

Abstract
Acute and chronic hypoxias are common cardiac diseases that lead often to arrhythmia and impaired contractility. At the cellular level it is unclear whether the suppression of cardiac Ca2+ channels (CaV1.2) results directly from oxygen deprivation on the channel protein or is mediated by intermediary proteins affecting the channel. To address this question we measured the early effects of hypoxia (5–60 s, 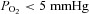 ) on Ca2+ current (ICa) and tested the involvement of protein kinase A (PKA) phosphorylation, Ca2+/calmodulin-mediated signalling and the haem oxygenase (HO) pathway in the hypoxic regulation of CaV1.2 in rat and cat ventricular myocytes and HEK-293 cells. Hypoxic suppression of ICa and Ca2+ transients was significant within 5 s and intensified in the following 50 s, and was reversible. Phosphorylation by cAMP or the phosphatase inhibitor okadaic acid desensitized ICa to hypoxia, while PKA inhibition by H-89 restored the sensitivity of ICa to hypoxia. This phosphorylation effect was specific to Ca2+, but not Ba2+ or Na+, permeating through the channel. CaMKII inhibitory peptide and Bay K8644 reversed the phosphorylation-induced desensitization to hypoxia. Mutation of CAM/CaMKII-binding motifs of the α1c subunit of CaV1.2 fully desensitized the Ca2+ channel to hypoxia. Rapid application of HO inhibitors (zinc protoporphyrin (ZnPP) and tin protoporphyrin (SnPP)) suppressed the channel in a manner similar to acute hypoxia such that: (1) ICa and IBa were suppressed within 5 s of ZnPP application; (2) PKA activation and CaMKII inhibitors desensitized ICa, but not IBa, to ZnPP; and (3) hypoxia failed to further suppress ICa and IBa in ZnPP-treated myocytes. We propose that the binding of HO to the CaM/CaMKII-specific motifs on Ca2+ channel may mediate the rapid response of the channel to hypoxia.
) on Ca2+ current (ICa) and tested the involvement of protein kinase A (PKA) phosphorylation, Ca2+/calmodulin-mediated signalling and the haem oxygenase (HO) pathway in the hypoxic regulation of CaV1.2 in rat and cat ventricular myocytes and HEK-293 cells. Hypoxic suppression of ICa and Ca2+ transients was significant within 5 s and intensified in the following 50 s, and was reversible. Phosphorylation by cAMP or the phosphatase inhibitor okadaic acid desensitized ICa to hypoxia, while PKA inhibition by H-89 restored the sensitivity of ICa to hypoxia. This phosphorylation effect was specific to Ca2+, but not Ba2+ or Na+, permeating through the channel. CaMKII inhibitory peptide and Bay K8644 reversed the phosphorylation-induced desensitization to hypoxia. Mutation of CAM/CaMKII-binding motifs of the α1c subunit of CaV1.2 fully desensitized the Ca2+ channel to hypoxia. Rapid application of HO inhibitors (zinc protoporphyrin (ZnPP) and tin protoporphyrin (SnPP)) suppressed the channel in a manner similar to acute hypoxia such that: (1) ICa and IBa were suppressed within 5 s of ZnPP application; (2) PKA activation and CaMKII inhibitors desensitized ICa, but not IBa, to ZnPP; and (3) hypoxia failed to further suppress ICa and IBa in ZnPP-treated myocytes. We propose that the binding of HO to the CaM/CaMKII-specific motifs on Ca2+ channel may mediate the rapid response of the channel to hypoxia.
Key points
Lack of oxygen to the heart, hypoxia, triggers a cascade of events that within minutes leads to irreversible tissue damage.
One of the initial steps at the onset of hypoxia is suppression of the cellular calcium channels that control the strength of the heartbeat.
The role of oxygen in energy production is well known, but it is not known how lack of oxygen within seconds may suppress the calcium channels.
We tested the involvement of different regulatory pathways that are known to modulate the calcium channel in response to various stimuli, and succeeded to link it to a haem oxygenase protein that may act as the primary oxygen sensor.
Our findings provide a better understanding of the initial events that occur during episodes of acute hypoxia induced by coronary thrombosis and bring into focus a new set of therapeutic considerations.
Introduction
O2 deprivation has significant detrimental effects on the myocardium that include ischaemic infarction, defective Ca2+ homeostasis and arrhythmia, leading eventually to cardiomyopathy and heart failure. It is generally thought that prolonged ischaemia leads to influx of Ca2+ on the Na+/Ca2+ exchanger, increasing the Ca2+ load of the sarcoplasmic reticulum (SR), which in turn is mobilized during reperfusion injury causing cell death (Piper et al. 2004). Conversely, the L-type Ca2+ current (ICa) is suppressed by cardiac hypoxia (Fearon et al. 1997) possibly reducing ICa-gated Ca2+ release, force development and phosphorylation by CaM-kinase II (Vittone et al. 2002). Thus, it may be critical to determine whether the suppression of Ca2+ channel by hypoxia serves as a regulatory mechanism that protects against Ca2+ overload during ischemia–reperfusion insult.
Previous studies of hypoxic modulation of ionic channels, on longer time scales and in different tissues, have yielded no consensus on the underlying mechanisms of oxygen sensing, the intermediate signalling pathways involved, and their convergence on the channel proteins. Rapid suppression of the L-type Ca2+ channel has been observed in smooth muscle (Franco-Obregon et al. 1995) and single glomus cells (Montoro et al. 1996) with either Ca2+ (ICa) or Ba2+ (IBa) as charge carrier, in the HEK-293 cell lines, where cytoplasmic residues 1786–1821 of the recombinant human cardiac α1C subunit were found to be involved (Fearon et al. 1997, 2000), and in rat ventricular myocytes (Movafagh & Morad, 2010). Some studies have tested the possibility that the hypoxic regulation of the Ca2+ channel may involve oxidation of intra- and extracellular thiol residues by reactive oxygen species (ROS) (Fearon et al. 1999; Hool, 2000; Hool & Corry, 2007). However, this idea has encountered some skepticism because (1) ROS production during hypoxia is either moderate or even decreased (Hool et al. 2005; Korge et al. 2008), (2) accumulation of mitochondrial superoxides is only moderate in the first 40–50 min of hypoxia, long after suppression of ICa (Hool, 2000), and (3) the evidence in support of involvement of extracellular thiol groups comes primarily from application of exogenous thiol-oxidizing or -reducing agents.
Here we have examined the possibility that the rapid hypoxic suppression of ICa might involve a more direct O2 sensing mechanism mediated e.g. by haem oxygenase (HO) or its reaction products (CO, Fe2+ and biliverdin). Mechanisms of this type have been associated with Ca2+-activated K+ channels in carotid bodies (Williams et al. 2004; Ortega-Saenz et al. 2007), anoxic activation of ganglionic nociceptive neurons (D’Agostino et al. 2009), and CO-mediated redox modulation of L-type Ca2+ channels (Scragg et al. 2008). The intriguing finding that calmodulin (CaM) binds to both HO-2, and the Ca2+ sensitive domain of the carboxyl tail of the α1C subunit of the CaV1.2 (Boehning et al. 2004), where Ca2+-dependent inactivation of the channel takes place (Zuhlke & Reuter, 1998; Erickson et al. 2001), provided further impetus to probe this possibility.
To test whether HO might serve as a rapid O2 sensor for the Ca2+ channel, we measured the response of the Ca2+ channel current only in the first 5–60 s of withdrawal of O2 (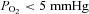 ) using experimental conditions with varying degrees of Ca2+-dependent inactivation and altered signalling by CaM and HO. We found that the suppression of the L-type Ca2+ channel in response to acute O2 loss has a rapid onset (>5 s) both in cardiomyocytes and in HEK-293 cells expressing the recombinant channel. The hypoxic response depended critically on Ca2+ influx through the channel, and on its suspected phosphorylation state, but not on release of Ca2+ from the SR. Using different isoforms of the recombinant Ca2+ channel, an 80 amino acid domain within the Ca2+/CaM/CaMKII binding motif of the C-terminus was identified as mediating the hypoxic response. Interestingly, the suppressive effects of withdrawal of O2 and HO-oxygenase inhibitors on the Ca2+ channel had similar profiles with respect to magnitude, kinetics, phosphorylation and ionic specificity, and were non-additive. Our data are consistent with the idea that HO may confer O2 sensitivity on the cardiac Ca2+ channel.
) using experimental conditions with varying degrees of Ca2+-dependent inactivation and altered signalling by CaM and HO. We found that the suppression of the L-type Ca2+ channel in response to acute O2 loss has a rapid onset (>5 s) both in cardiomyocytes and in HEK-293 cells expressing the recombinant channel. The hypoxic response depended critically on Ca2+ influx through the channel, and on its suspected phosphorylation state, but not on release of Ca2+ from the SR. Using different isoforms of the recombinant Ca2+ channel, an 80 amino acid domain within the Ca2+/CaM/CaMKII binding motif of the C-terminus was identified as mediating the hypoxic response. Interestingly, the suppressive effects of withdrawal of O2 and HO-oxygenase inhibitors on the Ca2+ channel had similar profiles with respect to magnitude, kinetics, phosphorylation and ionic specificity, and were non-additive. Our data are consistent with the idea that HO may confer O2 sensitivity on the cardiac Ca2+ channel.
Methods
Isolation of rat and cat cardiomyocytes
Protocols for experiments with rats (AR no. 2791) and cats (ACORP no. 454) were approved and supervised by the Institutional Animal Care and Use Committees (IACUC) of Georgetown University, the Medical University of South Carolina (A 3428–01), University of South Carolina (A 3049-01) and the Department of Veterans Affairs according to national and international guidelines.
