

Abstract
The study's purpose was to investigate if physical activity initiated with the start of high-fat feeding would oppose development of endothelial dysfunction, and if it does, then to determine some potential mechanisms. C57BL/6 female mice were randomly divided into three groups: (1) control low-fat diet (LF-SED; 15% of calories from fat), (2) high-fat diet (HF-SED; 45% of calories from fat), and (3) HF diet given access to a voluntary running wheel (HF-RUN). Our hypothesis was that HF-RUN would differ in multiple markers of endothelial dysfunction from HF-SED after 10 weeks of 45%-fat diet, but would not differ from LF-SED. HF-RUN differed from HF-SED in nine determinations in which HF-SED either had decreases in (1) acetylcholine (ACh)-induced and flow-induced vasodilatations in isolated, pressurized coronary arterioles, (2) heart phosphorylated endothelial nitric oxide synthase (p-eNOS/eNOS) protein, (3) coronary arteriole leptin (ob) receptor protein, (4) phosphorylated signal transducer and activator of transcription 3 (p-STAT3/STAT3) protein, and (5) coronary arteriole superoxide dismutase 1 protein; or had increases in (6) percentage body fat, (7) serum leptin, (8) coronary arteriole suppressor of cytokine signalling 3 (SOCS3) protein, and (9) coronary arteriole gp91phox protein. Higher endothelium-dependent vasodilatation by ACh or leptin was abolished with incubation of NOS inhibitor NG-nitro-l-arginine-methyl ester (l-NAME) in LF-SED and HF-RUN groups. Further, impaired ACh-induced vasodilatation in HF-SED was normalized by apocynin or TEMPOL to LF-SED and HF-RUN. These findings demonstrate multiple mechanisms (eNOS, leptin and redox balance) by which voluntary running opposes the development of impaired coronary arteriolar vasodilatation during simultaneous high-fat feeding.
Key points
Sedentary and high-fat diet lifestyles are associated with greater prevalence of obesity and type 2 diabetes in humans, both of which independently increase atherosclerosis.
High-fat diet in sedentary individuals produces endothelial dysfunction in blood vessels as a first step toward coronary arteriosclerosis.
We observed preservation of coronary arteriolar vascular function when mice began voluntary running in wheels at the start of a high-fat diet.
We further showed that mechanisms by which running opposed the detrimental effects of high-fat diet on vascular function included maintenance of eNOS phosphorylation, leptin sensitivity, and redox balance in mouse coronary arterioles.
The results provide evidence for how physical activity is an effective therapy to oppose the development of atherosclerosis in the first place.
Introduction
Atherosclerotic cardiovascular disease (CVD) is the number one cause of death in the United States and other developed nations (Lloyd-Jones et al. 2009). Ringseis & Eder (2010) contend that there is ‘a clear need for developing strategies to prevent atherosclerosis’. Obesity is an independent risk factor for CVD and stroke (Ezzati et al. 2007; Daniels et al. 2011). Physical inactivity itself causes endothelial dysfunction, CVD and stroke (Suvorava et al. 2004; Laufs et al. 2005; Hamburg et al. 2007). A 2011 American Heart Association Policy Statement utilizes what others have characterized as the prevention of the development of risk factors in the first place, termed ‘primordial prevention’ (Strasser, 1978; Weintraub et al. 2011). Importantly, in our opinion, approaches using primordial prevention provide insights into primary causes of CVD because of its processes for identification of variables that prevent a risk factor ‘in the first place’. The contention by some that ‘endothelial dysfunction is the first step toward coronary arteriosclerosis’ (Vanhoutte, 2009) places endothelial dysfunction as a primordial risk factor for atherosclerosis (Schwartz, 1980; Félétou & Vanhoutte, 2006; Vanhoutte, 2009).
Obesity-induced vascular dysfunction is tightly associated with (1) reduced bioavailability of vasodilators including nitric oxide (NO), endothelium-dependent hyperpolarizing factor (EDHF) and prostacyclin, and (2) enhanced action of vasoconstrictors such as endothelin-1 (ET-1), angiotensin II (Ang II), etc. in vascular endothelium (Fornoni & Raij, 2005; Kim et al. 2006; Jonk et al. 2007; Stapleton et al. 2008). In addition, reactive oxygen species (ROS) contribute to exacerbate endothelial dysfunction by reducing NO, which relaxes blood vessels, prevents platelet aggregation and adhesion, limits oxidation of low-density lipoprotein (LDL) cholesterol, inhibits proliferation of vascular smooth muscle cells, and decreases the expression of pro-inflammatory genes (Bjørbæk et al. 1998; Fenster et al. 2002; Gao et al. 2007; Park et al. 2010).
Leptin is a 16 kDa circulating hormone released primarily by adipocytes and exerts regulatory control on food intake and energy expenditure. Consequently, circulating leptin levels are directly proportional to adipose tissue and are markedly higher in obesity (Myers et al. 2008). Leptin is also directly related to vascular reactivities. Leptin receptors (Ob-R) have been shown to be expressed in endothelial cells from human, dogs and rodent rat models (Knudson et al. 2005), and activation of the leptin receptor induces NO-dependent vasodilatation (Kimura et al. 2000; Lembo et al. 2000; Winters et al. 2000; Vecchione et al. 2002). Decreased leptin sensitivity is one of the characteristics of HF-induced obesity (Lin et al. 2000; Levin & Dunn-Meynell, 2002) and hyperleptinaemia is associated with endothelial dysfunction through impaired NO production and an increase in oxidative stress (Beltowski et al. 2004; Beltowski, 2006; Korda et al. 2008). Physical activity normalizes elevated leptin level in obesity (Carhuatanta et al. 2011). However, no direct study exists to our knowledge testing whether leptin sensitivity for endothelial function is maintained normal when physical activity is initiated with HF diet.
The question that we posed is by what mechanisms voluntary running rescues endothelial dysfunction during the treatment of high-fat diet. Direct mechanisms as to how physical activity prevents or ameliorates obesity-induced endothelial dysfunction (Woo et al. 2004; Fornoni & Raij, 2005; Gill & Malkova, 2006; Hamburg et al. 2007) in primordial prevention have insufficiently been elucidated. Thus, we hypothesize that increased voluntary running opposes endothelial dysfunction in murine coronary microcirculation during simultaneous treatment of high-fat diet. We explored potential causal mechanisms by which voluntary running preserves endothelial function in coronary arterioles of mice fed a high-fat diet. To answer these questions, we evaluated: (1) whether free access into voluntary wheel running opposed HF diet-induced obesity and its associated endothelial dysfunction of murine coronary arterioles, (2) the mechanisms of impaired endothelial function in mice with HF diet and preserved endothelial function in mice that had combined treatment of HF diet and voluntary wheel running, and (3) whether physical activity opposed hyperleptinaemia and maintained leptin coronary arteriolar sensitivity in the HF-diet-induced obese mouse model.
Methods
Animal models
The procedures followed were in accordance with approved guidelines set by the Laboratory Animal Care Committee at University of Missouri. Female wild-type mice (C57BL/6J) were purchased from Harlan Laboratories (Indianapolis, IN, USA) at 10 weeks of age and were maintained on a low-fat diet (LF, 17% of calories from fat) or high-fat diet (HF, 45% of calories from fat) for 10 weeks. Our experimental design of primordial prevention (prevention of obesity and physical inactivity in the first place to prevent endothelial dysfunction in the first place) in mice has, to us, simple logic. First, adopt conventional housing without a running wheel with 17% fat diet as the reference group. Then add fat to make the diet 45% in a second group in a cage without a running wheel, which would mimic the majority of humans who consume a high-fat diet while not being physically inactive. The 45% diet was TestDiet 58G8 (Richmond, IN, USA) with energy sources of 18.6% protein, 45.0% fat (total diet contained 17.7% lard and 5.7% corn oil), and 36.4% carbohydrate (total diet contained 30.1% dextrin and 10.3% sucrose). Finally for the third group, initiate a 45% fat diet when activity is increased by addition of a wheel for voluntary running to test for endothelial mechanisms as to how physical inactivity has a causal role in the primordial prevention of endothelial dysfunction. HF diet mice were randomly assigned to the voluntary wheel running group (HF-RUN; those with free access to running wheels) and the sedentary group (HF-SED; those without access to the wheels). LF mice did not have access to running wheels (LF-SED). Mice assigned to running groups were immediately housed (at the age of 10 weeks) in cages equipped with a voluntary running wheel outfitted with a Sigma Sport BC 800 bicycle computer (Cherry Creek Cyclery, Foster Falls, VA, USA) for measuring daily running distance. Voluntary running was selected as approximating the more natural physical activity level of the animal. At the end of the 10 week treatment, citrate synthase activity was determined in red gastrocnemius muscle (Srere, 1969).
