

Abstract
We review the role of MRI in retinoblastoma and simulating lesions. Retinoblastoma is the most common paediatric intra-ocular tumour. It may be endophytic, exophytic or a diffuse infiltrating tumour. MRI can detect intra-ocular, extra-ocular and intracranial extension of the tumour. MRI is essential for monitoring patients after treatment and detection of associated second malignancies. It helps to differentiating the tumour from simulating lesions with leukocoria.
Retinoblastoma (RB) is the most common intraocular tumour of childhood. It is a highly malignant tumour of the primitive neural retina. RB is one of the most challenging problems in paediatric ophthalmology and radiology because it shows different patterns of growth, extension and recurrence. RB appears as a solid echogenic mass with high echogenic foci of calcifications on ultrasound studies. The tumour extension is not well delineated with ultrasound. MRI should be used to answer the key clinical questions that help in the selection of an appropriate line of treatment. It can detect the growth pattern of the tumour and determine the extension of the tumour, the involvement of the optic nerve and retrobulbar space, the presence of leptomeningeal spread or the existence of a second tumour. MRI is becoming an increasingly important tool for monitoring focal response to therapy. Also, it helps to differentiate RB from simulating lesions presenting with leukocoria [1-8].
The aim of this article is to review the role of MRI in RB.
Incidence
RB is the most common intraocular tumour in children and accounts for 3% of all cancers occurring in children. It occurs in 1 out of 18 000 to 30 000 live births worldwide. The average age at diagnosis is 18 months with 80% of cases occurring before the age of 3–4 years old. Approximately 30% are bilateral. Lesions may be synchronous, metachronous, unifocal or multifocal. No racial or sexual bias for the development of this cancer was found [9,10].
Genetics
RB is commonly sporadic (90%) but can be inherited (10%). Sporadic lesions usually result from spontaneous mutation. Inherited RB has an autosomal dominant pattern of inheritance with 80–100% penetrance. All bilateral and multifocal forms, as well as 10–15% of unilateral forms, are related to a constitutional mutation of the RB1 gene. The cause of hereditary RB is deletion or loss of function of the tumour suppressor gene RB1 on the long arm of chromosome 13 (13q14). All children with RB should have genetic analysis to establish the presence and site of the 13q mutation [2,5,10].
Pathology
RB is a tumour of neuroectodermal cells that become retinal photoreceptors under normal conditions. A characteristic finding of RB is a presence of Flexner-Wintersteiner rosettes, which are composed of cells arranged in a circular fashion around a well-defined lumen. The tumour cells can be poorly differentiated, undifferentiated or well differentiated [1-3,9].
Clinical presentation
The most common clinical sign of RB is leukocoria (60%) followed by strabismus (20%). Leukocoria is where the normal red reflex of the retina is replaced by a yellowish or greyish-white colour. Strabismus is a result of macular involvement by the tumour. It may also present with findings like vitreous haemorrhage, retinal detachment, angle-closure glaucoma, hyphaema, pseudohypopyon, iris heterochromia, proptosis and pseudo-orbital cellulites [2,8,10].
MR techniques
MRI of the globe is commonly done at 1.5 T. The head coil is commonly used; however, the application of the surface coil improves the signal-to-noise ratio (SNR) of the globe. The surface coil is a circular polarising coil with a diameter of 4 cm positioned 1 cm above the eye. Sedation with oral chloral hydrate used for infants and children 4 years of age and younger and intramuscular ketamine for children from 5–8 years of age is administrated [9,10].
Fast spin echo (FSE) T2 weighted imaging is used for the evaluation of the globe, but it has limited ability to detect calcification. High-resolution three-dimensional (3D) FSE T2 weighted imaging allows thin sections (0.4 mm) with high SNR that is sensitive to the detection of calcification. Gradient-echo T2 weighted imaging has been shown to be a more effective sequence to detect calcified structures. Pre- and post-contrast axial T1 weighted MRIs with and without fat suppression are obtained. Contrast-enhanced fat suppression T1 weighted MRI after intravenous injection of 0.1 mmol kg−1 gadopentate dimeglumine is done in axial and coronal planes as well as the parasagittal plane parallel to the long axis of the optic nerve. Finally, axial post-contrast T1 weighted imaging of the brain is obtained [11-14].