Freshly dissociated rat cardiomyocytes were prepared by enzymatic digestion as previously described (Knollmann et al. 2000) using one male Wistar rat (200–300 g) per experimental day. Briefly, each rat was anaesthetized with sodium pentobarbital (150 mg kg−1 i.p.) and the heart was rapid excised from the chest cavity and subjected to retrograde Langendorff perfusion using Tyrode solutions with different concentrations of Ca2+ and digestive enzymes (1.4 mg ml−1 collagenase and 0.16 mg ml−1 protease). The dispersed cells were equilibrated in Tyrode solution with 0.2 mm CaCl2 and were allowed 30 min to settle and adhere to coverslips before they were used in electrophysiological experiments within the next 4–8 h.
The preparation of cat ventricular myocytes was similar (Mann et al. 1991), but was performed under sterile conditions yielding cells that were kept in culture medium for up to 4 days before use. As with rats, the method of killing was exsanguinations following excision of the heart under deep anaesthesia (50 mg kg−1 ketamine hydrochloride and 5 mg kg−1 acepromazine maleate i.m.).
Expression of recombinant human Ca2+ channels in HEK-293 cells
was accomplished with the standard lipofectamine method (Lipofectamine 2000, Invitrogen) using cDNAs of all subunits (α1C77-YFP, β1, α2δ) with yellow fluorescent protein (YFP) for positive fluorescence identification (Chroma Technology, Bellows Falls, VT, USA; 470 nm excitation/525 nm emission). HEK-293 cells were cultured to 90% confluence in six-well plates using Dulbecco's modified Eagle's medium (DMEM) with 10% fetal bovine serum (FBS). The lipofectamine reagent was mixed with 1–6 μg of each cDNA, incubated for 20 min and dispensed into each well while swirling the culture plates. Cells were then incubated for 24 h allowing for gene expression to take place. The next day, cells were split onto coverslips at 10% confluence and allowed to incubate for another 48 h before being screened based on green fluorescent protein (GFP) fluorescence (60–70%) and used in voltage-clamp experiments similar to those conducted with cardiomyocytes. Mutant channels were expressed in HEK cells by replacing the α1c77 subunit with the α1c86 subunit. All cDNAs were generously provided by Dr Nikolai Soldatov (National Institute on Aging, NIH, Baltimore, MD, USA).
Electrophysiological experiments at different O2 pressures
Rat and cat cardiomyocytes and HEK-293 cells expressing recombinant Ca2+ channels were voltage-clamped in the whole-cell configuration on the stage of an inverted Nikon microscope using a Dagan amplifier and pCLAMP software (Axon Instruments, Union City, CA, USA). A rapid perfusion system was used to change between solutions equilibrated with different gasses or with different ionic composition. Epi-fluorescence detection was used for ratiometric measurements of intracellular Ca2+ in cardiomyocytes (Fura-2) and evaluation of protein expression in HEK-293 cells (YFP).
To measure Ca2+ current (ICa), patch pipettes with 2–5 MΩ resistance were filled with an internal solution containing (in mm): 120 Cs+, 120 Asp−, 20 TEACl, 2 EGTA, 20 Hepes and 5 MgATP, at pH 7.2 using CsOH. A series resistance compensation of ∼80% was applied and monitored throughout recordings. The liquid junction potential was corrected before seal formation. In many experiments, 0.2 mm cyclic adenosine monophosphate (cAMP) was added to the patch pipette solutions to prevent rundown of ICa and fully activate the SR Ca2+-ATPase via PKA-mediated phosphorylation. The extracellular Tyrode solution contained (in mm): 140 NaCl, 4.7 mm KCl or CsCl, 2–5 CaCl2, 1 MgCl2, 10 glucose and 10 Hepes (pH 7.4). For measurement of Ba2+ current (IBa), CaCl2 was replaced with 2–10 mm of BaCl2. To allow permeation of Na+ through the Ca2+ channels (INa), 2 mm EGTA was used to chelate traces of Ca2+ and Mg2+. To inhibit K+ currents, membrane currents were recorded with an extracellular solution where K+ was replaced by Cs+. A holding potential of −50 mV was chosen to inactivate Na+ channels.
The current through the Ca2+ channel was generally measured using 25–50 ms depolarizing pulses from −50 to +10 mV applied every 5 or 10 s. Pulses of 200 ms duration were used for simultaneous measurements of ICa and intracellular Ca2+ (Fig. 2). Current–voltage (I–V) relations were obtained with depolarizing pulses ranging from −50 to +80 mV in increments of 10 mV. To generate I–V relations that were free of secondary effects of prolonged hypoxia and rundown, we limited exposure to hypoxic solutions to 2 min. The measured currents were filtered at 1 or 10 KHz, digitized at 10 or 100 kHz, and plotted and analysed in terms of magnitude and time constants of decay using Origin 6.0 (OriginLab Corp., Northampton, MA, USA) and pCLAMP 9.0 software.
Figure 2. The suppression of ICa by hypoxia is accompanied by a similar suppression of the ICa-triggered intracellular Ca2+ transients.
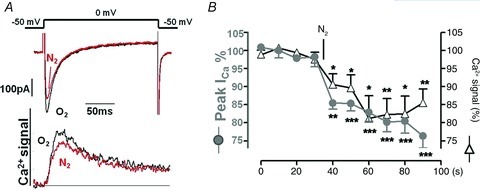
A, simultaneous measurements of ICa and Ca2+ transients measured with 0.4 mm K4Fura-2 in the patch pipette in non-cAMP dialysed rat cardiomyocytes before (O2) and after 40 s of hypoxia (N2). B, time course of suppression ICa and Ca2+ transients during exposure to hypoxic solution.
To study the effects of anoxia on ionic currents, the voltage-clamped single cells were superfused with external solutions equilibrated with atmospheric O2 (normoxic) or 100% N2 (hypoxic). Solution exchange took place within 50 ms using an electronically controlled perfusion system equipped with five barrels loaded with control and hypoxic and Tyrode solutions containing varying electrolyte concentrations and pharmacological agents (Cleemann & Morad, 1991). The O2 pressure was measured with a needle probe which registered >5 mmHg for the hypoxic solutions both in the bubbled reservoirs and near the port for solution outflow into the main chamber. Due to size of the O2 electrode (1.2 mm diameter) it measured the O2 pressure near the much smaller outflow tube (280 μm diameter) with a delay of 1–2 s. However, considering that the voltage clamped cell (∼100 μm in length) was located near the outflow port (100–150 μm) and entirely within its rapidly flowing stream (∼7 cm s−1, Belmonte & Morad, 2008), it is plausible that changes in O2 pressure at the surface of the cell occur on the same time scale (<50 ms) as measured when the Ca2+ channels were subjected to ionic interventions. In another set of experiments we verified rapid changes in local 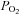 by using cells that were plated onto cover slips with an oxygen-sensitive fluorescent coating (Supplemental Fig. 1).
by using cells that were plated onto cover slips with an oxygen-sensitive fluorescent coating (Supplemental Fig. 1).
Simultaneous measurement of Ca2+ transients and ICa were performed with a dialysing solution containing 0.5 mm of the cell impermeant fluorescent ratiometric Ca2+ indicator K4Fura-2 (Invitrogen) together with 0.21 mm Ca2+, but no EGTA, yielding an intracellular concentration of 100 nm of free Ca2+. As previously described (Cleemann & Morad, 1991) a mirror vibrating at 1150 Hz was used to alternate between excitation wavelengths centred at 340 and 405 nm. The plotted Ca2+ signal (Fig. 2) is the ratio (F340/F405) of the corresponding fluorescence intensities measured at wavelengths above 515 nm.
When dialysing compounds of relatively high molecular weight into voltage-clamped cells, we used an equilibration period of 5–10 min between break-in and the first recordings of membrane current. Such compounds include K4Fura-2, the PKA inhibitor PH-89, CaM-binding domain peptide290−309, and the HO inhibitors zinc protoporphyrin (ZnPP) or tin protoporphyrin (SnPP). All experiments were done at room temperature (23–25°C), except those with cat cardiomyocytes that were conducted at 30–32°C.
Reagents
The CAMKII inhibitor CK59 was dissolved in dimethyl sulphoxide (DMSO) and was applied extracellularly at a concentration of 10 μm (Sigma-Aldrich, St Louise, MO, USA). CaM-binding domain peptide290−309 (Calbiochem Inc.) and the PKA inhibitor H-89 were internally dialysed using concentrations of 0.1 mm and 10 μm, respectively in the dialysing solution. The Ca2+ channels were subjected to phosphorylating conditions using 0.2 mm cAMP or 60 nm okadaic acid (OA, Sigma-Aldrich). (S)-(–)-Bay K8644 (1,4-dihydro-2,6-dimethyl-3-nitro-4-(2-trifluoromethylphenyl)-pyridine-5-carboxylic acid methyl ester) was from Sigma-Aldrich. Zinc protoporphyrin IX (ZnPP) and tin protoporphyrin IX (SnPP, Frontier Scientific Inc., Logan, UT, USA) were dissolved in DMSO and applied to external solutions in a maximal concentration of 0.005% of DMSO or dialysed into the cell for 10 min before recording of membrane current. K4Fura-2 was purchased from Invitrogen. Protease enzyme, collagenase A, was obtained from Roche Applied Sciences (Indianapolis, IN, USA).
Statistics
Student's t test was used to compare means, which are presented plus/minus their standard errors (SEM), with n indicating the number of experiments. Significance levels are indicated with an increasing number of asterisks (*P < 0.05, **P < 0.01, ***P < 0.001) as opposed to being not significant (NS, P > 0.05).