Dual-energy X-ray absorptiometry (DEXA)
Whole-body composition was measured using a Hologic QDR-1000/w DEXA machine calibrated for mice.
Serum assays
Blood was drawn from vena cava of mice and serum was collected after the blood was centrifuged. Serum insulin (ALPCO, Insulin (Mouse) Ultrasensitive EIA) and leptin (ALPCO, Leptin (Mouse/Rat) EIA and ALPCO) were measured using commercially available ELISA kits according to the manufacturer's instructions (ALPCO, Salem, NH, USA).
Functional assessment of isolated coronary arterioles
The techniques for identification and isolation of coronary arterioles were previously described in detail (Park et al. 2008, 2010). Briefly, hearts from three groups of animals, LF-SED, HF-SED and HF-RUN mice, were carefully dissected free and placed in cold (4°C) physiological saline solution (PSS) that contained 145.0 mm NaCl, 4.7 mm KCl, 2.0 mm CaCl2, 1.17 mm MgSO4, 1.2 mm NaH2PO4, 5.0 mm glucose, 2.0 mm pyruvate, 0.02 mm EDTA, 3.0 mm Mops buffer, and 1 g per 100 ml BSA at pH 7.4. With a dissecting microscope (Olympus SZX12), each coronary arteriole (50–100 μm in internal diameter) was carefully isolated and was transferred to a Lucite chamber that contained PSS at room temperature. Each end of the arteriole was cannulated with a glass micropipette and secured with nylon suture. After cannulation of the arterioles, the microvessel chamber was transferred to the stage of an inverted microscope (Olympus IX51) equipped with a video camera (Hitachi KP-M2AN) and video caliper (Colorado Video, Boulder, CO, USA) and data acquisition system for recording of intraluminal diameter. The arterioles were initially pressurized to 60 cmH2O with two independent hydrostatic pressure reservoirs. Leaks were detected by pressurizing the vessel and then the vessel that is not fully pressurized due to leaking must be discarded. After developing a basal tone, the experimental interventions were performed.
The concentration–diameter relationships for an activator of endothelium-dependent vasodilatation, ACh (1 nmol l−1 to 10 μmol l−1), flow-induced dilatation (endothelium-dependent but agonist-independent; pressure difference (ΔP) was 4–60 mmH2O), the endothelium-independent NO donor sodium nitroprusside (SNP; 1 nmol l−1 to 10 μmol l−1) and leptin (1 nmol l−1 to 10 μmol l−1)-mediated dilatation was then established. ACh and leptin-induced vasodilatation was also performed in the presence of the NOS inhibitor NG-nitro-l-arginine-methyl ester (l-NAME; 10 μmol l−1, 20 min) to determine whether a NO-mediated mechanism is involved in the differences of endothelium-dependent vasodilatation among LF-SED, HF-SED and HF-RUN mice. To determine the role of the superoxide radical (O2−) and NAD(P)H oxidase, which is major source of O2− in the vasculature, in altered vasoactive response in coronary arterioles, the ACh-mediated vasodilatory function was examined in the presence of the O2− scavenger TEMPOL (a membrane-permeant O2− dismutase mimetic; 1 mmol l−1, 60 min incubation), and NAD(P)H oxidase inhibitor apocynin (10 μmol l−1, 30 min), respectively. At the end of each experiment, the vessel was exposed to 100 μmol l−1 SNP to obtain its maximal diameter at 60 cmH2O intraluminal pressure (Park et al. 2008, 2010).
Protein expression measurement by Western blot analyses
Hearts or coronary arterioles were separately homogenized and sonicated in lysis buffer (Cellytic MT Mammalian Tissue Lysis/Extraction Reagent, Sigma). Protein concentrations were assessed (BCA Protein Assay Kit, Pierce), and equal amounts of protein (10 or 20 μg) were separated by SDS-PAGE and transferred to nitrocellulose membranes (Hybond, Amersham). Primary antibodies for phospho-(Ser 1177)-eNOS (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA, 1:100), eNOS (Santa Cruz, 1:200), leptin receptor (Ob-R, Santa Cruz, 1:500), phospho-(Tyr 705)-STAT3 (Santa Cruz 1:100), STAT3 (Abcam Inc., Cambridge, MA, USA, 1:200), SOCS3 (Abcam, 1:200), SOD1 (EMD Chemical, 1:200), SOD2 (EMD Chemical, Philadelphia, PA, USA, 1:1000), p22phox (Santa Cruz, 1:500), p47phox (Santa Cruz, 1:500), p67phox (Santa Cruz, 1:500), gp91phox (BD Biosciences, San Jose, CA, USA, 1:500) and GAPDH (IMGENEX, San Diego CA, USA, 1:1000) and corresponding secondary antibodies (1:1000∼1:2000) to each primary antibodies were used. Signals were visualized by enhanced chemiluminescence (Santa Cruz), scanned with a Fuji LAS3000 densitometer, and quantified by Multigauge software (Fuji film). The relative amounts of protein expression were quantified and normalized to those of the corresponding internal reference glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and then normalized to the corresponding LF-SED, which was set to a value of 1.0. Due to limited tissue, only enough protein was isolated to run one lane per sample. This was repeated across multiple blots. Consequently among blots, a common protein was not present, thus preventing determination of SEMs of the normalizing groups.
Data analysis
All diameter changes to pharmacological agonists were normalized to the control diameter. All data are presented as means ± SEM. Statistical comparisons of vasomotor responses between groups were performed with two-way analysis of variance (ANOVA) for repeated measure and intergroup differences were tested with the Bonferonni inequality. The significance of intergroup differences observed in molecular studies was evaluated by one-way ANOVA using software SPSS v11.5. Significance was accepted at P < 0.05.
Results
Animal characteristics
Table 1 shows that body weight, percentage body fat, fat mass and lean mass of the mice were significantly higher in HF-SED than that in LF-SED mice and voluntary wheel running in HF mice (HF-RUN) was effective in prevention of the increase in body weight, percentage body fat, fat mass and lean mass. Non-fasting insulin and blood glucose levels tended to be higher in HF-SED mice compared with LF-SED and HF-RUN mice but they were not statistically significant. Average daily running distance of mice in HF-RUN was 17.52 km and their citrate synthase activity from red gastrocnemius was significantly higher than in LF-SED or HF-SED groups, showing the efficacy of voluntary wheel running (Table 1).
Table 1.
Characteristics of animals
| LF-SED | HF-SED | HF-RUN | |
|---|---|---|---|
| n | 11 | 11 | 10 |
| Body weight (g) | 23.84 ± 0.97 | 30.44 ± 1.72* | 24.55 ± 0.61# |
| % Body fat | 15.55 ± 2.50 | 41.75 ± 3.87* | 17.88 ± 1.11# |
| Fat mass (g) | 3.07 ± 0.57 | 13.58 ± 1.91* | 4.35 ± 0.34# |
| Lean mass (g) | 19.97 ± 0.69 | 16.75 ± 0.40* | 19.80 ± 0.43# |
| Insulin (ng ml−1) | 0.71 ± 0.18 | 1.40 ± 0.84 | 0.84 ± 0.17 |
| Blood glucose (mg dl−1) | 147.33 ± 13.05 | 171.33 ± 10.65 | 163.60 ± 9.23 |
| Red gastrocnemius | |||
| Citrate synthase activity (nmol μg−1 min−1) | 494.62 ± 39.88 | 547.38 ± 63.40 | 699.36 ± 47.84*# |
| Running distance (km daily) | 17.52 ± 0.85 |
Body weight, % body fat and fat mass were higher in HF-SED mice compared with LF-SED mice but they were significantly lower in HF-RUN mice than in HF-SED mice and comparable to LF-SED mice. Insulin and blood glucose level were not significantly different among groups. Citrate synthase activity was enhanced in red gastrocnemius of HF-RUN compared with LF-SED or HF-SED. Data are expressed as means ± SEM. *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED.