The additional sequences included short tau inversion-recovery (STIR) that can detect optic nerve infiltration. Constructive interference in steady state (CISS) sequence allow performance of multiplanar reconstruction to better demonstrate tumour extension [14,15]. Diffusion weighted images of the globe are obtained using a multislice spin-echo type of echo-planar imaging sequence with a gradient factor, b, of 0 and 1000 s mm−2. Application of parallel imaging has the potential to produce better quality diffusion weighted MRI and apparent diffusion coefficient (ADC) maps.
3 T MRI with dedicated multichannel head and neck coils will result in substantially higher contrast and SNR compared with 1.5 T scanner [16]. It has a higher ability to visualise findings on T2 weighted images such as vitreous seedings, higher contrast and SNR on post-contrast T1 weighted images, and increased ability for 3D acquisitions with thin sections and higher resolution to better evaluate the optic nerve and the orbit.
MRI appearance
At T2 weighted imaging, the tumour is usually dark compared with the vitreous. The partially calcified areas may appear as hypointense foci within the tumour on gradient-echo T2 weighted and 3D FSE T2 weighted images. On T1 weighted imaging, RB is slightly hyperintense to the vitreous. The vitreous may be abnormally bright on T1 weighted images because of increased globulin content and a decreased ratio of albumin to globulin that occurs with malignancy. The tumour shows moderate to marked enhancement. On enhanced T1 weighted images, finely dispersed areas of very low signal intensity became visible inside the tumour that correspond to areas of calcification [9-13] (Figure 1). The tumour shows restricted diffusion on diffusion weighted imaging at high b values. It exhibits low ADC values in contrast to the high intensity of the vitreous in the ADC maps. RB is a round cell type tumour that is typically poorly differentiated, tightly packed and has a high nuclear-cytoplasmic ration, which probably accounts for the low ADC value (Figure 2).
Figure 1.

Retinoblastoma (RB). (a) Axial high-resolution three-dimensional T2 weighted image shows low signal intensity left intraocular mass with a few signal void regions that correspond to areas of calcification. (b) Coronal T1 weighted image shows a mass in the lower part of the left globe with relative high signal intensity. Note the smaller RB in the upper part of the right lobe. (c) Fat suppressed contrast T1 weighted image shows intense inhomogeneous pattern of contrast enhancement of the mass with non-enhanced regions of calcification.
Figure 2.

Retinoblastoma (RB). (a) Axial T2 weighted image shows low signal intensity and a right intraocular mass with few signal void calcified regions. (b) Apparent diffusion coefficient (ADC) map shows restricted diffusion of the lesion with low ADC value (image courtesy of Dr Mauricio Castillo).
Anterior eye segment (AES) enhancement may be seen in patients with RB (Figure 3). AES enhancement is a hallmark of advanced RB because its degree correlates with tumour volume and optic nerve invasion. The degree of abnormal AES enhancement may be moderate (36%) or strong (33%). The degree of abnormal AES enhancement reflects angiogenesis, hyperaemia and inflammation in the iris [17-19].
Figure 3.
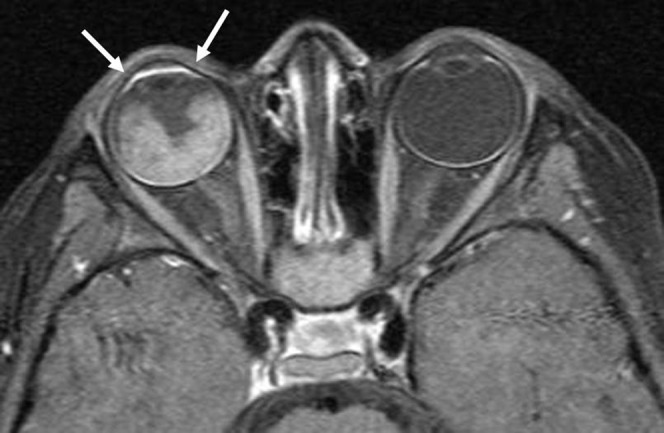
Anterior segment eye enhancement. Axial contrast T1 weighted image shows enhancement (arrows) of anterior segment in a patient with retinoblastoma (RB), which may be due to extension of RB or a tumour-induced angiogenesis in the iris.
