaction with the channel pore (Mullins et al. 2009).
How does the pore open and close?
As described above, the CRAC channel pore opens to allow Ca2+ influx in response to STIM1 binding. The conformational motion underlying the opening and closing of the pore, collectively called gating, is carefully regulated to provide the dynamic Ca2+ signals required for activating a wide range of cellular functions. Although our understanding of the moving parts of the CRAC channel pore that allow pore opening and closing still remains rudimentary, results from site-directed mutagenesis and cysteine scanning studies have begun to shed light on this issue.
One clue has come from mutational analysis of a highly conserved Gly residue, located in the middle of the predicted TM1 helix (Zhang et al. 2011). Mutation of G98 to Ala results in non-functional channels, suggesting a critical role for G98 in channel gating. More significantly, introducing a Pro at this position, which should produce a inflexible kink in the helix, results in constitutively open channels that are active even in the absence of STIM1. Likewise, mutation to Asp (G98D) causes STIM1-independent constitutive channel activity (Zhang et al. 2011). Interestingly, the G98D and G98P channel mutants are non-selective and do not display the signature high selectivity of CRAC channels, indicating that the mutations significantly disrupt the CRAC channel selectivity filter. Taken together, these results are consistent with the notion that G98 may function as a gating hinge to induce movement of TM1 and open the channel gate located elsewhere along the ion conduction pathway.
But where is the channel gate located? Cahalan and colleagues have proposed that R91, located at the cytoplasmic rim of TM1, could function as the gate (Zhang et al. 2011). R91C mutant channels can be induced to form intersubunit disulfide bonds in response to diamide, a potent oxidizing agent. Further, application of diamide to R91C Orai1 channels results in pore occlusion that could be relieved by the reducing agent bis(2-mercaptoethyl)sulfone (BMS) (Zhang et al. 2011). Importantly, the ability of diamide to induce the formation of intersubunit disulfide bonds is eliminated in the constitutively active R91C/G98D channels, suggesting that the channel pore is substantially widened by the G98D mutation (Zhang et al. 2011). Combined with ample physical evidence that CRAC channels have a narrow pore, these results indicated that cysteine residues at 91 from adjacent positions come close enough to form disulfide bridges. On the basis of this evidence, R91 is suggested to comprise part of the physical gate at the inner mouth of the channel (Zhang et al. 2011). It is important to note, however, that a broad range of mutations at R91, including charge neutralizing and reversal substitutions, do not perturb store-operated gating (Derler et al. 2009a). Moreover, the ability of R91C channels to form disulfide bonds is not unique to R91 but is seen in Cys substitutions at several pore lining positions including E106, V102 and G98 (McNally et al. 2009; Zhou et al. 2010b). Hence, additional studies are needed to clarify the precise mechanism of gating by R91 in this model.
In an alternative approach to uncover the molecular participants in TM1 for activation gating, we examined differences in the accessibility of thiol reagents between open and closed channels to Cys residues introduced at specific pore positions (McNally et al. 2012). In this approach, the positions of accessible pore lining residues are determined, as near as possible, to the closed gate. In the closed state of the pore, only residues located exteriorly to the closed gate should be accessible to reagents added to the extracellular medium. This pattern of accessibility is then compared to the accessibility in open channels to obtain a reasonable first approximation of the location of the gate (Karlin & Akabas, 1998). The results of this comparison indicated that the CRAC channel gate is located towards the extracellular region of the pore, juxtaposed in a pore region that includes the residue V102 (Fig 2; McNally et al. 2012).
Figure 2. The pore-lining residue G98C in Orai1 is modified by MTSEA in a state-dependent manner.

A, a HEK293 cell expressing G98C/E106D Orai1 and STIM1 was exposed to two applications of MTSEA, the first at rest prior to whole-cell break-in, and the second following activation of ICRAC by passive store depletion. The degree of MTSEA blockade for open and closed states of the channel was determined from the relief of blockade elicited by the reducing agent, bis(2-mercaptoethyl)sulfone (BMS) at the times indicated by the arrows. B, summary of MTSEA blockade in open and closed G98C/E106D Orai1 and D110C Orai1 (adapted from McNally et al. 2012).