Results
Current through the cardiac Ca2+ channel is rapidly suppressed by acute hypoxia
Direct effects of acute O2 withdrawal on the Ca2+ channel were studied in different preparations using conditions that allowed different ions (Ca2+, Ba2+ or Na+) to permeate through the channel in the absence and presence of PKA activation. To achieve rapid changes in extracellular 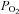 , the stream of solution surrounding the voltage-clamped cell was rapidly switched from one saturated with 100% O2 to another with 100% N2 (
, the stream of solution surrounding the voltage-clamped cell was rapidly switched from one saturated with 100% O2 to another with 100% N2 (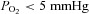 ) and peak ICa was measured during repeated depolarizations from −50 to +10 mV. In freshly dissociated rat ventricular cells at room temperature, the hypoxic solution produced ∼10% suppression of ICa within the first 5 s followed by a gradually increasing suppression that levelled off at 23 ± 3% (n = 15) in 45–60 s (Fig. 1A open circles, summarized in the bar graph, Fig. 1C). Current–voltage relations measured before (filled circles) and 1–2 min after (open circles) the onset of hypoxia showed that the suppression of ICa was equally pronounced at all potentials with no detectable change in either its activation range (Fig. 1B) or the rate of its inactivation (Inset panels). Similar suppressions of ICa (28 ± 3%, n = 9) were also measured in HEK-293 cells expressing all the recombinant subunits of the Ca2+ channel (filled circles in Fig. 1A and inset tracings), and, once again, with no significant change in the inactivation kinetics (inset panel). Short-term cultured feline cardiomyocytes (examined at 30–32°C) were similarly suppressed by O2 withdrawals (24 ± 6%, n = 3).
) and peak ICa was measured during repeated depolarizations from −50 to +10 mV. In freshly dissociated rat ventricular cells at room temperature, the hypoxic solution produced ∼10% suppression of ICa within the first 5 s followed by a gradually increasing suppression that levelled off at 23 ± 3% (n = 15) in 45–60 s (Fig. 1A open circles, summarized in the bar graph, Fig. 1C). Current–voltage relations measured before (filled circles) and 1–2 min after (open circles) the onset of hypoxia showed that the suppression of ICa was equally pronounced at all potentials with no detectable change in either its activation range (Fig. 1B) or the rate of its inactivation (Inset panels). Similar suppressions of ICa (28 ± 3%, n = 9) were also measured in HEK-293 cells expressing all the recombinant subunits of the Ca2+ channel (filled circles in Fig. 1A and inset tracings), and, once again, with no significant change in the inactivation kinetics (inset panel). Short-term cultured feline cardiomyocytes (examined at 30–32°C) were similarly suppressed by O2 withdrawals (24 ± 6%, n = 3).
Figure 1. Suppression of current through the L-type Ca2+ channel during brief periods of hypoxia.
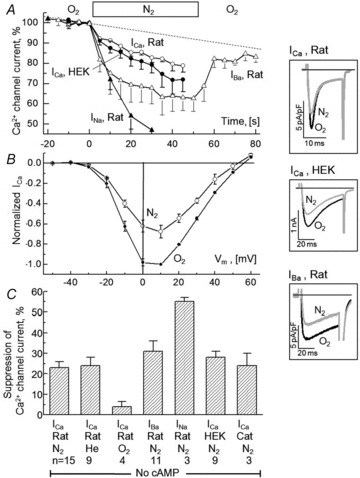
A, time course of hypoxia-induced changes in the peak inward current in rat cardiomyocytes where the permeating ion was Ca2+ (ICa), Ba2+ (IBa) or Na+ (INa) and in HEK cells where the recombinant channel conducted Ca2+ (ICa). B, current–voltage relations for ICa in rat ventricular cardiomyocytes measured under normoxic control conditions (O2) and after 1–2 min exposure to hypoxia (N2). C, average suppression of peak inward current measured with different permeating ions (Ca2+ vs. Ba2+ vs. Na+) and preparations (rat vs. cat cardiomyocytes vs. HEK-cell expression system) after 45–60 s superfusion with solutions equilibrated with N2, He or O2. The inset panels show that currents activated by depolarization from −60 to +10 mV displayed different rates of inactivation, but similar degrees of suppression during hypoxia (ICa vs. IBa in rat cardiomyocytes vs. ICa in HEK cells). All illustrated experiments were conducted in the absence of cAMP or other interventions that might evoke cAMP-dependent phosphorylation. The experiments with cat cardiomyocytes were performed at 30–32°C, the rest at room temperature, 23–25°C.
To minimize the effects of Ca2+ channel run-down, only the effects of the first 45–60 s of withdrawal of O2 were quantified and statistically analysed. On this time scale, ICa in rat cardiomyocytes showed a small degree of run-down (4 ± 3%, n = 4, Fig. 1C) in fully oxygenated solutions. This was much smaller than the effect of hypoxia, but did preclude full recovery after re-oxygenation (not shown). To exclude possible direct effects of N2 on ICa, He was substituted for N2 resulting in similar suppressive effects (24 ± 4%, n = 9).
Substitution of Ba2+ for Ca2+, as the charge carrier through the calcium channel, in cardiomyocytes resulted in suppressive effects of hypoxia that were somewhat larger (31 ± 5%, n = 11) and faster (Fig. 1A, Δ; see also Supplemental Fig. 1) compared to those with Ca2+ as charge carrier. The characteristic slower inactivation kinetics of IBa, which is known not to trigger SR Ca2+ release (Nabauer et al. 1989), remained also insensitive to withdrawal of O2 (inset panel). Readmission of O2 resulted in rapid and nearly complete recovery of IBa (Fig. 1A, open triangles) although some run-down appeared to be present (dotted line). Interestingly, the suppressive effect of hypoxia on the channel was significantly enhanced (52 ± 2%, n = 3) in the first minute of O2 withdrawal when Na+ was the permeating cation through the Ca2+ channel (in ‘zero’ Ca2+ solution with 2 mm EGTA, Fig. 1A, filled triangles).
The similarity in the suppressive effects of hypoxia on the Na+, Ba2+ and Ca2+ currents through the channel suggests that the suppressive effect of hypoxia must be independent of the Ca2+-triggering property of ICa on the SR. To test the role of the SR Ca2+ stores more directly, Fura-2 dialysed myocytes were used to measure ICa-triggered Ca2+-transients. Figure 2 shows that the hypoxia-induced suppression of ICa was accompanied by parallel decrease in Ca2+ transients, without any changes in either baseline [Ca2+]i or the release kinetics, suggesting that the hypoxic suppression of ICa is likely to result in a weakening of the heartbeat, thus reducing the requirements for metabolic energy.
The results thus far suggest that hypoxia in the absence of PKA stimulation uniformly and rapidly suppresses the Ca2+ channels both in freshly isolated rat and cat cardiomyocytes and in HEK-293 cells expressing the human CaV1.2, irrespective of the O2 substituents (N2 or He) or permeating cations (Ca2+, Ba2+ or Na+).
PKA phosphorylation desensitizes the Ca2+-transporting channel to hypoxia
Ca2+ channels are phosphorylated by the protein kinase A (PKA)/cAMP cascade causing altered gating kinetics and enhanced current. In rat ventricular myocytes dialysed with 0.2 mm cAMP, peak ICa at +10 mV increased from 2–6 pA pF−1 to 8–15 pA pF−1. Figure 3A shows that phosphorylation induced by cAMP significantly reduced the suppressive effects of hypoxia on ICa from 23 ± 3% to 6.8 ± 2% (Fig. 3A and C; P = 0.005). Similarly, inclusion of okadaic acid (an inhibitor of protein phosphatases PP1 and PP2a) in the patch pipette also reduced the response of ICa to hypoxia to 6.7 ± 2% (Fig. 3C; OA, P < 0.01).
Figure 3. Phosphorylation of the Ca2+ channel makes it insensitive to hypoxia when the permeating ion is Ca2+ (ICa), but not when it is Ba2+ (IBa) or Na+ (INa).
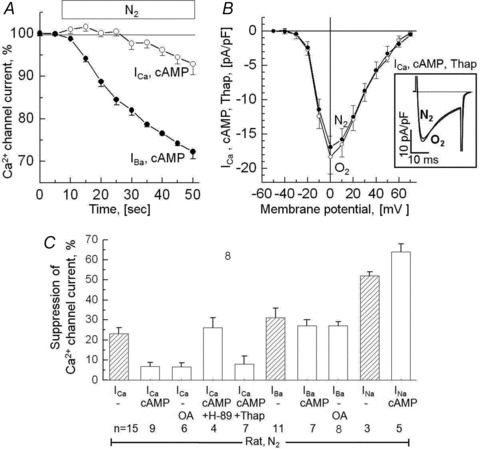
A, time course of ICa and IBa during 40 s hypoxia (N2) in rat ventricular myocytes dialysed with 0.2 mm cAMP. B, the I–V relation is insensitive to hypoxia even when SR Ca2+ stores are depleted by pre-incubation with 1 μm thapsigargin (Thap). The inset panel shows sample currents recorded with depolarization to +10 mV. C, bar graph showing the reduction in the currents through the Ca2+ channel during 45–60 s of hypoxia in cells dialysed with 0.2 mm cAMP or incubated with 1 μm of the phosphatase inhibitor okadaic acid (OA). Results obtained with the PKA inhibitor H-89 and thapsigargin (Thap) are included. Results obtained under non-phosphorylating conditions are reproduced for comparison from Fig. 1C as hatched bars. All illustrated results were obtained using rat ventricular cells and hypoxic solutions equilibrated with 100% N2.