The tumour may be unifocal or multifocal (Figure 4) within the same eye. MRI measurements of axial length, equatorial diameter and eye volume are significantly smaller in eyes with RB than in normal eyes. In addition, in patients with RB, the larger the tumour volume, the smaller the eye [20].
Figure 4.
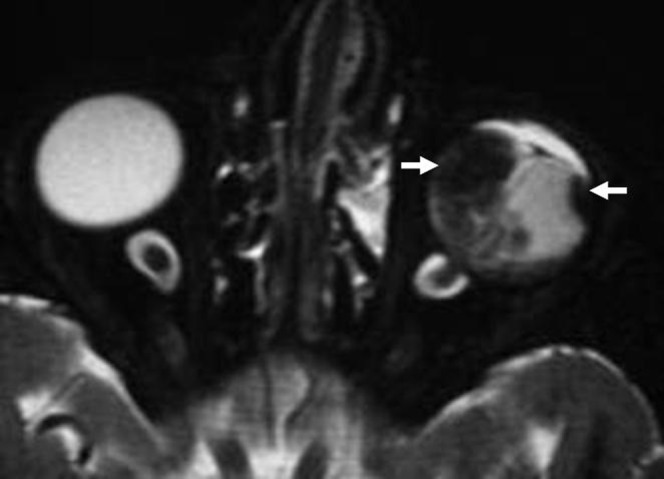
Multifocal retinoblastoma (RB). Axial three-dimensional T2 weighted image of the globe at 3 T shows multifocal RBs (arrows) seen in the left globe.
Growth pattern of retinoblastoma
Endophytic growth
The tumour arises from inner layers of the retina and grows into the vitreous (Figure 5). Small clusters of viable tumour cells may detach from the mass, producing multiple floating tumour islands throughout the globe called vitreous seeding. The presence of a vitreous seed has a poor prognostic value. In up to 63% of patients, vitreous seeding may be identified as T1 bright and T2 dark foci in the vitreous cavity [1,11,14].
Figure 5.
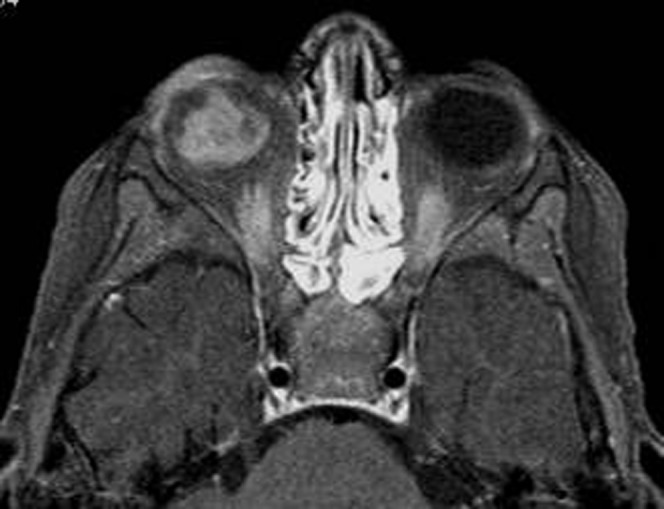
Endophytic retinoblastoma. Contrast T1 weighted image shows intense contrast enhancement of the right intraocular mass.
Exophytic growth
The tumour starts in the outer layers and grows in the subretinal space, which causes non-rhegmatogeneous retinal detachment with subretinal exudate and possible subretinal tumour seeding (Figure 6) [2,13,14].