If V102 is a component of the gating mechanism, mutations at this locus might be expected to destabilize channel gating. In agreement with this possibility, several mutations of V102 produce constitutively active Orai1 channels that are open even in the absence of STIM1 (McNally et al. 2012). These mutations include substitutions to several mildly hydrophobic and polar amino acids such as Cys, Gly, Ala, Ser and Thr. However, substitutions of V102 to strongly hydrophobic residues including Leu, Ile and Met produce only STIM1-dependent activation, as seen in wild-type Orai1. These results indicated that mutations of V102 destabilize activation gating, resulting in constitutively open CRAC channels. Coincidently, V102 appears to be located in an unusually narrow region of the pore as would be expected for the location of the gate: Cd2+ trapping in V102C channels occurs with a very high affinity (∼300 nm) and these channels display a high propensity to form intersubunit disulfide bonds (McNally et al. 2009; Zhou et al. 2010b). Moreover, as expected for a residue nestled in a narrow region of the pore, introducing a variety of bulky residues such as Trp, Tyr and Phe produce non-functional Orai1 channels (McNally et al. 2012), probably because of pore occlusion. The strong dependence on hydrophobicity at position V102 for store-operated gating is reminiscent of the behaviour of the nicotinic ACh receptor and the large mechanosensitive ion channel (MscL), which appear to use rings of hydrophobic residue (Leu, Val and/or Ile) as gates in the middle of the ion conduction pathway to inhibit the flux of hydrated ions (Chang et al. 1998; Miyazawa et al. 2003; Doyle, 2004). Based on these findings, it seems attractive to hypothesize that V102 comprises a hydrophobic gate in the extracellular pore region to regulate ion conduction, with mutations of the gate to less hydrophobic residues resulting in a leaky gate.
So where then is the activation gate? Is it at R91 at the lower end of TM1? Or is it at V102 towards the extracellular region of the pore, close to the selectivity filter at E106? And how can the apparent dichotomy between the conclusions of the two studies described above be reconciled? One possibility is that there are two activation gates whose actions are synergized to regulate pore opening. However, given the narrow dimensions of the pore, it is also probable that pore closure does not occur at a defined locus, but is rather, distributed along much of the length of the pore, with V102 and R91 forming the external/internal boundaries of the closed pore. In this scenario, destabilizing mutations at the external boundary at V102 could affect the stability of the entire pore and cause a leaky channel. Of course, given the many limitations of the mutagenesis and cysteine accessibility approaches employed in the studies, it is also plausible that neither location is the gate and that the observed effects arise simply due to general effects on the stability of the closed-to-open conformations of the channel. Ultimately, resolution of this question would have to involve additional functional and structural analyses to illuminate the molecular details of the pore structure in different channel states.
STIM1 regulates CRAC channel ion selectivity
A surprising functional aspect of CRAC channels revealed by analysis of the ion selectivity of V102X mutants was that STIM1 not only controls CRAC channel gating, but also bestows many fundamental features that have historically defined the fingerprint of the CRAC channel pore (McNally et al. 2012). In the absence of STIM1, V102C mutant channels exhibit poor Ca2+ selectivity and allow permeation of Cs+ and several other large cations that are normally impermeable through CRAC channels (Fig. 3). Unexpectedly, interaction of mutant channels with STIM1 effectively corrects the aberrant ion selectivity of the STIM1-free mutant channels, restoring high Ca2+ selectivity to the poorly selective STIM1-free channels (McNally et al. 2012). In fact, STIM1 was found to modulate CRAC channel permeability for a broad range of cations including Ca2+, Ba2+, Sr2+ and Cs+ (Fig. 3). These changes are accompanied by alterations in the pore geometry, and specifically, narrowing of the CRAC channel pore to a state that more closely resembles the dimensions seen in wild-type (WT) Orai1 channels. Moreover, tuning of Orai1 ion selectivity by STIM1 was not unique to the V102X mutant channels, but was also seen in WT Orai1 channels as the amount of STIM1 bound to Orai1 was increased (McNally et al. 2012), suggesting that the V102X mutations simply unmask a native intermediate channel activation state due to a leaky gate (Fig. 4).
Figure 3. STIM1 modulates the ion selectivity of the constitutively active V102C Orai1 channels.
