

Abstract
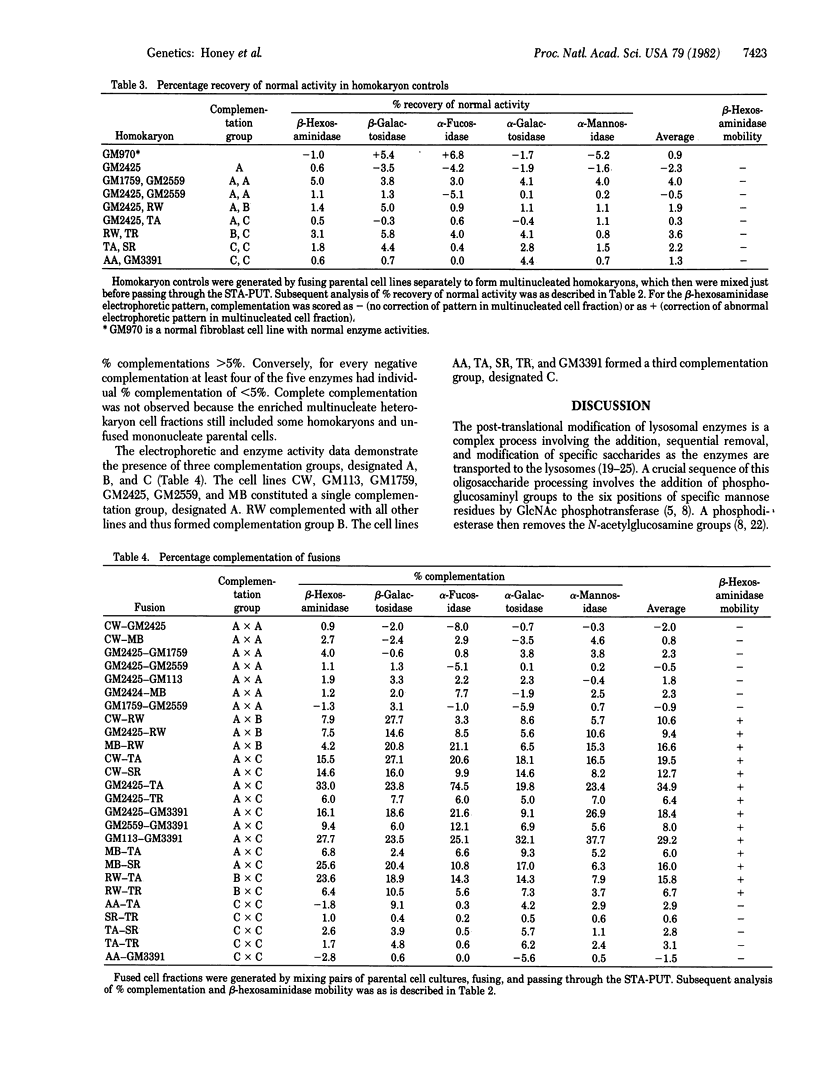
Mucolipidosis III (ML III), or pseudo-Hurler polydystrophy, is an inherited childhood disorder characterized biochemically by low activities and abnormal electrophoretic patterns of multiple lysosomal enzymes in fibroblasts. The primary deficiency of ML III has been proposed to be in UDP-N-acetylglucosamine:lysosomal enzyme N-acetylglucosamine-1-phosphotransferase. However, variation in this enzyme and in other biochemical properties of different ML III lines has been observed. Therefore, we investigated genetic heterogeneity within the disorder by complementation analysis. Heterokaryon cell fractions were generated by fusing together ML III fibroblast lines. When pairs of cells complemented, correction of lysosomal enzyme activities and electrophoretic patterns was observed. Twelve fibroblast lines from 10 sibships were analyzed and three distinct complementation groups were characterized. One complementation group represents the classical ML III disorder. A single cell line identifies a second complementation group. The cell lines comprising a third complementation group have a number of biochemical characteristics different from classical ML III and may represent a genetically distinct disorder.
Full text
PDF