Physical activity opposed obesity-induced coronary arteriolar endothelial dysfunction
ACh- or flow-induced endothelium-dependent vasodilatation (Fig. 1A and B) was attenuated in coronary arterioles in HF-SED mice compared with LF-SED. Voluntary wheel running completely opposed impaired ACh-induced vasodilatation of arterioles from HF obese mice, HF-SED, (Fig. 1A and B). However, dilatations in response to the NO donor SNP in LF-SED, HF-SED and HF-RUN were not statistically different (Fig. 1C).
Figure 1. Isolated coronary arterioles were dilated in response to ACh in a concentration-dependent manner.
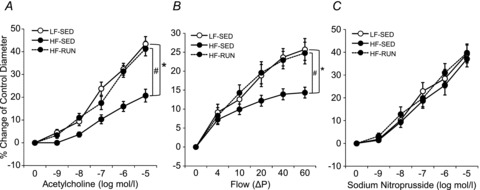
A, ACh-induced vasodilatation was significantly attenuated in HF-SED (n = 11) mice compared with LF-SED mice (n = 7). However, voluntary wheel running prohibited the development of impaired ACh-induced vasodilatation in coronary arterioles during high-fat feeding in HF-RUN (n = 8). B, flow-induced vasodilatation was lower in HF-SED mice compared with LF-SED mice, but it was opposed by voluntary wheel running in high-fat fed mice (HF-RUN). C, the endothelium-independent vasodilator, SNP (NO donor)-induced vasodilatation was not significantly different among the 3 mice groups. Data are expressed as means ± SEM. *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED.
In order to test whether NO is responsible for the difference of endothelium-dependent dilatation in Fig. 1A, we measured ACh-induced dilatation in the presence of the NOS inhibitor, l-NAME. l-NAME significantly attenuated vasodilatation to ACh in LF-SED and HF-RUN (Fig. 2A) to the level of the obese HF-SED with and without l-NAME. The dilatations of HF-SED vessels incubated with l-NAME were not altered (P > 0.05) in HF-SED (Fig. 2A). In addition, eNOS protein expression was measured to confirm whether alteration of eNOS protein expression was associated with a change in endothelium-dependent dilatation, and we found no change in eNOS protein expression with the HF diet or physical activity. However, eNOS phosphorylation at Ser1177 was significantly reduced in HF-SED and physical activity in HF-RUN opposed this decrease in eNOS phosphorylation (Fig. 2B).
Figure 2. Role of NO in endothelial function altered by high fat diet and voluntary wheel running.
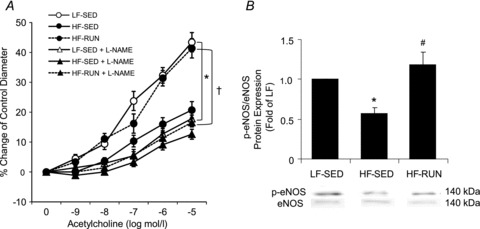
A, ACh-induced vasodilatation was significantly blunted by l-NAME so that the difference of ACh-induced vasodilatory function was abolished among the groups. B, p-eNOS/eNOS protein in mice hearts was significantly decreased in HF-SED mice compared with LF-SED mice. Voluntary running during high-fat diet opposed the lowering of p-eNOS/eNOS in mice hearts. Data are expressed as means ± SEM. *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED, †P < 0.05 vs. HF-RUN.
Role of leptin in obesity-induced vascular dysfunction
Absolute serum leptin level (Fig. 3A) and relative serum leptin level normalized to total body fat mass (Fig. 3B) were markedly higher in HF-SED than them in LF-SED. Voluntary wheel running in mice with HF diet opposed the rise of both of serum level and serum/fat mass. Western blot analysis of Ob-R protein expression in arterioles from LF-SED, HF-SED and HF-RUN mice (Fig. 3C) revealed a significantly lower level of Ob-R protein in HF-SED mice compared with LF-SED and HF-RUN mice.
Figure 3.
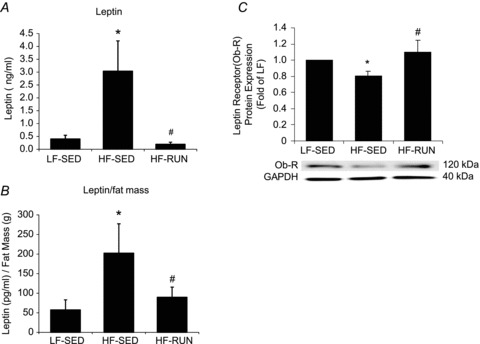
A, serum leptin level was significantly higher in HF-SED mice than that in LF-SED. Voluntary running during high-fat feeding opposed the development of the significant rise in serum leptin. B, circulating level of leptin normalized to total body fat mass (leptin/fat mass) was higher in HF-SED than in LF-SED. However, voluntary wheel running during high-fat feeding opposed the reduction in the leptin/fat mass ratio seen in high fat fed mice (HF-SED). C, the protein expression of leptin receptor (Ob-R) in coronary arterioles was significantly lower in HF-SED mice vs. LF-SED mice. Voluntary wheel running opposed the development of decreased leptin receptor protein in coronary arterioles of HF fed mice. Data are expressed as means ± SEM. *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED.
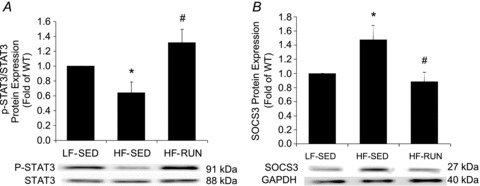
To explore whether leptin sensitivity is altered by high fat diet and/or physical activity, Western blot analysis was performed to measure phosphorylation of STAT3 and protein expression of SOCS3, a negative regulator of STAT3 phosphorylation. Figure 4A shows diminished STAT3 phosphorylation at Tyr 705 in HF-SED mice compared with LF-SED and HF-RUN mice. Conversely, approximately 35% higher SOCS3 protein expression in HF-SED mice than in LF-SED mice was observed (Fig. 4B). Noteworthy, physical activity opposed a decrease in STAT3 phosphorylation and an increase in SOCS3 protein expression in HF mice coronary arterioles.
Figure 4. Leptin caused vasodilatation of mice coronary arterioles in a concentration-dependent manner.
A, leptin-induced vasodilatation was significantly decreased in HF-SED mice vs. HF-SED mice. However, voluntary wheel running opposed the development of a fall in leptin-induced vasodilatation caused by high fat diet. B, l-NAME blunted leptin-mediated vasodilatation in LF-SED and HF-RUN mice to levels observed in HF-SED mice with or without l-NAME. Voluntary running by HF-Run group opposed the development of the loss in leptin sensitivity for coronary vasodilatation as compared to LF-SED. Data are expressed as means ± SEM, *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED.
Leptin-induced vasodilatory function was also diminished in HF-SED mice compared with both LF-SED and HF-RUN, which did not differ (Fig. 5A). The leptin-induced dilatation was significantly attenuated with l-NAME treatment in LF-SED and HF-RUN to the level of HF-SED, and l-NAME treatment did not further lower leptin-induced vasodilatation (Fig. 5B).
Figure 5.