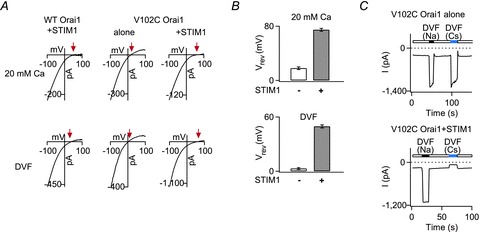
A, current–voltage relationships of currents produced by WT Orai1 + STIM1, V102C Orai1 alone, and V102C Orai1 + STIM1 in 20 mm Ca2+ or divalent-free (DVF) Ringer solutions. The red arrows indicate the reversal potentials in each case. B, summary of reversal potentials (Vrev) of V102C Orai1 currents in the presence and absence of STIM1. C, currents elicited by replacing the standard extracellular Ringer solution with Na+- or Cs+-based DVF solutions in V102C Orai1 channels expressed either alone or together with STIM1. In the absence of STIM1, large Cs+ currents are seen in V102C Orai1 channels. By contrast very little Cs+ conduction is observed in the presence of STIM1 (adapted from McNally et al. 2012).
Figure 4. A schematic representation of the conformational transitions of resting, open and V102C mutant CRAC channels in response to STIM1 binding.
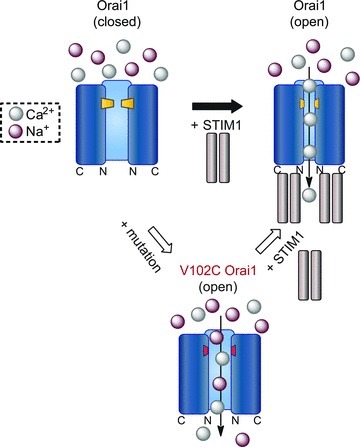
STIM1 binding alters the pore structure of wild-type (WT) Orai1 channels to open the channel gate and produce a highly Ca2+ selective channel with a narrow pore. The V102C Orai1 mutation destabilizes the channel gate to unmask a constitutively active CRAC channel with poor Ca2+ selectivity and wide pore, probably due to a leaky gate. Interaction of V102C Orai1 mutant channels with STIM1 restores high Ca2+ selectivity and a narrow pore to the mutant channels, mimicking the conformational transition that occurs in WT Orai1.
A potential explanation for the different ion selectivities of STIM1-bound and -free V102 mutant channels is that these channels have different subunit stoichiometries, as proposed in a recent model for resting and active WT Orai channels (Penna et al. 2008). Because STIM1-free Orai complexes are dimers in this model, the inevitable change in pore structure resulting from STIM1-induced assembly of Orai dimers to tetramers could naturally lead to very different permeation properties. However, analysis of the Cd2+ blockade in concatemeric Orai1 channels, in which the number of Cys-bearing protomers was varied, indicated that the subunit stoichiometries of the STIM1-free and STIM1-bound mutant channels are identical (McNally et al. 2012). This result argues that the distinct permeation properties of the STIM1-bound and -free V102X channels are due to different pore structures of fully assembled, tetrameric channels rather than an alternate subunit stoichiometry. Whether this finding can be extrapolated to WT Orai1 channels remains to be established, but given that WT Orai1 ion selectivity is also tuned by the amount of STIM1 bound to the channels (McNally et al. 2012), it is tempting to speculate that basal WT Orai1 channels also exist as preformed tetramers under resting conditions.