To probe whether the insensitivity of the Ca2+ channel to hypoxia was related to PKA-dependent phosphorylation rather than to other cAMP-mediated pathways, a subset of cells that were dialysed with cAMP were then exposed to10 μm H-89, a PKA inhibitor. Figure 3C shows that cells dialysed with cAMP, but exposed to H-89, recovered their sensitivity to hypoxia (26 ± 5% suppression, n = 4, P = 0.002, vs. 6.7%). Thus, there was no significant difference between cells dialysed with cAMP-free and those dialysed with cAMP and exposed to H-89 (Fig. 3C). It appears therefore that PKA-mediated phosphorylation protects the Ca2+ channels against acute hypoxia.
Phosphorylation by PKA is known to affect the gating of the Ca2+ channel not only directly, but also indirectly through enhanced Ca2+ release caused by phosphorylation of the ryanodine receptors and phospholamban of the SR (Reuter & Scholz, 1977; Yamaoka & Kameyama, 2003). To examine whether the observed PKA effects were related, in part, to modulation of SR Ca2+ release, we incubated cAMP dialysed cells with 1 μm thapsigargin, which is known to deplete the SR Ca2+ content by inhibiting the SR Ca2+-ATPase (Treiman et al. 1998). Figure 3B shows that, even though thapsigargin greatly slowed the inactivation kinetics of ICa (changing τ from 5.4 ± 0.5 ms to 15.3 ± 0.8 ms; cf. insets in Figs 1 and 3), it did not restore the O2 sensitivity of phosphorylated Ca2+ transporting channels (n = 7). These data imply that the phosphorylation-induced protection against hypoxia is independent of release of Ca2+ from the SR.
Since Ba2+ transporting Ca2+ channels fail to trigger Ca2+ release (Nabauer et al. 1989), but are suppressed by acute hypoxia (Fig. 1C), we examined whether phosphorylation could also protect the Ba2+ transporting channels against acute hypoxia. Surprisingly, phosphorylation failed to desensitize the Ba2+-transporting channels to hypoxia. Statistical analysis revealed significant differences in the response of phosphorylated channels to O2 withdrawal when the permeating ion was Ba2+ or Na+ instead of Ca2+ (Fig. 3C) suggesting that phosphorylation-induced protection against hypoxia required entry of Ca2+ through the channel.
In ventricular cells dialysed with okadaic acid, to achieve channel phosphorylation without the use of cAMP, the desensitizing effects of phosphorylation continued to be specific to permeation of Ca2+ (Fig. 3A). Thus the data obtained with different charge carriers (Ca2+ vs. Ba2+ vs. Na+) and different means of channel phosphorylation (cAMP vs. inhibition of phosphatases vs. PKA-inhibitors), suggest that the degree of hypoxic suppression of the Ca2+ channel is somewhat dependent on the permeating cationic species (Na+ > Ba2+≍ Ca2+), while the PKA-induced desensitization against hypoxia required strictly the permeation of Ca2+, but, surprisingly, Ca2+ released from the SR did not substitute for the permeating Ca2+.
Role of calmodulin (CaM) and CaM kinase II (CAMKII) in the O2 sensing of the Ca2+ channel
cAMP is thought to increase the endogenous activity of CaMKII, by a sequence of intermediary steps that include PKA-mediated phosphorylation of inhibitor 1 and protein phosphatase PP1 leading to Ca2+- and CaM-dependent auto-phosphorylation at T286 (Fig. 4A) (Bradshaw et al. 2003; Huke & Bers, 2007). The activity of CAMKII seems to depend on higher local rather than global cytosolic concentrations of Ca2+ during excitation–contraction coupling (Song et al. 2008). This mechanism may be, in part, responsible for the PKA-induced desensitization of the Ca2+-, but not Ba2+- or Na+-transporting channel to acute hypoxia. To test this possibility we first used inhibitory peptide290−309 (PEP), which comprises the CaM-binding domain of CaMKII and by competitive binding of available CaM may block the activation of CaMKII (Payne et al. 1988) and other CaM-dependent processes tested in subsequent experiments. Following 8 min of dialysis with an internal solution containing inhibitory peptide and 0.2 mm cAMP, the voltage-clamped cardiomyocytes were exposed to hypoxic solution. Figure 4 shows that the presence of the peptide in the cytosol (PEP) restored the sensitivity of the cAMP-stimulated channels to the suppressive effects of hypoxia. The suppression of ICa was gradual (panel B, filled circles) and reached 26 ± 5% after 60–90 s of hypoxia (panel C). For comparison the dashed curve in Fig. 4B show the time course of the much smaller effect of hypoxia on ICa in cAMP-dialysed myocytes in the absence of the inhibitory peptide (6.8 ± 2%, cf. Fig. 3A).
Figure 4. Inhibition of Ca2+-dependent inactivation of ICa by calmodulin domain peptide290−309 and Bay K is associated with enhanced suppression of ICa by anoxia.
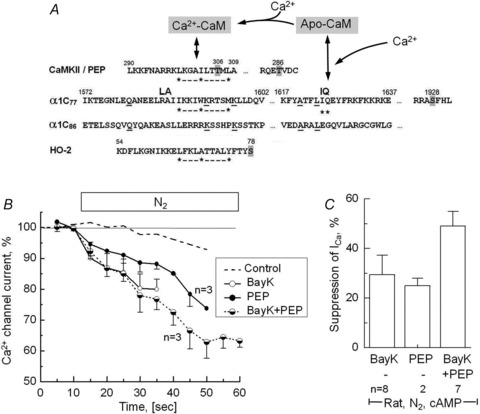
A, putative CaM-binding (IQ and hydrophobic 1–5–10 motifs) and phosphorylation sites (grey highlight) in the partial AA sequences of CaMKII, peptide290−309 (PEP), wild-type and mutant α1c subunits of the Ca2+ channel (α1c77 and α1c86) and haem oxygenase (HO-2) (Colbran, 1993; Rhoads & Friedberg, 1997; Soldatov et al. 1997; Boehning et al. 2004). B, time course of suppression of ICa upon removal of extracellular O2 in cAMP-dialysed rat cardiomyocytes that in addition were dialysed with 100 μm PEP, treated with 1 μm Bay K8644 (BayK) or both. For comparison purposes, ICa in cardiac control cells dialysed with 0.2 mm of cAMP is reproduced from Fig. 3A (dashed line). C, comparison of the anoxic suppressions of ICa in cardiomyocytes measured after 60–90 s of hypoxia (during recording of I–V relations). A, amino acid sequences of α1c77 and α1c86 subunits.
We also examined whether pharmacological inhibition of both Ca2+-dependent inactivation (using Ca2+ channel agonist (S)-(–)Bay K8644; Adachi-Akahane et al. 1999) and Ca2+-dependent facilitation (using CaMKII inhibiting peptide290−309) would further enhance the suppressive effects of hypoxia on ICa. Figure 4B shows, unexpectedly, that Ca2+ channels that had been rendered insensitive to hypoxia by cAMP, once again became sensitive to hypoxia when the myocytes were exposed to the ICa enhancing effects of Bay K (open circles). Experimentally this was then followed by measurements of the voltage dependence of current, which confirmed that the insensitivity of ICa to hypoxia in cAMP-dialysed myocytes was reversed by Bay K causing 29 ± 8% suppression at all voltages tested, somewhat similar to the effect of the CaM inhibitory peptide290−309. The suppression in the presence of Bay K or the inhibitory peptide was comparable (separately causing 29 ± 8%vs. 26 ± 5% inhibition), roughly summing to about 49 ± 6% when Bay K and inhibitory peptide were used in combination. Our data therefore suggest that pharmacological inhibition of Ca2+-dependent inactivation and facilitation can reverse the PKA-induced protection of the Ca2+ channel against hypoxia, even when Ca2+ is the charge carrier, and implicates a critical role for calmodulin in the O2 sensing pathway.
In probing the role of CaM-dependent signalling in O2 sensing we next targeted the CaM-binding motifs that is located on the carboxyl-terminal tail of the CaV1.2 channel (Fig. 4A) (Soldatov et al. 1998). In this set of experiments the native α1c77 channel was replaced by its isoform α1c86 channel with alternative splicing at exons 40–41, involving residues 1572–1651 that include both pre-IQ and IQ domains of the CaM binding motif (Soldatov et al. 1997) (Fig. 4A). This mutant channel lacks Ca2+-dependent inactivation, but shows similar fast voltage-dependent inactivation kinetics for both ICa and IBa (Soldatov et al. 1997; Erickson et al. 2003). Figure 5 confirms the rapid voltage-dependent inactivation of Ba2+ currents in α1c86 (Fig. 5B) and shows, in addition, that the mutant channel is completely insensitive to acute hypoxia (Fig. 5C). While peak Ba2+ current in α1c77 channels was markedly suppressed by hypoxia by an average of 52% (Fig. 5D; n = 8, P < 0.001 at 10 mV), both IBa, and ICa in the mutant channel were equally resistant to hypoxic suppression (Fig. 5D, n = 7). There were also no significant changes in the I–V relations of α1c86 channel in response to hypoxia (n = 4, P = 0.39, data not shown). Together these findings support the idea that the CaM association site of the Ca2+ channel C-terminus is critically important in hypoxic regulation of ICa.
Figure 5. The cytoplasmic CaM/CaMKII binding sites of the α1C subunit of the Ca2+ channel is required for its hypoxia-induced suppression.