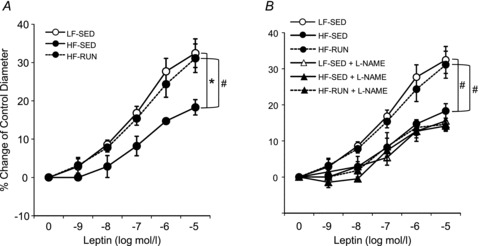
A, phosphorylation of STAT3 is reduced in HF-SED compared with LF-SED. However, voluntary running opposed the reduced phosphorylation of STAT3 found in HF mice. B, SOCS3 protein was increased in HF-SED. HF-RUN had no development of increased SOCS3 in mouse coronary arterioles. Data are expressed as means ± SEM. *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED.
Role of oxidative stress in obesity-induced vascular dysfunction
To establish the role of oxidative stress, more specifically O2− production, in endothelial dysfunction induced by HF diet, we administered the O2− scavenger TEMPOL in coronary arterioles from HF-SED and HF-RUN mice. TEMPOL restored impaired vasodilatation to ACh in HF-SED to the higher levels of the LF-SED and HF-RUN, with LF-SED not differing from HF-RUN (Fig. 6A). TEMPOL did not alter vascular function in HF-RUN mice. To potentially explain this finding, the protein expressions of SOD1 and SOD2 were measured to test whether these defensive mechanisms to O2− production were altered by HF diet and physical activity. SOD1 protein expression was attenuated in HF-SED, but this reduction was opposed by physical activity in HF-RUN (Fig. 6B). SOD2 protein expression was not different among the three groups of mice, LF-SED, HF-SED and HF-RUN mice (Fig. 6C).
Figure 6.
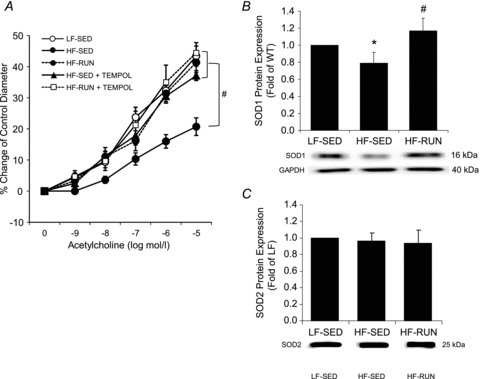
A, the open circle symbol for LF-SED was essentially overlapped by the filled symbols for HF-RUN, HF-RUN + TEMPOL, and HF-SED + TEMPOL, which did not differ from one another. In contrast, HF-SED was significantly less than all other groups. TEMPOL restored endothelium-dependent vasodilatation in arterioles from HF-SED mice, but there was no effect of TEMPOL on the arterioles from HF-RUN. B, SOD1 protein expression in HF-SED mice was lower than that in LF-SED mice. Voluntary running with high fat diet (HF-RUN) opposed the development of less SOD1 protein found in the HF-SED group. C, however, protein expression of SOD2 was not difference among 3 groups, LF-SED, HF-SED and HF-RUN mice. Data are expressed as means ± SEM. *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED.
To investigate a role of NAD(P)H oxidase in the prevention of HF-induced obesity and endothelial dysfunction, the role of the NAD(P)H oxidase inhibitor apocynin (Fig. 7A) and protein expression of NAD(P)H oxidase subunits p22phox, p47phox, p67phox and gp91phox (Fig. 7B–E) were determined. ACh-mediated dilatation was enhanced with apocynin incubation in the arterioles from HF-SED, but it was not altered in HF-RUN mice. Protein expression of gp91phox was significantly greater in HF-SED mice compared with LF-SED mice (Fig. 7E). However, physical activity in HF-RUN opposed the rise in gp91phox expression in HF-SED. In contrast, protein expressions of p22phox, p47phox and p67phox were not different among LF-SED, HF-SED and HF-RUN mice.
Figure 7. The open circle symbol for LF-SED was essentially overlapped by the filled symbols for HF-RUN, HF-RUN + Apocynin, and HF-SED + Apocynin, which did not differ from one another. In contrast, HF-SED was significantly less.
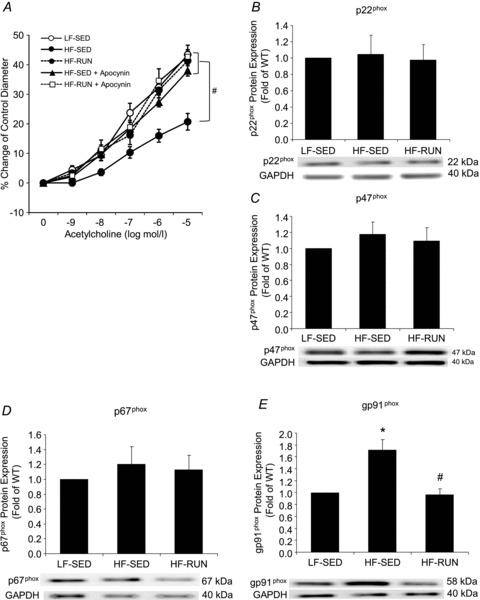
A, apocynin restored endothelium-dependent vasodilatation in arterioles from HF-SED mice, although apocynin did not change it in HF-RUN mice relative to HF-RUN only. Protein expression of NAD(P)H oxidase subunits, p22phox (B), p47phox (C) and p67phox (D) had no significant differences among 3 groups of LF-SED, HF-SED and HF-RUN. Protein expression of NAD(P)H oxidase subunits, gp91phox (E) was significantly higher in HF-SED compared with LF-SED mice. Voluntary wheel running opposed the development of enhanced gp91phox protein of coronary arterioles from high fat diet fed mice. Data were expressed as means ± SEM. *P < 0.05 vs. LF-SED, #P < 0.05 vs. HF-SED.
Discussion
Endothelial dysfunction has been claimed to be the first step toward atherosclerosis (Schwartz, 1980; Félétou & Vanhoutte, 2006; Vanhoutte, 2009). We have studied the role of physical activity in the primordial prevention of endothelial dysfunction caused by high-fat diet induction of obesity. Primordial prevention would require simultaneous application of voluntary running in wheels and high-fat diet. Stated alternatively, would the application of one risk factor (chylomicron remnants from high-fat diet; Dalla-Riva et al. 2010) for endothelial dysfunction be ameliorated by removal of a second risk factor (physical inactivity; Suvorava et al. 2004; Laufs et al. 2005; Hamburg et al. 2007) for endothelial dysfunction? The newer experimental strategy was employed for three reasons. First, primordial prevention is prevention of risk factors, such as endothelial dysfunction, in the first place (Strasser, 1978; Weintraub et al. 2011). The logic, in our opinion, is simple. Prevention of endothelial dysfunction, which leads to atherosclerosis (Félétou & Vanhoutte, 2006; Vanhoutte, 2009), would lessen the odds of progressing to atherosclerosis. Second, infants and children could initiate high feeding at the time when physical activity is restricted. For example, some schooling restricts physical activity from summer levels coupled with school lunches containing high-fat. Limited insight is available for interaction of the simultaneous initiation of high-fat diet with high levels of physical activity. Third, if you make an animal obese by high-fat diet first, in most cases later access to wheels is associated with minimal voluntary running. The major findings of the present study are that: (1) high-fat diet in the absence of obesity produced by voluntary running is not necessarily a risk factor for vascular dysfunction in our C57BL/6J female mouse model; and (2) the primordial prevention observed was associated with mechanisms of maintained eNOS phosphorylation, leptin sensitivity and redox balance.