Taken together, these results reveal that STIM1-free Orai1 channels are intrinsically poorly Ca2+ selective. Instead, high Ca2+ selectivity and several other distinguishing features of CRAC channels (low Cs+ permeability and a narrow pore) are bestowed to the otherwise poorly Ca2+ selective Orai1 channels by STIM1 (Fig. 4). Why STIM1 has such a powerful effect on ion selectivity is unknown, but given that the Orai1 N-terminus bears a STIM1 binding site in close proximity to the pore-forming TM1 segment (Fig. 1A), it is not difficult to envision that STIM1 binding to the N-terminus could exert powerful effects on the energetic stability of the selectivity filter. This finding has many important implications for the molecular mechanisms governing ion selectivity, the nature of Ca2+ signals arising from the opening of Orai1 channels under different conditions, and even the identity of native cation currents activated by non-STIM1 Orai1 activators. The ability of CRAC channels to conduct Na+ under certain conditions may expand their potential functions to include novel modes by which they encode and process cellular information. Because emerging evidence suggests that CRAC channels, aside from activation by STIM1, can also be activated in a STIM1-independent fashion by other ligands, including the small molecule 2-APB (DeHaven et al. 2008; Peinelt et al. 2008; Schindl et al. 2008; Zhang et al. 2008; Yamashita et al. 2011), and the Golgi Ca2+-ATPase SPCA2 (Feng et al. 2010), these findings raise the possibility that Orai1 channels may function either as highly Ca2+ selective channels or non-selective channels depending on the nature of the upstream activation signal. In addition, the tight coupling of permeation and gating described in this study offers a new perspective on ligand–ion channel interactions that contradicts conventional ion channel postulates on the separation of these processes. The picture that emerges is of a hydrophobic gate (V102) located in close proximity to the selectivity filter (E106); the proximity of the two structures most likely results in a variety of conformational alterations in the selectivity filter during gating. The close coupling of permeation and gating in CRAC channels provides support for the emerging viewpoint in the ion channel literature that there is much more happening in the vicinity of the selectivity filter with respect to gating than initially imagined (Contreras et al. 2008; Thompson & Begenisich, 2012).
Conclusions
Thus far, approaches for elucidating the CRAC channel's permeation and gating properties have consisted of examining alterations in channel properties resulting from site-directed mutagenesis or examining the pattern of solvent accessibility in combination with Cys scanning. While these approaches have given us a strong framework for building a structural model of the pore, many fundamental aspects of permeation and gating remain unclear. How does STIM1 regulate CDI and the ion selectivity of CRAC channels? What are the roles of the STIM1 binding sites in the Orai1 C- and N-termini for permeation, selectivity, and channel gating? How do the candidate gate residues, V102 and R91, regulate the opening and closing of the pore? What are the conformational changes in the pore involved in pore opening? More structure–function studies are needed to achieve a deeper understanding of these issues. However, ultimately, the clarity of a high resolution X-ray crystal structure of the pore is required for a full understanding of the structural basis of selectivity and permeation. In conjunction with related approaches such as NMR spectroscopy and spin labelling, these approaches may make it possible to elucidate the dynamics of the CRAC channel ion transport cycle, and ultimately herald the development of newer and more specific CRAC channel blockers for treating a range of immune syndromes.
Acknowledgments
The authors would like to thank members of the laboratory for helpful discussions. The work described in this review was supported by NIH grant NS057499 to M.P.
References