Images in this article

Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Den Tandt W. R., Lassila E., Philippart M. Leroy's l-cell disease: markedly increased activity of plasma acid hydrolases. J Lab Clin Med. 1974 Mar;83(3):403–408. [PubMed] [Google Scholar]
- Erickson A. H., Blobel G. Early events in the biosynthesis of the lysosomal enzyme cathepsin D. J Biol Chem. 1979 Dec 10;254(23):11771–11774. [PubMed] [Google Scholar]
- Goldstone A., Koenig H. Physicochemical modifications of lysosomal hydrolases during intracellular transport. Biochem J. 1973 Feb;132(2):267–282. doi: 10.1042/bj1320267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasilik A., Neufeld E. F. Biosynthesis of lysosomal enzymes in fibroblasts. Phosphorylation of mannose residues. J Biol Chem. 1980 May 25;255(10):4946–4950. [PubMed] [Google Scholar]
- Hasilik A., Neufeld E. F. Biosynthesis of lysosomal enzymes in fibroblasts. Synthesis as precursors of higher molecular weight. J Biol Chem. 1980 May 25;255(10):4937–4945. [PubMed] [Google Scholar]
- Hasilik A., Waheed A., von Figura K. Enzymatic phosphorylation of lysosomal enzymes in the presence of UDP-N-acetylglucosamine. Absence of the activity in I-cell fibroblasts. Biochem Biophys Res Commun. 1981 Feb 12;98(3):761–767. doi: 10.1016/0006-291x(81)91177-3. [DOI] [PubMed] [Google Scholar]
- Hohmann L. K., Shows T. B. Complementation of genetic disease: a velocity sedimentation procedure for the enrichment of heterokaryons. Somatic Cell Genet. 1979 Nov;5(6):1013–1029. doi: 10.1007/BF01542657. [DOI] [PubMed] [Google Scholar]
- Honey N. K., Miller A. L., Shows T. B. The mucolipidoses: identification by abnormal electrophoretic patterns of lysosomal hydrolases. Am J Med Genet. 1981;9(3):239–253. doi: 10.1002/ajmg.1320090310. [DOI] [PubMed] [Google Scholar]
- Hoogeveen A. T., Verheijen F. W., d'Azzo A., Galjaard H. Genetic heterogeneity in human neuraminidase deficiency. Nature. 1980 Jun 12;285(5765):500–502. doi: 10.1038/285500a0. [DOI] [PubMed] [Google Scholar]
- Kaplan A., Achord D. T., Sly W. S. Phosphohexosyl components of a lysosomal enzyme are recognized by pinocytosis receptors on human fibroblasts. Proc Natl Acad Sci U S A. 1977 May;74(5):2026–2030. doi: 10.1073/pnas.74.5.2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly T. E., Thomas G. H., Taylor H. A., Jr, McKusick V. A., Sly W. S., Glaser J. H., Robinow M., Luzzatti L., Espiritu C., Feingold M. Mucolipidosis III (pseudo-Hurler polydystrophy): Clinical and laboratory studies in a series of 12 patients. Johns Hopkins Med J. 1975 Oct;137(4):156–175. [PubMed] [Google Scholar]
- Kress B. C., Miller A. L. Urinary lysosomal hydrolases in mucolipidosis II and mucolipidosis III. Biochem J. 1979 Feb 1;177(2):409–415. doi: 10.1042/bj1770409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOWRY O. H., ROSEBROUGH N. J., FARR A. L., RANDALL R. J. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951 Nov;193(1):265–275. [PubMed] [Google Scholar]
- Leroy J. G., O'Brien J. S. Mucolipidosis II and III: different residual activity of beta-galactosidase in cultured fibroblasts. Clin Genet. 1976 May;9(5):533–539. doi: 10.1111/j.1399-0004.1976.tb01608.x. [DOI] [PubMed] [Google Scholar]
- Norwood T. H., Zeigler C. J., Martin G. M. Dimethyl sulfoxide enhances polyethylene glycol-mediated somatic cell fusion. Somatic Cell Genet. 1976 May;2(3):263–270. doi: 10.1007/BF01538964. [DOI] [PubMed] [Google Scholar]
- Pittman R. C., Williams J. C., Miller A. L., Steinberg D. Acid acylhydrolase deficiency in I-cell disease and pseudo-Hurler polydystrophy. Biochim Biophys Acta. 1979 Dec 18;575(3):399–409. doi: 10.1016/0005-2760(79)90109-7. [DOI] [PubMed] [Google Scholar]
- Reitman M. L., Kornfeld S. UDP-N-acetylglucosamine:glycoprotein N-acetylglucosamine-1-phosphotransferase. Proposed enzyme for the phosphorylation of the high mannose oligosaccharide units of lysosomal enzymes. J Biol Chem. 1981 May 10;256(9):4275–4281. [PubMed] [Google Scholar]
- Reitman M. L., Varki A., Kornfeld S. Fibroblasts from patients with I-cell disease and pseudo-Hurler polydystrophy are deficient in uridine 5'-diphosphate-N-acetylglucosamine: glycoprotein N-acetylglucosaminylphosphotransferase activity. J Clin Invest. 1981 May;67(5):1574–1579. doi: 10.1172/JCI110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shows T. B., Mueller O. T., Honey N. K., Wright C. E., Miller A. L. Genetic heterogeneity of I-cell disease is demonstrated by complementation of lysosomal enzyme processing mutants. Am J Med Genet. 1982 Jul;12(3):343–353. doi: 10.1002/ajmg.1320120312. [DOI] [PubMed] [Google Scholar]
- Tabas I., Kornfeld S. Biosynthetic intermediates of beta-glucuronidase contain high mannose oligosaccharides with blocked phosphate residues. J Biol Chem. 1980 Jul 25;255(14):6633–6639. [PubMed] [Google Scholar]
- Torreblanca J., Antelo M. C., García García S., García Consuegra J., Collado F. Sialidosis tipo II o mucolipidosis III. A propósito de tres observaciones. An Esp Pediatr. 1979 Feb;12(2):113–122. [PubMed] [Google Scholar]
- Varki A. P., Reitman M. L., Kornfeld S. Identification of a variant of mucolipidosis III (pseudo-Hurler polydystrophy): a catalytically active N-acetylglucosaminylphosphotransferase that fails to phosphorylate lysosomal enzymes. Proc Natl Acad Sci U S A. 1981 Dec;78(12):7773–7777. doi: 10.1073/pnas.78.12.7773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A., Kornfeld S. Identification of a rat liver alpha-N-acetylglucosaminyl phosphodiesterase capable of removing "blocking" alpha-N-acetylglucosamine residues from phosphorylated high mannose oligosaccharides of lysosomal enzymes. J Biol Chem. 1980 Sep 25;255(18):8398–8401. [PubMed] [Google Scholar]
- Waheed A., Pohlmann R., Hasilik A., von Figura K. Subcellular location of two enzymes involved in the synthesis of phosphorylated recognition markers in lysosomal enzymes. J Biol Chem. 1981 May 10;256(9):4150–4152. [PubMed] [Google Scholar]