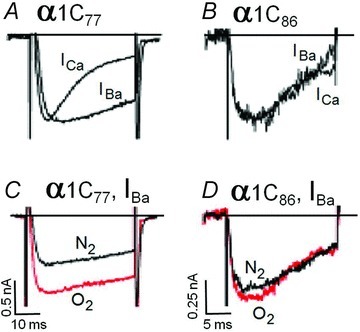
The measured Ca2+ and Ba2+ currents (ICa and IBa) show that Ca2+-dependent inactivation (A) and anoxic suppression (C) are present in HEK-293 cell expressing the α1c77 of the L-type Ca2+ channel, but not in cells expressing the mutant α1c86 subunit lacking CaM/CAMKII binding sites (B and D; cf. Fig. 4B). The currents in panels A and B were normalized to emphasize differences and similarities in kinetics. Extracellular Ba2+ at 20 mm was used to enhance the currents shown in panels C and D. All measurements were done under non-phosphorylating conditions.
Role of haem oxygenase in the hypoxic regulation of the cardiac Ca2+ channel
Since haem oxygenase binds oxygen and has been implicated in the O2-dependent regulation of Ca2+-activated K+ channels (Williams et al. 2004; Ortega-Saenz et al. 2007), and has been reported to form a Ca2+-sensitive complex with CaM (Fig. 4A; Boehning et al. 2004), we considered the possibility that this protein might be involved in the O2-dependent regulation of the cardiac Ca2+ channel by, for instance, interacting with a CaM/CaMKII binding site near the inner mouth of the channel. Figure 6A shows that rapid application of 10–100 nm of the HO inhibitor ZnPP reversibly suppressed ICa without changing its kinetics. To exclude the possibility that contaminating free Zn2+ in ZnPP solutions may cause the Ca2+ channel suppression, 10 nm ZnCl2 (an upper estimate of possible free Zn2+ released from 100 nm ZnPP in solution) was added before the application of the 100 nm ZnPP-containing solutions. Figure 6B shows that 10 nm of Zn2+ hardly affected ICa, while ZnPP again blocked ICa rapidly and reversibly. Application of ZnPP for 1–2 min blocked IBa equally at all voltages tested (Fig. 6C) with ∼64.3% suppression at 0 mV (n = 3). Similarly, another HO inhibitor, SnPP, caused 48 ± 20% suppression of IBa at 100 nm concentrations (n = 4, data not shown). In cells dialysed for 9 min with ZnPP through the patch pipette, IBa was consistently smaller than in control experiments with cells that were equilibrated for the same duration with control internal solutions. In this case, peak ICa was suppressed by 69 ± 10% at 0 mV and by similar amounts at other potentials (Fig. 6D, n = 4). These findings are consistent with the idea that HO modulates L-type Ca2+ channels at an intracellular binding site.
Figure 6. Suppression the Ca2+ channel by inhibition of haem oxygenase.
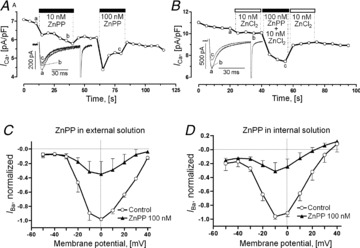
A, brief exposures to the haem oxygenase inhibitor ZnPP in concentrations of 10 and 100 nm cause rapid, reversible, dose-dependent suppression of ICa. B, effect of 10 nm ZnCl2 or 100 nm ZnPP on ICa. Insets in panels A and B show traces of ICa measured at the indicated times (a, b and c). C and D, normalized I–V relations of IBa measured in control cells (Control, open circles) and in cells exposed to 100 nm ZnPP (C,filled triangles) that was applied either in the external solution (C) or via the patch pipette by 9 min of dialysis (D). All experiments were performed on rat ventricular cardiomyocytes under normoxic and non-phosphorylating conditions.
Does PKA phosphorylation desensitize Ca2+- but not Ba2+-transporting channels to HO-inhibitors?
We compared the effects of HO inhibitors to those of hypoxia in rat ventricular myocytes with and without dialysis of either 0.2 mm cAMP or 60 nm okadaic acid (OA). Under these conditions the suppression of ICa produced by 50 s exposures to 100 nm ZnPP was reduced by the phosphorylating interventions from 21.7 ± 4.3% (n = 5) in control cells to 6.2 ± 2.9% (n = 6) in cells dialysed with cAMP and to 7.4 ± 7.5% (n = 4) in cells with OA (Fig. 7A and E). In comparison IBa continued to be suppressed by ZnPP when the channel was phosphorylated (Fig. 7B and E). The suppression of IBa by ZnPP was somewhat enhanced in OA-dialysed cells (69.1 ± 13.2%, n = 4) compared to control 43.7 ± 4.7%, n = 4). Figure 7C shows that the CAMKII inhibitor CK59 (10 μm), which strongly reduced ICa in cells dialysed with 0.2 mm cAMP, also restored the suppressive effect of ZnPP (double arrow). These effects of phosphorylation on the ZnPP-induced suppression of ICa and IBa are summarized Fig. 7E in a format that facilitates comparison with the hypoxia-induced effects (cf. Fig. 3C). The similarity in the responses suggest that HO inhibitors and the effects of hypoxia on L-type Ca2+ channels may be mediated by the same channel entity. Consistent with this idea, we found that acute hypoxia failed to further suppress the Ca2+ channel in the presence of 1 μm ZnPP (Fig. 7D). Similarly there was no additive effect when ZnPP was applied after the current through the channel had been suppressed by hypoxia (Supplemental Fig. 2).
Figure 7. Inhibition of the Ca2+ channel in rat cardiomyocytes by haem oxygenase inhibitors with respect to phosphorylation, permeating ion and hypoxia.

A and B, effect of 100 nm ZnPP on ICa (A) and IBa (B) measured in the absence of phosphorylating interventions (no cAMP, filled circles) and with dialysis of either 0.2 mm cAMP (cAMP, open circles) or 60 nm okadaic acid (OA, open triangles). C, pretreatment with an inhibitor of CaMKII (10 μm CK59) restored the suppressive effect (double arrow) of 100 nm ZnPP on ICa in cardiomyocytes dialysed with 0.2 mm cAMP. Notice that in this, as in all our experiments, the suppression of ICa was quantified based on a normalization that yielded 100% immediately before the exposure to hypoxia or ZnPP. D, hypoxia (N2) had no additional suppressive effects after IBa had been suppressed by 1 μm ZnPP. E, summary of the quantitative effects of 100 nm ZnPP on ICa and IBa, as measured in panels A–C. This bar graph has the same layout as used for hypoxic effects in Fig. 3C, but shows data obtained with a different protein kinase inhibitor (CK59 vs. H-89).
Discussion
Several studies have attempted to identify the mechanisms by which L-type Ca2+ channels are regulated by hypoxia including redox-modification of thiol containing residues and involvement of residues 1786–1821 of the C-terminus of the channel (Fearon et al. 1997; Hool, 2000; Yatani & Kamp, 2000). Here we propose that the CaM/CaMKII binding domain of the L-type Ca2+ channel is a critical site in rapid O2 sensing. Furthermore, our experiments suggest that HO may mediate the rapid sensing of O2 loss. Consistent with this idea, we found that the pharmacology of the fast suppressive effect of hypoxia on cardiac L-type Ca2+ channels to be similar to that of HO inhibitors (cf. Figs 3 and 7). Our finding of rapid suppression of ICa within the first 5 s of O2 withdrawal (Figs 1A and 2B), long before accumulation of metabolic products and O2 radicals, suggests a direct role for the Ca2+-channel protein as a likely first step in sensing of O2. In native channels and without PKA stimulation this suppressive effect of hypoxia was independent of permeating cations (Na+, Ba2+, Ca2+), but became specific to permeation of Ba2+ and Na+ in PKA-phosphorylated myocytes (Fig. 1C vs. 3C). Interestingly, the suppressive effect of withdrawal of O2 on the Ca2+ transporting channel was almost completely abolished by phosphorylating interventions, but surprisingly the requirement for permeating Ca2+ could not be substituted by release of Ca2+ from the SR (Fig. 3B). The dependence on permeating Ca2+ held under different conditions in native and recombinant Ca2+ channels, except when myocytes were exposed to the Ca2+ agonist drug Bay K8644 (Fig. 4B). This finding, though appearing peripheral to the major thrust of the proposed O2 sensing pathway, maybe relevant in the light of finding that Bay K8644 suppresses the Ca2+-dependent inactivation (CDI) property of the channel (Adachi-Akahane et al. 1999) (perhaps by interfering with the binding of Ca2+ to CaM). The specificity for Ca2+ also implicates a critical role for CaM in the Ca2+ channel O2 sensing. Consistent with this idea, the CaM-inhibitory peptide reversed the protective effects of phosphorylation in cAMP-dialysed myocytes, making the channel once again responsive to withdrawal of O2 (Fig. 4B). Further, possible involvement of CaMKII phosphorylation became apparent based on experiments showing that (1) an isoform of α1C channel, deleted from CaM/CaMKII binding motifs, was insensitive to O2 withdrawal (Fig. 5), and (2) inhibition of CaMKII phosphorylation by CK59 in cAMP dialysed myocytes restored the channel sensitivity to O2 (Fig. 3C). Even though these findings implicated the Ca2+-regulatory domains of Ca2+ channel in the hypoxic response, we could not identify a candidate AA sequence that would bind O2. The possible involvement of haem oxygenase became likely when we considered that this molecule had structural and functional similarities with CaMKII as to allow it to interact with the carboxyl-tail of the Ca2+ channel (Colbran, 1993; Boehning et al. 2004; Xu et al. 2010). Supporting this idea were the findings that (1) HO inhibitors (ZnPP and SnPP) also rapidly suppressed ICa (Fig. 6), (2) the blocking effect of HO inhibitors was abolished in cAMP-dialysed myocytes only if Ca2+, but not Ba2+ was the permeating cation (Fig. 7A, B and D), and (3) the suppressive effects of hypoxia and HO-2 inhibitors were not additive, i.e. either intervention blocked the suppressive effect of the other (Fig. 7C). A possible pathway for O2 sensing pathway, based on these findings, is illustrated in Fig. 8. Though HO-2 inhibitors have the same pharmacology and phosphorylation fingerprints as hypoxia, making HO-2 a likely candidate for O2 binding, it remains to be determined how this interaction leads to suppression of the Ca2+-channel current. Future experiments, possibly through mutagenesis, or genetic engineering, may provide more insight on the detailed molecular signalling of O2 sensing.