- Ashmole I, Duffy SM, Leyland ML, Morrison VS, Begg M, Bradding P. CRACM/Orai ion channel expression and function in human lung mast cells. J Allergy Clin Immunol. 2012;129:1628–1635.e2. doi: 10.1016/j.jaci.2012.01.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aussel C, Marhaba R, Pelassy C, Breittmayer JP. Submicromolar La3+ concentrations block the calcium release-activated channel, and impair CD69 and CD25 expression in CD3- or thapsigargin-activated Jurkat cells. Biochem J. 1996;313:909–913. doi: 10.1042/bj3130909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakowski D, Parekh AB. Monovalent cation permeability and Ca2+ block of the store-operated Ca2+ current ICRAC in rat basophilic leukemia cells. Pflugers Arch. 2002;443:892–902. doi: 10.1007/s00424-001-0775-8. [DOI] [PubMed] [Google Scholar]
- Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-Aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002;16:1145–1150. doi: 10.1096/fj.02-0037rev. [DOI] [PubMed] [Google Scholar]
- Cai X. Molecular evolution and structural analysis of the Ca2+ release-activated Ca2+ channel subunit, Orai. J Mol Biol. 2007;368:1284–1291. doi: 10.1016/j.jmb.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Chang G, Spencer RH, Lee AT, Barclay MT, Rees DC. Structure of the MscL homolog from Mycobacterium tuberculosis: a gated mechanosensitive ion channel. Science. 1998;282:2220–2226. doi: 10.1126/science.282.5397.2220. [DOI] [PubMed] [Google Scholar]
- Contreras JE, Srikumar D, Holmgren M. Gating at the selectivity filter in cyclic nucleotide-gated channels. Proc Natl Acad Sci U S A. 2008;105:3310–3314. doi: 10.1073/pnas.0709809105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW., Jr Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem. 2008;283:19265–19273. doi: 10.1074/jbc.M801535200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Fahrner M, Carugo O, Muik M, Bergsmann J, Schindl R, Frischauf I, Eshaghi S, Romanin C. Increased hydrophobicity at the N-terminus/membrane interface impairs gating of the SCID-related ORAI1 mutant. J Biol Chem. 2009a;284:15903–15915. doi: 10.1074/jbc.M808312200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derler I, Fahrner M, Muik M, Lackner B, Schindl R, Groschner K, Romanin C. A Ca2+ release-activated Ca2+ (CRAC) modulatory domain (CMD) within STIM1 mediates fast Ca2+-dependent inactivation of ORAI1 channels. J Biol Chem. 2009b;284:24933–24938. doi: 10.1074/jbc.C109.024083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirksen RT. Checking your SOCCs and feet: the molecular mechanisms of Ca2+ entry in skeletal muscle. J Physiol. 2009;587:3139–3147. doi: 10.1113/jphysiol.2009.172148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA. Molecular insights into ion channel function (Review) Mol Membr Biol. 2004;21:221–225. doi: 10.1080/09687680410001716844. [DOI] [PubMed] [Google Scholar]
- Feng M, Grice DM, Faddy HM, Nguyen N, Leitch S, Wang Y, Muend S, Kenny PA, Sukumar S, Roberts-Thomson SJ, Monteith GR, Rao R. Store-independent activation of Orai1 by SPCA2 in mammary tumors. Cell. 2010;143:84–98. doi: 10.1016/j.cell.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci. 2011;31:3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins BJ, Irrinki KM, Mallilankaraman K, Lien YC, Wang Y, Bhanumathy CD, Subbiah R, Ritchie MF, Soboloff J, Baba Y, Kurosaki T, Joseph SK, Gill DL, Madesh M. S-glutathionylation activates STIM1 and alters mitochondrial homeostasis. J Cell Biol. 2010;190:391–405. doi: 10.1083/jcb.201004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover PJ, Lewis RS. Stoichiometric requirements for trapping and gating of Ca2+ release-activated Ca2+ (CRAC) channels by stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci U S A. 2011;108:13299–13304. doi: 10.1073/pnas.1101664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. J Physiol. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF. ‘Quantal’ Ca2+ release and the control of Ca2+ entry by inositol phosphates–a possible mechanism. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- Ji W, Xu P, Li Z, Lu J, Liu L, Zhan Y, Chen Y, Hille B, Xu T, Chen L. Functional stoichiometry of the unitary calcium-release-activated calcium channel. Proc Natl Acad Sci U S A. 2008;105:13668–13673. doi: 10.1073/pnas.0806499105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- Lansman JB, Hess P, Tsien RW. Blockade of current through single calcium channels by Cd2+, Mg2+, and Ca2+. Voltage and concentration dependence of calcium entry into the pore. J Gen Physiol. 