NO-mediated endothelial function is maintained by voluntary running in HF-induced obesity
In the present study, as expected, HF diet results in obesity (Table 1) and impairs endothelium (ACh)-dependent coronary arteriolar vasodilatory function in mice (Fig. 1). Reduced NO bioavailability through decreased phosphorylation of eNOS is believed to be the primary defect that links to endothelial dysfunction in HF-induced obesity (Roberts et al. 2000; Jonk et al. 2007; Bourgoin et al. 2008). The current study also shows attenuated ACh-induced vasodilatation in coronary arterioles, which is NO dependent, along with reduced eNOS phosphorylation in obese mice as a contributing mechanism to impaired endothelial function (Fig. 2). However, our data are unique to coronary arterioles, showing that simultaneous initiation of HF diet and voluntary wheel running opposed the development of obesity (Table 1), as well as coronary endothelial dysfunction (Fig. 1A and B) through NOS signalling mechanisms (Fig. 2). Similarly, the metabolic syndrome-induced vascular dysfunction can be reversed in skeletal muscle resistance arterioles of obese Zucker rats (Frisbee et al. 2006) and human brachial artery (De Filippis et al. 2006) by regular exercise and/or physical activity through improved NO production (Jonk et al. 2007).
Maintenance of leptin-induced endothelial function by voluntary running
Consistent with previous studies, our study showed approximately 5-fold higher level of circulating leptin in obese HF-SED mice and prevention of this high serum level of leptin by voluntary wheel running in leaner HF-RUN mice (Fig. 3A). While circulating leptin is proportionally correlated with adiposity (Scarpace & Zhang, 2009), physical activity reduces high circulating leptin level in obesity (Carhuatanta et al. 2011). The linear relationship between circulating leptin level and a given fat mass is higher in HF-SED mice, suggesting reduced leptin responsiveness (increased leptin resistance) in the HF-SED group (Fig. 3B). Voluntary running during HF diet preserved the ratio of leptin at the level of LF-SED, implying maintenance of leptin sensitivity if voluntary running occurs simultaneously with HF diet. Taken together, findings from the serum leptin provided the preliminary support to pursue determinations of leptin sensitivity in coronary arterioles. The clinical relevance of pursuing leptin sensitivity changes is that endothelial dysfunction is often viewed as a precursor to atherosclerotic disease (Ross, 1999; Lusis, 2000) and that hyperleptinemia has been suggested to be an independent risk factor for cardiovascular disease (Leyva et al. 1998; Söderberg et al. 1999; Wallace et al. 2001; Ren, 2004).
From the above, we hypothesized that voluntary running would maintain indices of leptin sensitivity in coronary arterioles of mice provided wheels for voluntary running when fed HF diet. We first hypothesized that Ob-R would be present on the coronary arterioles, as Knudson et al. (2005) reported that the long form of Ob-R is present in coronary arteries and that hyperleptinaemia induces significant coronary endothelial dysfunction. The Ob-R antibody employed in our studies detected both long and short forms of Ob protein. While Ob-R protein in coronary arterioles was depressed in HF-SED, as compared to LF-SED, voluntary running during HF feeding (HF-RUN) opposed the decline in OB-R protein, maintaining it at the LF-SED level in coronary arterioles (Fig. 3C). Our observation would imply leptin resistance with HF diet could be prevented by preservation of Ob-R if voluntary physical activity were to be initiated with HF diet.
In addition to Ob-R protein density, the Janus kinase (JAK)/STAT signal transduction pathway is one of the main signalling cascades activated by leptin (Frühbeck, 2006) and is blunted with leptin resistance (Tune & Considine, 2007). The JAK/STAT pathway of cytokine signalling is under the negative-feedback control of SOCS (suppressors of cytokine signalling) proteins (Frühbeck, 2006). Peripheral leptin administration to ob/ob, but not db/db, mice rapidly induces SOCS3 mRNA in hypothalamus (Bjørbæk et al. 1998). The induction of SOCS3 protein expression attenuates JAK kinase activity, which in turn, lowers JAK phosphorylation of STAT3. In mammalian cell lines SOCS3 blocks leptin-induced signal transduction (Bjørbæk et al. 1998). We hypothesized that voluntary running while consuming a HF diet would prevent changes in SOCS3 and STAT3 signalling that occur in HF-SED mice that do not have access to running wheels. Mice in the group fed HF diet without voluntary running had increased SOCS3 protein (Fig. 4B). Voluntary running opposed the increase in SOCS3 protein of coronary arteriole when mice were fed HF diet. Further, the ratio of phospho-(Tyr 705)-STAT3 to STAT3 protein in coronary arterioles was lower in HF-SED, implying diminished JAK-STAT signalling, but this ratio was maintained in HF-RUN compared to LF-SED (Fig. 4A). Together, the prevention in the directional changes for three proteins that are associated with leptin resistance led to the next experiment to determine a dose-dependent leptin-induced vasodilatation of coronary arterioles. However, because exogenous leptin was not used, the differences in JAK/STAT and SOCS3 levels may be due to resistance in signalling of other cytokines, which also signal through JAK/STAT pathways. Exogenous leptin-induced pharmacological vasodilatation in coronary arterioles shows significant coronary endothelial dysfunction in obese Zucker rats (Knudson et al. 2005). The obesity-related leptin resistance in coronary arterioles was confirmed by reduced percentage dilatation of coronary arterioles at a given concentration of leptin that occurred in HF-SED mice (Fig. 5A). However for the first time, leptin resistance was shown to be preventable if HF diet was accompanied by voluntary running of mice. The lowest leptin dose (16.0 ng ml−1) in Fig. 5 is within the high range for serum leptin values (ng ml−1) of 1–4 (3 month old normal; Rana et al. 2011; Van Schothorst et al. 2011), 4–10 (6–12 month old normal; Rana et al. 2011), 10 (high glycemic index; Van Schothorst et al. 2011), and 12 (high-fat; Rana et al. 2011) published in recent papers. Nonetheless, no significant difference in vasodilatation existed at our 16.0 ng ml−1 leptin dose between HF-SED and either HF-RUN or LF-SED. The finding agrees with other conclusions. Sweeney stated in a review published in 2010 that ‘while the literature contains several indications that leptin mediates endothelial dysfunction, it should be stressed that endothelial dysfunction is typically observed at supraphysiological leptin concentrations’ (Sweeney, 2010). Tune & Considine (2007) have concluded, ‘leptin-induced, endothelial-dependent vasodilatation is a purely pharmacological phenomenon.’ A conundrum thus exists from our observations. On the one hand at the protein level, values for Ob-R, SOSC-3, and phospho-(Tyr 705)-STAT3 to STAT3 protein in coronary arterioles are consistent with the presence of leptin resistance with obesity (Figs 3C and 4). On the other hand at the functional level, leptin resistance manifested as attenuated vasodilatation is only present at pharmacological levels (Knudson et al. 2005) (Fig. 5A).
The clinical impact of leptin and physical activity on coronary arterioles is that impaired vasodilatory function of arterioles would lower blood flow, supplying less oxygen to cardiomyocytes. A potential clinical outcome of the prevention of leptin resistance by voluntary running can be deduced from a prospective study of 27,055 apparently healthy women reported by Mora et al. (2007). They estimated only 59% and 36% of cardiovascular disease (CVD) and coronary heart disease (CHD) could be attributed to conventional cardiovascular risk factors (baseline levels of haemoglobin A1c, traditional lipids, novel lipids, creatinine, homocysteine and inflammatory/hemostatic biomarkers) in this prospective study (Mora et al. 2007). Thus, other risk factors remain to be found and accepted. Prevention of leptin resistance in obese conditions could be one of the unexplained risk factors causing CVD and CHD. For example, the JAK/STAT pathway has been suggested to play an important role in the protection against myocardial ischaemia–reperfusion injury (Smith et al. 2010). Prevention of coronary arteriolar leptin resistance by increasing coronary perfusion could in part support an epidemiological observation.