Figure 8. Proposed model for modulation of L-type Ca2+ channel by oxygen.
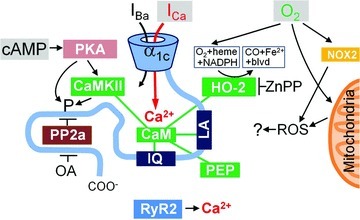
The carboxy-tail of the α1C subunit includes LA and IQ motifs for calmodulin- (CaM) mediated interactions and a binding site for protein phosphatase 2a (PP2a). CaM is shown as also interacting with CaMKII, haem oxygenase-2 (HO-2) and inhibitory peptide290−309 (PEP). The primary inputs to the system are thought to be: cAMP leading to PKA-mediated phosphorylation (left), permeation of Ca2+ through the channel to a restricted space (middle), and molecular oxygen (O2) that binds to haem/HO. PKA may act directly or via CaMKII and may have multiple phoshporylation targets (including HO-2). Similarly okadaic acid (OA) may promote phosphorylation inhibiting different phosphatases. ROS generation by mitochondria or membrane bound NOX proteins, CO-mediated signalling and ICa-triggered release of Ca2+ from the SR are thought to be of little or no importance for the rapid suppression of the Ca2+ channel at the onset of hypoxia.
L-type Ca2+ channels as primary oxygen sensor during acute cardiac hypoxia
In our studies we mostly compared baseline values of ICa in experimental cells to their values during the first minute of hypoxia. This approach raises the question as to what extent rundown of ICa may have contributed to its suppression during hypoxia. Based on control experiments without hypoxia (Fig. 1C), readmission of oxygen (Fig. 1A), and washout of HO inhibitors (Figs 6A and B and 7D) we estimate a rundown of ∼5% during the first 50–60 s of repetitive ICa measurements. Corrections of this magnitude are warranted when considering the hypoxic suppressions of ICa that were consistently 20–30% under non-phosphorylating conditions (Figs 1 and 2). The observed rundown may have masked the effectiveness of cAMP-induced desensitization of ICa (Figs 3 and 7), but it is not of sufficient magnitude to invalidate the experiments testing protein kinase inhibitors (H-89, C-59, Figs 3C and 7D), peptide290−309 (Fig. 4), or the α1c86 isoform of the channel (Fig. 5) and it is of little importance when considering the larger and faster suppressions of IBa and INa.
Our experimental approach was designed to simulate the response of cardiac ICa to acute hypoxia under different degrees of β-adrenergic stimulation and PKA-mediated phosphorylation (Herzig et al. 1993). The macromolecular signalling complex that provides effective regulation of ICa appears to be located at the C-terminus of α1C subunit and includes the adenylate cyclase/PKA signalling complex and protein phosphatases PP2a and PP2b (Xu et al. 2010). We propose that the haem oxygenase may be integrated into this system perhaps via the regulatory pathways that start with (1) cAMP, (2) permeation of Ca2+ through the channel, and (3) molecular oxygen (O2) all converging on calmodulin (Fig. 8).
Roles of CaM and CaMKII in the hypoxic regulation of the cardiac Ca2+channel
It is a novel idea that CaM and CaMKII play a critical role in the hypoxia-induced suppression of the Ca2+ channel (Movafagh & Morad, 2010). Our observations implicating these signalling molecules are that (1) O2-dependent suppression of ICa is absent in HEK cells expressing mutant α1c86 channel lacking the CaM/CaMKII binding domain (Fig. 5); (2) in cardiac myocytes the suppressive effect of hypoxia, absent in cAMP-phosphorylated cells, could be restored by dialysing peptide290−309 (Fig. 4); (3) given the reported interactions between HO-2 and CaM/CaMKII (Boehning et al. 2004), there is striking similarity in the suppressive effects of hypoxia (Fig. 3C) and HO inhibitors on ICa (ZnPP, Fig. 7E); and (4) CaMKII inhibitor, CK59, restored ZnPP-induced suppression of ICa in cardiac cells dialysed with cAMP (Fig. 7C). Considering the previously proposed signalling pathway for cAMP, Ca2+ channel, and CaM/CaMKII (as simplified in the schematic diagram of Fig. 8) and the findings reported here, we feel it is reasonable to include an O2-signalling pathway that interacts with CaM/CaMKII signalling as suggested in Fig. 8.
PKA phosphorylation and desensitization of O2 sensing
The finding that PKA phosphorylation significantly desensitizes the channel to acute hypoxia (Fig. 3) was somewhat unexpected. Hool et al. (2000, 2005, 2007) reported that hypoxia enhances β-adrenergic sensitivity of ICa via specific activation of a PKC isoform and suggested that O2 loss enhances PKA phosphorylation. Although we do not find enhancement of ICa of the phosphorylated channel on withdrawal of O2, the channel did become less sensitive to O2 withdrawal (Fig. 3A) and the O2 sensitivity of the channel could be restored by the PKA inhibitor H-89 in cAMP dialysed cells (Fig. 3C). It is unlikely that these findings are related to activation of PKC, and they do not address whether crosstalk between the PKA and PKC phosphorylation pathways (Yang et al. 2009) is of importance for the hypoxic modulation of ICa or if different phosphorylation patterns might explain the hypoxic enhancement of ICa found by Hool et al. Possibly the use of isoproterenol in combination with strong intracellular Ca2+ buffering (10 mm EGTA/1 mm Ca2) may have blunted CaM/CaMKII signalling in the experiments reported by these authors. Furthermore, in our experiments, the desensitization induced by PKA phosphorylation of the channel was only present when cAMP levels were elevated by either internal dialysis of cAMP or by phosphatase inhibitors, and not by basal cAMP levels (Fig. 1A).
The role of Ca2+-dependent signalling in the hypoxic response
The endogenous activity of CaMKII depends on its auto-phosphorylation at T286, which is in turn modulated by the phosphatase PP1. Stimulation of β-adrenergic receptors suppresses PP1 activity via PKA-dependent phosphorylation of the PP1 inhibitor I-1 at T35 (Bradshaw et al. 2003; Huke & Bers, 2007). Thus, PKA-induced auto-phosphorylation of CaMKII may be the pathway by which PKA controls the channel's sensitivity to hypoxia. Our data clearly show that the desensitization of the phosphorylated Ca2+ channel to hypoxia requires permeation of Ca2+, but not Ba2+ or Na+, through the channel, consistent with CaMKII activity requiring high local rather than global cytosolic Ca2+ concentrations (Zuhlke et al. 1999). The specific requirement for Ca2+ influx, but not SR Ca2+ release, in desensitizing the phosphorylated channel to O2 withdrawal was unexpected considering the report that SR Ca2+ release accelerates the decay kinetics of ICa (Adachi-Akahane et al. 1996), but at the same time emphasizes the specificity of interactions of Ca2+ in the nano-domains of the C-terminus as evident by re-sensitization of the phosphorylated channel to hypoxia by dialysis of CaMKII inhibitory peptide290−309 suggesting that CaMKII phosphorylation desensitizes the channel to O2 loss. Since the primary site of CaMKII-induced Ca2+-dependent facilitation is within the C-terminus (residues 1654–1659) and mutation of the IQ region abolishes such effects (Hudmon et al. 2005), we posit that the inhibitory peptide290−309 may prevent the binding of CaMKII to the IQ region (Payne et al. 1988) thus allowing the recovery of O2 sensitivity of the channel. Mutation of residues 1572–1651 (α1c86 channel isoform), which includes the entire CaM/CaMKII-binding motifs of the C-terminus (Fig. 4), completely desensitizes the channel to O2 withdrawal. A different region of the C-terminus has previously been implicated in the hypoxia-induced suppression of the cardiac Ca2+ channel (AA 1786–1821; Fearon et al. 2000) and is now known to bind a protein phosphatase (AA 1795–1818, PP2a; Xu et al. 2010) that antagonizes PKA-mediated phosphorylation at S1928 (Fig. 8, left; Davare et al. 2000). This is consistent with the notion that modulation of ICa by O2 and HO inhibitors is governed by the balance between kinases and phosphatases (Figs 3 and 7).
Curiously, hypoxia does not change the kinetics of the measured currents (Figs 1 and 3) despite the fact that the removal of the CaMKII/CaM binding (where IQ motif is placed) impairs responses elicited by hypoxia on the Cav1.2 currents. This may be related to the possible role of this site in transmitting O2 sensing to the pore region of the channel, but not serving as the O2 sensor itself.