1986;88:321–347. doi: 10.1085/jgp.88.3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KP, Yuan JP, Zeng W, So I, Worley PF, Muallem S. Molecular determinants of fast Ca2+-dependent inactivation and gating of the Orai channels. Proc Natl Acad Sci U S A. 2009;106:14687–14692. doi: 10.1073/pnas.0904664106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepple-Wienhues A, Cahalan MD. Conductance and permeation of monovalent cations through depletion-activated Ca2+ channels (ICRAC) in Jurkat T cells. Biophys J. 1996;71:787–794. doi: 10.1016/S0006-3495(96)79278-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS. The molecular choreography of a store-operated calcium channel. Nature. 2007;446:284–287. doi: 10.1038/nature05637. [DOI] [PubMed] [Google Scholar]
- Lewis RS. Store-operated calcium channels: new perspectives on mechanism and function. Cold Spring Harb Perspect Biol. 2011;3:pii: a003970. doi: 10.1101/cshperspect.a003970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Liu L, Deng Y, Ji W, Du W, Xu P, Chen L, Xu T. Graded activation of CRAC channel by binding of different numbers of STIM1 to Orai1 subunits. Cell Res. 2011;21:305–315. doi: 10.1038/cr.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Lu J, Xu P, Xie X, Chen L, Xu T. Mapping the interacting domains of STIM1 and Orai1 in Ca2+ release-activated Ca2+ channel activation. J Biol Chem. 2007;282:29448–29456. doi: 10.1074/jbc.M703573200. [DOI] [PubMed] [Google Scholar]
- Liou J, Fivaz M, Inoue T, Meyer T. Live-cell imaging reveals sequential oligomerization and local plasma membrane targeting of stromal interaction molecule 1 after Ca2+ store depletion. Proc Natl Acad Sci U S A. 2007;104:9301–9306. doi: 10.1073/pnas.0702866104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr Biol. 2005;15:1235–1241. doi: 10.1016/j.cub.2005.05.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis A, Zierler S, Peinelt C, Fleig A, Penner R. A single lysine in the N-terminal region of store-operated channels is critical for STIM1-mediated gating. J Gen Physiol. 2010;136:673–686. doi: 10.1085/jgp.201010484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litjens T, Harland ML, Roberts ML, Barritt GJ, Rychkov GY. Fast Ca2+-dependent inactivation of the store-operated Ca2+ current (ISOC) in liver cells: a role for calmodulin. J Physiol. 2004;558:85–97. doi: 10.1113/jphysiol.2004.065870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- Loussouarn G, Phillips LR, Masia R, Rose T, Nichols CG. Flexibility of the Kir6.2 inward rectifier K+ channel pore. Proc Natl Acad Sci U S A. 2001;98:4227–4232. doi: 10.1073/pnas.061452698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luik RM, Wang B, Prakriya M, Wu MM, Lewis RS. Oligomerization of STIM1 couples ER calcium depletion to CRAC channel activation. Nature. 2008;454:538–542. doi: 10.1038/nature07065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma HT, Venkatachalam K, Parys JB, Gill DL. Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J Biol Chem. 2002;277:6915–6922. doi: 10.1074/jbc.M107755200. [DOI] [PubMed] [Google Scholar]
- McNally BA, Somasundaram A, Yamashita M, Prakriya M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482:241–245. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNally BA, Yamashita M, Engh A, Prakriya M. Structural determinants of ion permeation in CRAC channels. Proc Natl Acad Sci U S A. 2009;106:22516–22521. doi: 10.1073/pnas.0909574106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MJ, Mahaut-Smith MP, Grinstein S. The role of intracellular Ca2+ in the regulation of the plasma membrane Ca2+ permeability of unstimulated rat lymphocytes. J Biol Chem. 1991;266:10872–10879. [PubMed] [Google Scholar]
- Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J Biol Chem. 2006;281:24979–24990. doi: 10.1074/jbc.M604589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Orai1 subunit stoichiometry of the mammalian CRAC channel pore. J Physiol. 2008;586:419–425. doi: 10.1113/jphysiol.2007.147249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- Muik M, Frischauf I, Derler I, Fahrner M, Bergsmann J, Eder P, Schindl R, Hesch C, Polzinger B, Fritsch R, Kahr H, Madl J, Gruber H, Groschner K, Romanin C. Dynamic coupling of the putative coiled-coil domain of ORAI1 with STIM1 mediates ORAI1 channel activation. J Biol Chem. 2008;283:8014–8022. doi: 10.1074/jbc.M708898200. [DOI] [PubMed] [Google Scholar]