Voluntary running opposes ROS-induced endothelial dysfunction of coronary arterioles in HF-induced Obesity
The NAD(P)H oxidase inhibitor apocynin, the O2− dismutase mimetic TEMPOL, or voluntary running, each alone, rescued attenuated vasodilatation of coronary arterioles that occurs in obese, HF fed mice. Previous publications have reported rescue of obesity-induced endothelial dysfunction by apocynin or TEMPOL in sedentary mice (Picchi et al. 2006; Gao et al. 2007) and rescue by exercise (Hägg et al. 2004; Kojda & Hambrecht, 2005; Jonk et al. 2007). Here we show that the simultaneous application of TEMPOL or apocynin produced no further vasodilatory effect than applied alone in the animals with voluntary running during high-fat feeding (Figs 6A and 7A), which proves that running indeed acted through antioxidant actions of upregulation of SOD1 protein and inhibition of the increase in NADPH-oxidase subunit gp91phox (Figs 6 and 7). The question remaining is identification of molecules involved in the prevention of coronary arteriole endothelial dysfunction by voluntary running at the molecular level.
One candidate, superoxide dismutase (SOD), has an essential role in the survival of aerobic organisms and prevention of pathological conditions (Miao & St. Clair, 2009). In our study HF-SED mice had decreases in SOD1, but not in SOD2, protein. Uniquely, voluntary running opposed the HF diet-mediated decrease in SOD1 protein in coronary arterioles, but running had no effect on SOD2 protein. SOD1 is a cytoplasmic protein that binds copper and zinc ions, and that scavenges free superoxide radicals by converting them to molecular oxygen and hydrogen peroxide in the body, while SOD2 is a mitochondrial protein. The characteristics of their protein structure, metal cofactor requirements, and cellular compartmentalization are distinctly different for the SOD isoforms (Miao & St. Clair, 2009). It is known that several transcription factors play unique regulatory roles in the expression of superoxide dismutase isoforms (Miao & St. Clair, 2009). The diverging effects on the two SOD isoforms are intriguing, and could be the direction of a future study.
The benefits of voluntary running on antioxidant proteins extended to the subunits of NAD(P)H oxidase, which is major source of O2− in the vasculature. Of the four subunits tested that compose NAD(P)H oxidase, only gp91phox protein increased by HF diet alone, and voluntary running opposed its increase (Fig. 7). gp91phox (Nox2) is the catalytic subunit that transfers electrons from NAD(P)H to O2. An overexpression of endothelial gp91phox levels in transgenic mice contributes to angiotensin II-induced endothelial dysfunction, vascular remodelling and hypertension (Murdoch et al. 2011). The prevention by voluntary running of gp91phox protein from increasing in HF-fed mice may be a mechanism related to running's prevention of obesity, a shear effect of the increased blood flow, or both (Milovanova et al. 2008).
Limitations exist in the current study. The lack of a low fat voluntary running group does not allow comparisons of the magnitude of the effect of voluntary running between high-fat and low-fat groups, which might reveal whether there is an interaction between diet and voluntary running in their vascular and molecular effects. Such a comparison is not feasible because the magnitude of distances of voluntary running differs between groups. High-fat feeding increases the magnitude of running distances in mice relative to low-fat mice. For example, Meek et al. (2010) reported that a Western high-fat diet had little or no effect on wheel running in control Hsd:ICR mice, but increased revolutions per day by as much as 75% in their lines of high voluntary running mice, mainly through increased time spent running. In addition, we have observed that C57BL/6 female mice fed a 45% fat diet voluntarily run 2.5 times longer distances than randomly selected cohorts fed a 17% fat diet (unpublished observations). Another limitation is the lack of an experimental group of physical exercise as an intervention once diet-induced obesity was established to see whether vascular dysfunction was in fact reversed by exercise. A future experiment will be to test the role of voluntary running in the treatment of existing obesity, as opposed to the current study's testing for primordial prevention of obesity in the first place.
Conclusion
In conclusion, high-fat diet in the absence of obesity, which was produced by voluntary running, is not necessarily a risk factor for vascular dysfunction in this model. More importantly, voluntary running initiated with HF diet opposed endothelial dysfunction in the first place in coronary arterioles of C57BL/6J mice. The observation allowed determination of mechanisms as to how physical activity opposes endothelial dysfunction by high-fat diet. It remains to be seen whether prevention of endothelial dysfunction was indirect from the absence of obesity with sufficient voluntary running, or a direct effect from vascular shear stress, or both. Importantly, if endothelial dysfunction is the first step toward coronary arteriosclerosis, as emphasized by Vanhoutte, (2009), then the only way for atherosclerosis to occur would be in the presence of normal endothelial function. The increased physical activity begun with initiation of the HF diet maintained NOS signalling, leptin responsiveness/sensitivity, and redox balance in coronary arterioles at the levels found in low-fat diet cohorts. The NOS inhibitor l-NAME was equally effective in producing endothelial dysfunction in LF-SED and HF-RUN groups with increasing leptin dosages, implying the preventive effect of running at high leptin levels during HF diet is via its maintenance of eNOS actions. Further, voluntary running was an effective NAD(P)H oxidase inhibitor at the level of gp91phox subunit or O2− dismutase mimetic via SOD1 in preventing endothelial dysfunction during HF feeding in the mouse model used in this study. These novel findings provide an important new extension of insights into the pleiotropic mechanisms of physical activity to include leptin-mediated vascular function in coronary microcirculation. They also reinforce physical activity as an effective primordial preventive strategy to oppose cardiovascular disease associated with diet- and sedentary life style-induced obesity.
Acknowledgments
This work was supported by grants from Pfizer Atorvastatin Research Award (2004-37), American Heart Association Scientist Development Grant (110350047A) and NIH grant (RO1-HL077566) to Dr Cuihua Zhang; and by gifts to Frank Booth. We acknowledge Mr Scott Naples for a support in monitoring of voluntary running and animal care. Nathan Jenkins and Jaume Padilla offered editorial comments.
Glossary
- ACh
acetylcholine
- Ang II
angiotensin II
- EDHF
endothelium dependent hyperpolarizing factor
- eNOS
endothelial nitric oxide synthase
- ET-1
endothelin-1
- JAK
Janus kinase
- l-NAME
NG-nitro-l-arginine-methyl ester
- LDL
low-density lipoprotein
- NO
nitric oxide
- O2
superoxide radical
- Ob-R
leptin receptors
- SNP
sodium nitroprusside
- SOCS3
suppressor of cytokine signalling 3
- SOD
superoxide dismutase
- STAT3
signal transducer and activator of transcription 3
Author contributions
Y.P. contributed to conception and design of the experiments, collection, analysis and interpretation of data, and drafting the article and revising it critically for important intellectual content. F.W.B contributed to revising the article critically for important intellectual content. S.L. contributed to collection, analysis and interpretation of data. M.J.L. contributed to collection, analysis and interpretation of data and revising the manuscript critically. C.Z. contributed to conception and design of the experiments, analysis and interpretation of data and revising the critically critically for important intellectual content. Y.P., F.W.B., S.L., and M.J.L. approved the final version for publication.