Possible role of haem oxygenase as an O2 sensor of the Ca2+ channel
HO-2 has been suggested to serve as the O2 sensor for potassium channels in the carotid bodies (Williams et al. 2004; Lahiri et al. 2006). Significant levels of the constitutively active isoform of HO-2 are expressed in cardiomyocytes, and are enhanced by β-adrenergic agonists (Ding et al. 2011). It is further known that HO-2 complexes with CaM (Boehning et al. 2004), which led us to examine whether it may serve as an O2 sensor feeding into the CaM/CaMKII signalling pathway. In support of this idea we found that HO-inhibitors (1) rapidly suppressed ICa with kinetics similar to those of hypoxic suppression, and (2) blocked completely the suppressive effects of hypoxia on ICa (Figs 6 and 7), consistent with a unitary locus of action for hypoxia and HO inhibitors. We posit that in PKA-activated myocytes Ca2+ influx through the channel phosphorylates CaMKII allowing it to associate with CaM and possibly preventing HO/CaM binding, and the resultant suppression of ICa. In this scheme, dephosphorylated CaMKII would facilitate CaM association with HO and facilitate modulation of ICa by rapid changes in O2 concentrations. This may help to explain why the deletion of CaM/CaMKII binding motifs in the α1c86 mutant channel renders it insensitive to O2 removal. These considerations implicate CaMKII as a modulatory partner of the O2 sensing site, rather than the oxygen sensor itself. Indeed, if activated CaMKII were needed for hypoxia-induced effects on Cav1.2, the use of other cations to replace Ca2+ would not have resulted in an increased O2 sensing at all. In this scheme the sensing of O2 by HO is thought to be transmitted rapidly and directly to the Ca2+ channel via the CaM/CamKII system. In an alternative role, HO is thought to suppress the Ca2+ channel by generating CO thereby stimulating mitochondrial ROS production and redox modulation of the carboxy-tail of α1C (Scragg et al. 2008).
Do reactive oxygen species (ROS) mediate the rapid O2 sensing of the channel?
Since other groups have proposed that the modulation of ICa by O2 depends on a redox mechanism (Fearon et al. 1999; Hool, 2000; Hool & Corry, 2007) we examined this possibility by treating the cells with intra- or extracellular reduced glutathione (0.5–10 mm) to examine the role of ROS in the early onset of ICa suppression, but found no significant alterations in time course or magnitude of the O2 withdrawal response (Movafagh & Morad, unpublished data). This led us to conclude that reduced thiol residues were unlikely to be responsible for the fast suppression of ICa within the first 5 s of O2 withdrawal (Figs 1, 2 and 4B) and consequently we pursued experiments testing the role of haem oxygenase in rapid O2 sensing. In sharp contrast to rapid suppression of ICa, ROS generation by mitochondria proceeds too slowly (Hool et al. 2005; Korge et al. 2008) to be compatible with the kinetics of ICa suppression. Alternatively it may be argued that ICa could be modulated by ROS generated by an extra-mitochondrial O2-dependent pathway, e.g. involving a membrane-bound NADPH oxidase, like NOX-2, that generates superoxide that in turn triggers release of Ca2+ from the SR within a fraction of a second (Prosser et al. 2011). Inconsistent with such a possibility, we found no involvement of the SR Ca2+ release in the rapid hypoxic response (Fig. 3B). Further, NOX-2, though known to bind haem, is not modulated by haem analogues like ZnPP. Yet another redox mechanism to be considered involves CO, which is reported, to modulate the BK (Yi et al. 2010) and Ca2+ channels (Scragg et al. 2008). We did not examine the effects of CO, in part because it is a likely modulator of any haem-binding protein and therefore non-specific with respect to the identity the oxygen sensor of the Ca2+ channel. Thus, our data and the existing literature point to haem oxygenase as the likely oxygen sensor of the Ca2+ channel, but they do not exclude the possibility that other O2-sensitive proteins and processes are also involved in the cumulative effects of hypoxia.
Pathophysiological implications
Our data clearly show that Ca2+ channels of the heart respond to the decrease in O2 tension well within 50 s, suggesting a rapid detection mechanism for hypoxia. In general this early mechanism is embedded within the sequence of ischaemic events that occur over periods of minutes to hours. Under these conditions, early and transient suppression of ICa may work as a protective mechanism against subsequent Ca2+-mediated toxicity. In this regard inhibition of Ca2+ channels by nifedipine may reduce the degree of phospholamban phosphorylation by CaMKII within the first 5 min of ischaemia (Vittone et al. 2002). In our simultaneous measurements of ICa and Ca2+ transients in non-phosphorylated cells, we found an ∼10% decrease in Cai-transients after 40 s of hypoxia compared to an ∼20% decrease in ICa. The small hypoxia-induced decrease in released Ca2+ is consistent with the modest rapid suppression of ICa and its role in regulating Ca2+-induced Ca2+ release. Considering the high degree of cooperativity in activation of contraction by binding of Ca2+ to troponin and in cross bridge recycling, it is plausible that associated changes in energy demands may be considerably larger (Fabiato & Fabiato, 1975; Han et al. 2009; ter Keurs, 2012).
The pharmacological approach of using (S)-(–)-Bay K8644 in combination with the CaMKII inhibitor peptide290−309, where we found significant reduction of ICa even in phosphorylated channels, provides an intriguing conceptual possibility for using a similar clinical strategy, but such an approach is far from compelling given the arrhythmogenicity associated with Ca2+ channel agonists. Nevertheless, a mixed DHP agonist/antagonists therapeutic approach in combination with a CaMKII inhibitor could be a potential therapeutic avenue to consider.
Acknowledgments
We thank and Santosh K. Mani and Dr Donald R. Menick for providing the feline cardiomyocytes. Dr. Nikolai Soldatov generously provided all cDNAs. John Scaringi performed the experiments illustrated in Supplemental Figure 1. This work was supported by NIH grants HL-16152, 5 F32 HL088855 and 1R01 HL107600.
Glossary
- HO
haem oxygenase
- PEP
inhibitory peptide290−309
- PKA
protein kinase A
- ROS
reactive oxygen species
- SnPP
tin protoporphyrin
- YFP
yellow fluorescent protein
- ZnPP
zinc protoporphyrin
Author contributions
A.O.R and S.M. did almost all of the experimental work. All authors took part in design, analysis and preparation of the manuscript and have approved the manuscript. The work was done both at Georgetown University and the Medical University of South Carolina.
Disclosures
None.
Supplementary material
Supplemental Figure 1
Supplemental Figure 2
References
- Adachi-Akahane S, Cleemann L, Morad M. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. J Gen Physiol. 1996;108:435–454. doi: 10.1085/jgp.108.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi-Akahane S, Cleemann L, Morad M. BAY K 8644 modifies Ca2+ cross signaling between DHP and ryanodine receptors in rat ventricular myocytes. Am J Physiol Heart Circ Physiol. 1999;276:H1178–1189. doi: 10.1152/ajpheart.1999.276.4.H1178. [DOI] [PubMed] [Google Scholar]
- Belmonte S, Morad M. Shear fluid-induced Ca2+ release and the role of mitochondria in rat cardiac myocytes. Ann NY Acad Sci. 2008;1123:58–63. doi: 10.1196/annals.1420.007. [DOI] [PubMed] [Google Scholar]
- Boehning D, Sedaghat L, Sedlak TW, Snyder SH. Heme oxygenase-2 is activated by calcium-calmodulin. J Biol Chem. 2004;279:30927–30930. doi: 10.1074/jbc.C400222200. [DOI] [PubMed] [Google Scholar]
- Bradshaw JM, Kubota Y, Meyer T, Schulman H. An ultrasensitive Ca2+/calmodulin-dependent protein kinase II-protein phosphatase 1 switch facilitates specificity in postsynaptic calcium signaling. Proc Natl Acad Sci U S A. 2003;100:10512–10517. doi: 10.1073/pnas.1932759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleemann L, Morad M. Role of Ca2+ channel in cardiac excitation-contraction coupling in the rat: evidence from Ca2+ transients and contraction. J Physiol. 1991;432:283–312. doi: 10.1113/jphysiol.1991.sp018385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbran RJ. Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J Biol Chem. 1993;268:7163–7170. [PubMed] [Google Scholar]
- D’Agostino D, Mazza E, Jr, Neubauer JA. Heme oxygenase is necessary for the excitatory response of cultured neonatal rat rostral ventrolateral medulla neurons to hypoxia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R102–118. doi: 10.1152/ajpregu.90325.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Horne MC, Hell JW. Protein phosphatase 2A is associated with class C L-type calcium channels (Cav1.2) and antagonizes channel phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2000;275:39710–39717. doi: 10.1074/jbc.M005462200. [DOI] [PubMed] [Google Scholar]