- Mullins FM, Park CY, Dolmetsch RE, Lewis RS. STIM1 and calmodulin interact with Orai1 to induce Ca2+-dependent inactivation of CRAC channels. Proc Natl Acad Sci U S A. 2009;106:15495–15500. doi: 10.1073/pnas.0906781106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Borelly L, Somasundaram A, Yamashita M, Ren D, Miller RJ, Prakriya M. STIM1–Orai1 interactions and Orai1 conformational changes revealed by live-cell FRET microscopy. J Physiol. 2008;586:5383–5401. doi: 10.1113/jphysiol.2008.162503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell. 2009;136:876–890. doi: 10.1016/j.cell.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol. 2008;586:3061–3073. doi: 10.1113/jphysiol.2008.151365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinelt C, Vig M, Koomoa DL, Beck A, Nadler MJ, Koblan-Huberson M, Lis A, Fleig A, Penner R, Kinet JP. Amplification of CRAC current by STIM1 and CRACM1 (Orai1) Nat Cell Biol. 2006;8:771–773. doi: 10.1038/ncb1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna A, Demuro A, Yeromin AV, Zhang SL, Safrina O, Parker I, Cahalan MD. The CRAC channel consists of a tetramer formed by Stim-induced dimerization of Orai dimers. Nature. 2008;456:116–120. doi: 10.1038/nature07338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Potentiation and inhibition of Ca2+ release-activated Ca2+ channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP3 receptors. J Physiol. 2001;536:3–19. doi: 10.1111/j.1469-7793.2001.t01-1-00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J Gen Physiol. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M, Lewis RS. Regulation of CRAC channel activity by recruitment of silent channels to a high open-probability gating mode. J Gen Physiol. 2006;128:373–386. doi: 10.1085/jgp.200609588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather WA, McCleskey EW. Permeation and selectivity in calcium channels. Annu Rev Physiol. 2003;65:133–159. doi: 10.1146/annurev.physiol.65.092101.142345. [DOI] [PubMed] [Google Scholar]
- Schindl R, Bergsmann J, Frischauf I, Derler I, Fahrner M, Muik M, Fritsch R, Groschner K, Romanin C. 2-Amino-ethoxydiphenyl borate alters selectivity of Orai3 channels by increasing their pore size. J Biol Chem. 2008;283:20261–20267. doi: 10.1074/jbc.M803101200. [DOI] [PubMed] [Google Scholar]
- Scrimgeour N, Litjens T, Ma L, Barritt GJ, Rychkov GY. Properties of Orai1 mediated store-operated current depend on the expression levels of STIM1 and Orai1 proteins. J Physiol. 2009;587:2903–2918. doi: 10.1113/jphysiol.2009.170662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikanth S, Jung HJ, Kim KD, Souda P, Whitelegge J, Gwack Y. A novel EF-hand protein, CRACR2A, is a cytosolic Ca2+ sensor that stabilizes CRAC channels in T cells. Nat Cell Biol. 2010;12:436–446. doi: 10.1038/ncb2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stathopulos PB, Li GY, Plevin MJ, Ames JB, Ikura M. Stored Ca2+ depletion-induced oligomerization of stromal interaction molecule 1 (STIM1) via the EF-SAM region: An initiation mechanism for capacitive Ca2+ entry. J Biol Chem. 2006;281:35855–35862. doi: 10.1074/jbc.M608247200. [DOI] [PubMed] [Google Scholar]
- Stathopulos PB, Zheng L, Li GY, Plevin MJ, Ikura M. Structural and mechanistic insights into STIM1-mediated initiation of store-operated calcium entry. Cell. 2008;135:110–122. doi: 10.1016/j.cell.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Su Z, Shoemaker RL, Marchase RB, Blalock JE. Ca2+ modulation of Ca2+ release-activated Ca2+ channels is responsible for the inactivation of its monovalent cation current. Biophys J. 2004;86:805–814. doi: 10.1016/S0006-3495(04)74156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa R, Cheng H, Beck A, Ishikawa J, Launay P, Kubota H, Kinet JP, Fleig A, Yamada T, Penner R. A pyrazole derivative potently inhibits lymphocyte Ca2+ influx and cytokine production by facilitating transient receptor potential melastatin 4 channel activity. Mol Pharmacol. 2006;69:1413–1420. doi: 10.1124/mol.105.021154. [DOI] [PubMed] [Google Scholar]