References
- Beltowski J. Role of leptin in blood pressure regulation and arterial hypertension. J Hypertens. 2006;24:789–801. doi: 10.1097/01.hjh.0000222743.06584.66. [DOI] [PubMed] [Google Scholar]
- Beltowski J, Wojcicka G, Marciniak A, Jamroz A. Oxidative stress, nitric oxide production, and renal sodium handling in leptin-induced hypertension. Life Sci. 2004;74:2987–3000. doi: 10.1016/j.lfs.2003.10.029. [DOI] [PubMed] [Google Scholar]
- Bjørbæk C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- Bourgoin F, Bachelard H, Badeau M, Melancon S, Pitre M, Lariviere R, Nadeau A. Endothelial and vascular dysfunctions and insulin resistance in rats fed a high-fat, high-sucrose diet. Am J Physiol Heart Circ Physiol. 2008;295:H1044–H1055. doi: 10.1152/ajpheart.00516.2008. [DOI] [PubMed] [Google Scholar]
- Carhuatanta KAK, Demuro G, Tschöp MH, Pfluger PT, Benoit SC, Obici S. Voluntary exercise improves high-fat diet-induced leptin resistance independent of adiposity. Endocrinology. 2011;152:2655–2664. doi: 10.1210/en.2010-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalla-Riva J, Garonna E, Elliott J, Botham KM, Wheeler-Jones CP. Endothelial cells as targets for chylomicron remnants. Atheroscler Suppl. 2010;11:31–37. doi: 10.1016/j.atherosclerosissup.2010.04.001. [DOI] [PubMed] [Google Scholar]
- Daniels SR, Pratt CA, Hayman LL. Reduction of risk for cardiovascular disease in children and adolescents. Circulation. 2011;124:1673–1686. doi: 10.1161/CIRCULATIONAHA.110.016170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Filippis E, Cusi K, Ocampo G, Berria R, Buck S, Consoli A, Mandarino LJ. Exercise-induced improvement in vasodilatory function accompanies increased insulin sensitivity in obesity and type 2 diabetes mellitus. J Clin Endocrinol Metab. 2006;91:4903–4910. doi: 10.1210/jc.2006-1142. [DOI] [PubMed] [Google Scholar]
- Ezzati M, Hoorn S, Lopez A, Danaei G, Rodgers A, Mathers C, Murray C. Comparative quantification of mortality and burden of disease attributable to selected risk factors. In: Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJL, editors. Global Burden of Disease and Risk Factors. Washington(DC): World Bank; 2006. Global Burden of Disease and Risk Factors. Chapter 4. [PubMed] [Google Scholar]
- Félétou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture) Am J Physiol Heart Circ Physiol. 2006;291:H985–H1002. doi: 10.1152/ajpheart.00292.2006. [DOI] [PubMed] [Google Scholar]
- Fenster CP, Weinsier RL, Darley-Usmar VM, Patel RP. Obesity, aerobic exercise, and vascular disease: the role of oxidant stress. Obesity. 2002;10:964–968. doi: 10.1038/oby.2002.131. [DOI] [PubMed] [Google Scholar]
- Fornoni A, Raij L. Metabolic syndrome and endothelial dysfunction. Curr Hypertens Rep. 2005;7:88–95. doi: 10.1007/s11906-005-0080-6. [DOI] [PubMed] [Google Scholar]
- Frisbee JC, Samora JB, Peterson J, Bryner R. Exercise training blunts microvascular rarefaction in the metabolic syndrome. Am J Physiol Heart Circ Physiol. 2006;291:H2483–H2492. doi: 10.1152/ajpheart.00566.2006. [DOI] [PubMed] [Google Scholar]
- Frühbeck G. Intracellular signalling pathways activated by leptin. Biochem J. 2006;393:7–20. doi: 10.1042/BJ20051578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Belmadani S, Picchi A, Xu X, Potter BJ, Tewari-Singh N, Capobianco S, Chilian WM, Zhang C. Tumor necrosis factor-α induces endothelial dysfunction in Leprdb mice. Circulation. 2007;115:245–254. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- Gill JMR, Malkova D. Physical activity, fitness and cardiovascular disease risk in adults: interactions with insulin resistance and obesity. Clin Sci. 2006;110:409–425. doi: 10.1042/CS20050207. [DOI] [PubMed] [Google Scholar]
- Hägg U, Andersson I, Naylor AS, Grönros J, Jonsdottir IH, Bergström G, Gan LM. Voluntary physical exercise-induced vascular effects in spontaneously hypertensive rats. Clin Sci. 2004;107:571–581. doi: 10.1042/CS20040171. [DOI] [PubMed] [Google Scholar]
- Hamburg NM, McMackin CJ, Huang AL, Shenouda SM, Widlansky ME, Schulz E, Gokce N, Ruderman NB, Keaney JF, Jr, Vita JA. Physical inactivity rapidly induces insulin resistance and microvascular dysfunction in healthy volunteers. Arterioscler Thromb Vasc Biol. 2007;27:2650–2656. doi: 10.1161/ATVBAHA.107.153288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonk AM, Houben AJHM, de Jongh RT, Serne EH, Schaper NC, Stehouwer CDA. Microvascular dysfunction in obesity: a potential mechanism in the pathogenesis of obesity-associated insulin resistance and hypertension. Physiology. 2007;22:252–260. doi: 10.1152/physiol.00012.2007. [DOI] [PubMed] [Google Scholar]
- Kim J-A, Montagnani M, Koh KK, Quon MJ. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888–1904. doi: 10.1161/CIRCULATIONAHA.105.563213. [DOI] [PubMed] [Google Scholar]
- Kimura K, Tsuda K, Baba A, Kawabe T, Boh-oka S-i, Ibata M, Moriwaki C, Hano T, Nishio I. Involvement of nitric oxide in endothelium-dependent arterial relaxation by leptin. Biochem Biophys Res Commun. 2000;273:745–749. doi: 10.1006/bbrc.2000.3005. [DOI] [PubMed] [Google Scholar]
- Knudson JD, Dincer UD, Zhang C, Swafford AN, Jr, Koshida R, Picchi A, Focardi M, Dick GM, Tune JD. Leptin receptors are expressed in coronary arteries, and hyperleptinemia causes significant coronary endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2005;289:H48–56. doi: 10.1152/ajpheart.01159.2004. [DOI] [PubMed] [Google Scholar]
- Kojda G, Hambrecht R. Molecular mechanisms of vascular adaptations to exercise. Physical activity as an effective antioxidant therapy? Cardiovasc Res. 2005;67:187–197. doi: 10.1016/j.cardiores.2005.04.032. [DOI] [PubMed] [Google Scholar]
- Korda M, Kubant R, Patton S, Malinski T. Leptin-induced endothelial dysfunction in obesity. Am J Physiol Heart Circ Physiol. 2008;295:H1514–H1521. doi: 10.1152/ajpheart.00479.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laufs U, Wassmann S, Czech T, Münzel T, Eisenhauer M, Böhm M, Nickenig G. Physical inactivity increases oxidative stress, endothelial dysfunction, and atherosclerosis. Arterioscler Thromb Vasc Biol. 2005;25:809–814. doi: 10.1161/01.ATV.0000158311.24443.af. [DOI] [PubMed] [Google Scholar]
- Lembo G, Vecchione C, Fratta L, Marino G, Trimarco V, d’Amati G, Trimarco B. Leptin induces direct vasodilation through distinct endothelial mechanisms. Diabetes. 2000;49:293–297. doi: 10.2337/diabetes.49.2.293. [DOI] [PubMed] [Google Scholar]
- Levin BE, Dunn-Meynell AA. Reduced central leptin sensitivity in rats with diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2002;283:R941–948. doi: 10.1152/ajpregu.00245.2002. [DOI] [PubMed] [Google Scholar]
- Leyva F, Godsland IF, Ghatei M, Proudler AJ, Aldis S, Walton C, Bloom S, Stevenson JC. Hyperleptinemia as a component of a metabolic syndrome of cardiovascular risk. Arterioscler Thromb Vasc Biol. 1998;18:928–933. doi: 10.1161/01.atv.18.6.928. [DOI] [PubMed] [Google Scholar]
- Lin S, Thomas TC, Storlien LH, Huang XF. Development of high fat diet-induced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord. 2000;24:639–646. doi: 10.1038/sj.ijo.0801209. [DOI] [PubMed] [Google Scholar]