- Ding B, Gibbs PE, Brookes PS, Maines MD. The coordinated increased expression of biliverdin reductase and heme oxygenase-2 promotes cardiomyocyte survival: a reductase-based peptide counters beta-adrenergic receptor ligand-mediated cardiac dysfunction. FASEB J. 2011;25:301–313. doi: 10.1096/fj.10-166454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson MG, Alseikhan BA, Peterson BZ, Yue DT. Preassociation of calmodulin with voltage-gated Ca2+ channels revealed by FRET in single living cells. Neuron. 2001;31:973–985. doi: 10.1016/s0896-6273(01)00438-x. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX, Yue DT. FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron. 2003;39:97–107. doi: 10.1016/s0896-6273(03)00395-7. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon IM, Palmer AC, Balmforth AJ, Ball SG, Mikala G, Schwartz A, Peers C. Hypoxia inhibits the recombinant α1C subunit of the human cardiac L-type Ca2+ channel. J Physiol. 1997;500:551–556. doi: 10.1113/jphysiol.1997.sp022041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon IM, Palmer AC, Balmforth AJ, Ball SG, Varadi G, Peers C. Modulation of recombinant human cardiac L-type Ca2+ channel α1C subunits by redox agents and hypoxia. J Physiol. 1999;514:629–637. doi: 10.1111/j.1469-7793.1999.629ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fearon IM, Varadi G, Koch S, Isaacsohn I, Ball SG, Peers C. Splice variants reveal the region involved in oxygen sensing by recombinant human L-type Ca2+ channels. Circ Res. 2000;87:537–539. doi: 10.1161/01.res.87.7.537. [DOI] [PubMed] [Google Scholar]
- Franco-Obregon A, Urena J, Lopez-Barneo J. Oxygen-sensitive calcium channels in vascular smooth muscle and their possible role in hypoxic arterial relaxation. Proc Natl Acad Sci U S A. 1995;92:4715–4719. doi: 10.1073/pnas.92.10.4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JC, Taberner AJ, Kirton RS, Nielsen PM, Smith NP, Loiselle DS. A unique micromechanocalorimeter for simultaneous measurement of heat rate and force production of cardiac trabeculae carneae. J Appl Physiol. 2009;107:946–951. doi: 10.1152/japplphysiol.00549.2009. [DOI] [PubMed] [Google Scholar]
- Herzig S, Patil P, Neumann J, Staschen CM, Yue DT. Mechanisms of β-adrenergic stimulation of cardiac Ca2+ channels revealed by discrete-time Markov analysis of slow gating. Biophys J. 1993;65:1599–1612. doi: 10.1016/S0006-3495(93)81199-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hool LC. Hypoxia increases the sensitivity of the L-type Ca2+ current to β-adrenergic receptor stimulation via a C2 region-containing protein kinase C isoform. Circ Res. 2000;87:1164–1171. doi: 10.1161/01.res.87.12.1164. [DOI] [PubMed] [Google Scholar]
- Hool LC, Corry B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxid Redox Signal. 2007;9:409–435. doi: 10.1089/ars.2006.1446. [DOI] [PubMed] [Google Scholar]
- Hool LC, Di Maria CA, Viola HM, Arthur PG. Role of NAD(P)H oxidase in the regulation of cardiac L-type Ca2+ channel function during acute hypoxia. Cardiovasc Res. 2005;67:624–635. doi: 10.1016/j.cardiores.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Hudmon A, Schulman H, Kim J, Maltez JM, Tsien RW, Pitt GS. CaMKII tethers to L-type Ca2+ channels, establishing a local and dedicated integrator of Ca2+ signals for facilitation. J Cell Biol. 2005;171:537–547. doi: 10.1083/jcb.200505155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huke S, Bers DM. Temporal dissociation of frequency-dependent acceleration of relaxation and protein phosphorylation by CaMKII. J Mol Cell Cardiol. 2007;42:590–599. doi: 10.1016/j.yjmcc.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knollmann BC, Knollmann-Ritschel BE, Weissman NJ, Jones LR, Morad M. Remodelling of ionic currents in hypertrophied and failing hearts of transgenic mice overexpressing calsequestrin. J Physiol. 2000;525:483–498. doi: 10.1111/j.1469-7793.2000.t01-1-00483.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korge P, Ping P, Weiss JN. Reactive oxygen species production in energized cardiac mitochondria during hypoxia/reoxygenation: modulation by nitric oxide. Circ Res. 2008;103:873–880. doi: 10.1161/CIRCRESAHA.108.180869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S, Roy A, Baby SM, Hoshi T, Semenza GL, Prabhakar NR. Oxygen sensing in the body. Prog Biophys Mol Biol. 2006;91:249–286. doi: 10.1016/j.pbiomolbio.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Mann DL, Urabe Y, Kent RL, Vinciguerra S, Cooper GT. Cellular versus myocardial basis for the contractile dysfunction of hypertrophied myocardium. Circ Res. 1991;68:402–415. doi: 10.1161/01.res.68.2.402. [DOI] [PubMed] [Google Scholar]
- Montoro RJ, Urena J, Fernandez-Chacon R, Alvarez de Toledo G, Lopez-Barneo J. Oxygen sensing by ion channels and chemotransduction in single glomus cells. J Gen Physiol. 1996;107:133–143. doi: 10.1085/jgp.107.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movafagh S, Morad M. L-type calcium channel as a cardiac oxygen sensor. vann N Y Acad Sci. 2010;1188:153–158. doi: 10.1111/j.1749-6632.2009.05095.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabauer M, Callewaert G, Cleemann L, Morad M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 1989;244:800–803. doi: 10.1126/science.2543067. [DOI] [PubMed] [Google Scholar]
- Ortega-Saenz P, Pascual A, Piruat JI, Lopez-Barneo J. Mechanisms of acute oxygen sensing by the carotid body: lessons from genetically modified animals. Respir Physiol Neurobiol. 2007;157:140–147. doi: 10.1016/j.resp.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Payne ME, Fong YL, Ono T, Colbran RJ, Kemp BE, Soderling TR, Means AR. Calcium/calmodulin-dependent protein kinase II. Characterization of distinct calmodulin binding and inhibitory domains. J Biol Chemy. 1988;263:7190–7195. [PubMed] [Google Scholar]
- Piper HM, Abdallah Y, Schafer C. The first minutes of reperfusion: a window of opportunity for cardioprotection. Cardiovasc Res. 2004;61:365–371. doi: 10.1016/j.cardiores.2003.12.012. [DOI] [PubMed] [Google Scholar]
- Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- Reuter H, Scholz H. The regulation of the calcium conductance of cardiac muscle by adrenaline. J Physiol. 1977;264:49–62. doi: 10.1113/jphysiol.1977.sp011657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- Scragg JL, Dallas ML, Wilkinson JA, Varadi G, Peers C. Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J Biol Chem. 2008;283:24412–24419. doi: 10.1074/jbc.M803037200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soldatov NM, Oz M, O’Brien KA, Abernethy DR, Morad M. Molecular determinants of L-type Ca2+ channel inactivation. Segment exchange analysis of the carboxyl-terminal cytoplasmic motif encoded by exons 40–42 of the human α1C subunit gene. J Biol Chem. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- Soldatov NM, Zuhlke RD, Bouron A, Reuter H. Molecular structures involved in L-type calcium channel inactivation. Role of the carboxyl-terminal region encoded by exons 40–42 in α1C subunit in the kinetics and Ca2+ dependence of inactivation. J Biol Chem. 1997;272:3560–3566. doi: 10.1074/jbc.272.6.3560. [DOI] [PubMed] [Google Scholar]
- Song Q, Saucerman JJ, Bossuyt J, Bers DM. Differential integration of Ca2+-calmodulin signal in intact ventricular myocytes at low and high affinity Ca2+-calmodulin targets. J Biol Chem. 2008;283:31531–31540. doi: 10.1074/jbc.M804902200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Keurs HE. The interaction of Ca2+ with sarcomeric proteins: role in function and dysfunction of the heart. Am J Physiol Heart Circ Physiol. 2012;302:H38–50. doi: 10.1152/ajpheart.00219.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiman M, Caspersen C, Christensen SB. A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca2+-ATPases. Trends Pharmacol Sci. 1998;19:131–135. doi: 10.1016/s0165-6147(98)01184-5. [DOI] [PubMed] [Google Scholar]
- Vittone L, Mundina-Weilenmann C, Said M, Ferrero P, Mattiazzi A. Time course and mechanisms of phosphorylation of phospholamban residues in ischemia-reperfused rat hearts. Dissociation of phospholamban phosphorylation pathways. J Mol Cell Cardiol. 2002;34:39–50. doi: 10.1006/jmcc.2001.1488. [DOI] [PubMed] [Google Scholar]
- Williams SE, Wootton P, Mason HS, Bould J, Iles DE, Riccardi D, Peers C, Kemp PJ. Hemoxygenase-2 is an oxygen sensor for a calcium-sensitive potassium channel. Science. 2004;306:2093–2097. doi: 10.1126/science.1105010. [DOI] [PubMed] [Google Scholar]
- Xu H, Ginsburg KS, Hall DD, Zimmermann M, Stein IS, Zhang M, Tandan S, Hill JA, Horne MC, Bers D, Hell JW. Targeting of protein phosphatases PP2A and PP2B to the C-terminus of the L-type calcium channel Cav1.2. Biochemistry. 2010;49:10298–10307. doi: 10.1021/bi101018c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka K, Kameyama M. Regulation of L-type Ca2+ channels in the heart: overview of recent advances. Mol Cell Biochem. 2003;253:3–13. doi: 10.1023/a:1026036931170. [DOI] [PubMed] [Google Scholar]
- Yang L, Doshi D, Morrow J, Katchman A, Chen X, Marx SO. Protein kinase C isoforms differentially phosphorylate Cav1.2 α1c. Biochemistry. 2009;48:6674–6683. doi: 10.1021/bi900322a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatani A, Kamp TJ. Tails of the L-type Ca2+ channel: to sense oxygen or not. Circ Res. 2000;87:535–536. doi: 10.1161/01.res.87.7.535. [DOI] [PubMed] [Google Scholar]
- Yi L, Morgan JT, Ragsdale SW. Identification of a thiol/disulfide redox switch in the human BK channel that controls its affinity for heme and CO. J Biol Chem. 2010;285:20117–20127. doi: 10.1074/jbc.M110.116483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]
- Zuhlke RD, Reuter H. Ca2+-sensitive inactivation of L-type Ca2+ channels depends on multiple cytoplasmic amino acid sequences of the α1C subunit. Proc Natl Acad Sci U S A. 1998;95:3287–3294. doi: 10.1073/pnas.95.6.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.