- Thompson J, Begenisich T. Selectivity filter gating in large-conductance Ca2+-activated K+ channels. J Gen Physiol. 2012;139:235–244. doi: 10.1085/jgp.201110748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr Biol. 2006a;16:2073–2079. doi: 10.1016/j.cub.2006.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M, Peinelt C, Beck A, Koomoa DL, Rabah D, Koblan-Huberson M, Kraft S, Turner H, Fleig A, Penner R, Kinet JP. CRACM1 is a plasma membrane protein essential for store-operated Ca2+ entry. Science. 2006b;312:1220–1223. doi: 10.1126/science.1127883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao B, Coste B, Mathur J, Patapoutian A. Temperature-dependent STIM1 activation induces Ca2+ influx and modulates gene expression. Nat Chem Biol. 2011;7:351–358. doi: 10.1038/nchembio.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Navarro-Borelly L, McNally BA, Prakriya M. Orai1 mutations alter ion permeation and Ca2+-dependent inactivation of CRAC channels: evidence for coupling of permeation and gating. J Gen Physiol. 2007;130:525–540. doi: 10.1085/jgp.200709872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Jin H, Cui X, Li S, Shen Y. Structural and mechanistic insights into the activation of stromal interaction molecule 1 (STIM1) Proc Natl Acad Sci USA. 2012;109:5657–5662. doi: 10.1073/pnas.1118947109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita M, Somasundaram A, Prakriya M. Competitive modulation of CRAC channel gating by STIM1 and 2-aminoethyldiphenyl borate (2-APB) J Biol Chem. 2011;286:9429–9442. doi: 10.1074/jbc.M110.189035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G. The moving parts of voltage-gated ion channels. Q Rev Biophys. 1998;31:239–295. doi: 10.1017/s0033583598003448. [DOI] [PubMed] [Google Scholar]
- Yeromin AV, Roos J, Stauderman KA, Cahalan MD. A store-operated calcium channel in Drosophila S2 cells. J Gen Physiol. 2004;123:167–182. doi: 10.1085/jgp.200308982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeromin AV, Zhang SL, Jiang W, Yu Y, Safrina O, Cahalan MD. Molecular identification of the CRAC channel by altered ion selectivity in a mutant of Orai. Nature. 2006;443:226–229. doi: 10.1038/nature05108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Kozak JA, Jiang W, Yeromin AV, Chen J, Yu Y, Penna A, Shen W, Chi V, Cahalan MD. Store-dependent and -independent modes regulating Ca2+ release-activated Ca2+ channel activity of human Orai1 and Orai3. J Biol Chem. 2008;283:17662–17671. doi: 10.1074/jbc.M801536200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Hu J, Amcheslavsky A, Zheng H, Cahalan MD. Mutations in Orai1 transmembrane segment 1 cause STIM1-independent activation of Orai1 channels at glycine 98 and channel closure at arginine 91. Proc Natl Acad Sci U S A. 2011;108:17838–17843. doi: 10.1073/pnas.1114821108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. Genome-wide RNAi screen of Ca2+ influx identifies genes that regulate Ca2+ release-activated Ca2+ channel activity. Proc Natl Acad Sci U S A. 2006;103:9357–9362. doi: 10.1073/pnas.0603161103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, Stauderman KA, Cahalan MD. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–905. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen XG, Xie C, Fitzmaurice A, Schoonover CE, Orenstein ET, Yang J. Functional architecture of the inner pore of a voltage-gated Ca2+ channel. J Gen Physiol. 2005;126:193–204. doi: 10.1085/jgp.200509292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Meraner P, Kwon HT, Machnes D, Oh-hora M, Zimmer J, Huang Y, Stura A, Rao A, Hogan PG. STIM1 gates the store-operated calcium channel ORAI1 in vitro. Nat Struct Mol Biol. 2010a;17:112–116. doi: 10.1038/nsmb.1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Ramachandran S, Oh-Hora M, Rao A, Hogan PG. Pore architecture of the ORAI1 store-operated calcium channel. Proc Natl Acad Sci U S A. 2010b;107:4896–4901. doi: 10.1073/pnas.1001169107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitt C, Strauss B, Schwarz EC, Spaeth N, Rast G, Hatzelmann A, Hoth M. Potent inhibition of Ca2+ release-activated Ca2+ channels and T-lymphocyte activation by the pyrazole derivative BTP2. J Biol Chem. 2004;279:12427–12437. doi: 10.1074/jbc.M309297200. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Mitogen-regulated Ca2+ current of T lymphocytes is activated by depletion of intracellular Ca2+ stores. Proc Natl Acad Sci U S A. 1993;90:6295–6299. doi: 10.1073/pnas.90.13.6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. J Gen Physiol. 1995;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]