- Lloyd-Jones D, Adams R, Carnethon M, De Simone G, Ferguson TB, Flegal K, Ford E, Furie K, Go A, Greenlund K, Haase N, Hailpern S, Ho M, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, McDermott M, Meigs J, Mozaffarian D, Nichol G, O’Donnell C, Roger V, Rosamond W, Sacco R, Sorlie P, Stafford R, Steinberger J, Thom T, Wasserthiel-Smoller S, Wong N, Wylie-Rosett J, Hong Y. American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:480–486. doi: 10.1161/CIRCULATIONAHA.108.191259. Heart disease and stroke statistics – 2009 update. [DOI] [PubMed] [Google Scholar]
- Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek TH, Eisenmann JC, Garland T. Western diet increases wheel running in mice selectively bred for high voluntary wheel running. Int J Obes. 2010;34:960–969. doi: 10.1038/ijo.2010.25. [DOI] [PubMed] [Google Scholar]
- Miao L, St. Clair DK. Regulation of superoxide dismutase genes: Implications in disease. Free Radic Biol Med. 2009;47:344–356. doi: 10.1016/j.freeradbiomed.2009.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milovanova T, Chatterjee S, Hawkins BJ, Hong N, Sorokina EM, DeBolt K, Moore JS, Madesh M, Fisher AB. Caveolae are an essential component of the pathway for endothelial cell signaling associated with abrupt reduction of shear stress. Biochim Biophys Acta. 2008;1783:1866–1875. doi: 10.1016/j.bbamcr.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mora S, Cook N, Buring JE, Ridker PM, Lee I-M. Physical activity and reduced risk of cardiovascular events. Circulation. 2007;116:2110–2118. doi: 10.1161/CIRCULATIONAHA.107.729939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdoch C, Alom-Ruiz S, Wang M, Zhang M, Walker S, Yu B, Brewer A, Shah A. Role of endothelial Nox2 NADPH oxidase in angiotensin II-induced hypertension and vasomotor dysfunction. Basic Res Cardiol. 2011;106:527–538. doi: 10.1007/s00395-011-0179-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- Park Y, Capobianco S, Gao X, Falck JR, Dellsperger KC, Zhang C. Role of EDHF in type 2 diabetes-induced endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2008;295:H1982–1988. doi: 10.1152/ajpheart.01261.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y, Yang J, Zhang H, Chen X, Zhang C. Effect of PAR2 in regulating TNF-α and NAD(P)H oxidase in coronary arterioles in type 2 diabetic mice. Basic Res Cardiol. 2010;106:111–123. doi: 10.1007/s00395-010-0129-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picchi A, Gao X, Belmadani S, Potter BJ, Focardi M, Chilian WM, Zhang C. Tumor necrosis factor-α induces endothelial dysfunction in the prediabetic metabolic syndrome. Circ Res. 2006;99:69–77. doi: 10.1161/01.RES.0000229685.37402.80. [DOI] [PubMed] [Google Scholar]
- Rana K, Fam BC, Clarke MV, Pang TP, Zajac JD, MacLean HE. Increased adiposity in DNA binding-dependent androgen receptor knockout male mice associated with decreased voluntary activity and not insulin resistance. Am J Physiol Endocrinol Metab. 2011;301:E767–778. doi: 10.1152/ajpendo.00584.2010. [DOI] [PubMed] [Google Scholar]
- Ren J. Leptin and hyperleptinemia – from friend to foe for cardiovascular function. J Endocrinol. 2004;181:1–10. doi: 10.1677/joe.0.1810001. [DOI] [PubMed] [Google Scholar]
- Ringseis R, Eder K. Fatty acids and signalling in endothelial cells. Prostaglandins Leukot Essent Fatty Acids. 2010;82:189–198. doi: 10.1016/j.plefa.2010.02.022. [DOI] [PubMed] [Google Scholar]
- Roberts CK, Vaziri ND, Wang XQ, Barnard RJ. Enhanced NO inactivation and hypertension induced by a high-fat, refined-carbohydrate diet. Hypertension. 2000;36:423–429. doi: 10.1161/01.hyp.36.3.423. [DOI] [PubMed] [Google Scholar]
- Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- Scarpace PJ, Zhang Y. Leptin resistance: a prediposing factor for diet-induced obesity. Am J Physiol Regul Integr Comp Physiol. 2009;296:R493–500. doi: 10.1152/ajpregu.90669.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S. Role of endothelial integrity in atherosclerosis. Artery. 1980;8:305–314. [PubMed] [Google Scholar]
- Smith CCT, Dixon RA, Wynne AM, Theodorou L, Ong S-G, Subrayan S, Davidson SM, Hausenloy DJ, Yellon DM. Leptin-induced cardioprotection involves JAK/STAT signaling that may be linked to the mitochondrial permeability transition pore. Am J Physiol Heart Circ Physiol. 2010;299:H1265–H1270. doi: 10.1152/ajpheart.00092.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg S, Ahrén B, Jansson JH, Johnson O, Hallmans G, Asplund K, Olsson T. Leptin is associated with increased risk of myocardial infarction. J Int Med. 1999;246:409–418. doi: 10.1046/j.1365-2796.1999.00571.x. [DOI] [PubMed] [Google Scholar]
- Srere PA. Citrate synthase. Methods Enzymol. 1969;13:3–11. [Google Scholar]
- Stapleton PA, James ME, Goodwill AG, Frisbee JC. Obesity and vascular dysfunction. Pathophysiology. 2008;15:79–89. doi: 10.1016/j.pathophys.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser T. Reflections on cardiovascular diseases. Inter Sci Reviews. 1978;3:225–230. [Google Scholar]
- Suvorava T, Lauer N, Kojda G. Physical inactivity causes endothelial dysfunction in healthy young mice. J Am Coll Cardiol. 2004;44:1320–1327. doi: 10.1016/j.jacc.2004.06.030. [DOI] [PubMed] [Google Scholar]
- Sweeney G. Cardiovascular effects of leptin. Nat Rev Cardiol. 2010;7:22–29. doi: 10.1038/nrcardio.2009.224. [DOI] [PubMed] [Google Scholar]
- Tune J, Considine R. Effects of leptin on cardiovascular physiology. J Am Soc Hypertens. 2007;1:231–241. doi: 10.1016/j.jash.2007.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schothorst EM, Bunschoten A, Verlinde E, Schrauwen P, Keijer J. Glycemic index differences of high-fat diets modulate primarily lipid metabolism in murine adipose tissue. Physiol Genomics. 2011;43:942–949. doi: 10.1152/physiolgenomics.00042.2011. [DOI] [PubMed] [Google Scholar]
- Vanhoutte P. Endothelial dysfunction: the first step toward coronary arteriosclerosis. Circ J. 2009;73:595–601. doi: 10.1253/circj.cj-08-1169. [DOI] [PubMed] [Google Scholar]
- Vecchione C, Maffei A, Colella S, Aretini A, Poulet R, Frati G, Gentile MT, Fratta L, Trimarco V, Trimarco B, Lembo G. Leptin effect on endothelial nitric oxide is mediated through Akt-endothelial nitric oxide synthase phosphorylation pathway. Diabetes. 2002;51:168–173. doi: 10.2337/diabetes.51.1.168. [DOI] [PubMed] [Google Scholar]
- Wallace AM, McMahon AD, Packard CJ, Kelly A, Shepherd J, Gaw A, Sattar N. Plasma leptin and the risk of cardiovascular disease in the West of Scotland Coronary Prevention Study (WOSCOPS) Circulation. 2001;104:3052–3056. doi: 10.1161/hc5001.101061. [DOI] [PubMed] [Google Scholar]
- Weintraub WS, Daniels SR, Burke LE, Franklin BA, Goff DC, Hayman LL, Lloyd-Jones D, Pandey DK, Sanchez EJ, Schram AP, Whitsel LP. Value of primordial and primary prevention for cardiovascular disease. Circulation. 2011;124:967–990. doi: 10.1161/CIR.0b013e3182285a81. [DOI] [PubMed] [Google Scholar]
- Winters B, Mo Z, Brooks-Asplund E, Kim S, Shoukas A, Li D, Nyhan D, Berkowitz DE. Reduction of obesity, as induced by leptin, reverses endothelial dysfunction in obese (Lepob) mice. J Appl Physiol. 2000;89:2382–2390. doi: 10.1152/jappl.2000.89.6.2382. [DOI] [PubMed] [Google Scholar]
- Woo KS, Chook P, Yu CW, Sung RYT, Qiao M, Leung SSF, Lam CWK, Metreweli C, Celermajer DS. Effects of diet and exercise on obesity-related vascular dysfunction in children. Circulation. 2004;109:1981–1986. doi: 10.1161/01.CIR.0000126599.47470.BE. [DOI] [PubMed] [Google Scholar]