Figure 6.
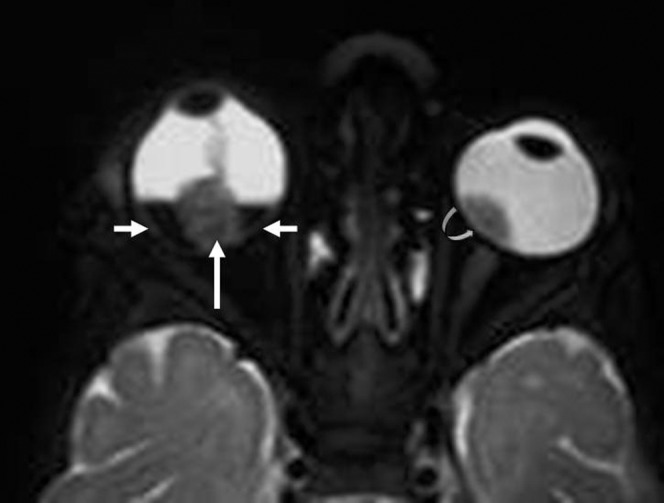
Exophytic retinoblastoma (RB). Axial T2 weighted image shows right RB (long arrow) associated with dark sub-retinal fluid (small arrows). Note the left RB (curved arrow).
Diffuse infiltrating growth
The tumour grows along the retina, appearing as a placoid mass, simulating inflammatory or haemorrhagic conditions. This is a rare (1–2%) form of RB that presents at advanced age (6 years) and more frequently in boys (M:F = 1.8:1). It is unilateral and sporadic. The absence of a discrete mass and lack of calcium deposits make diagnosis difficult. Cells may be discharged into the vitreous and seed the anterior chamber, mimicking an inflammatory process (pseudohypopyon). It commonly presents with pseudohypopyon (59%) and can be associated with leukocoria (24%). On MRI, there is often retinal detachment without a discrete mass lesion. Diffuse irregular and nodular thickening of the detached retinal leaflets is evident, with abnormal enhancement that often extends to the anterior eye segment (Figure 7) [3,21].
Figure 7.
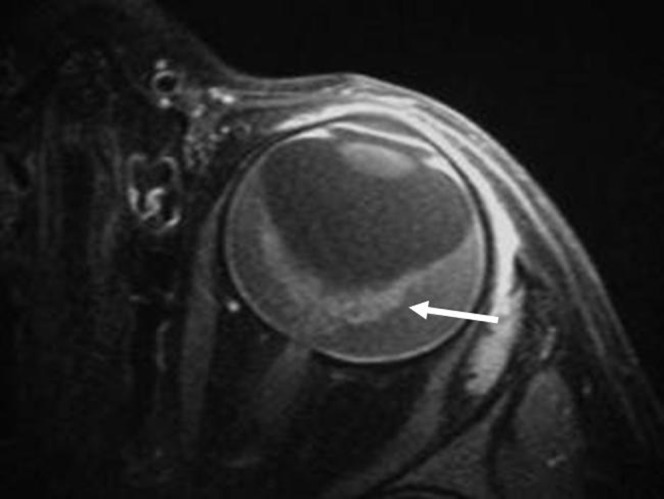
Diffuse retinoblastoma. Contrast T1 weighted image at 3 T with surface coil shows diffuse infiltrative placode lesion with retinal detachment (arrow).
Phthisis bulbi is a term to describe a shrunken non-functioning globe with extensive intraocular calcifications. RB rarely presents (2%) with phthisis bulbi in one eye and buphthalmos of the other eye owing to secondary glaucoma [1].
Extension of retinoblastoma
RB commonly spreads by direct extension; however, haematogenous and lymphatic dissemination have been reported. In the staging of RB, MRI should include evaluation of intraocular extension (choroids or sclera) and extraocular (optic nerve or orbital invasion) or intracranial (leptomeningeal or brain metastases) tumour spread [22-26].
Intraocular extension
Focal thickening or irregularity of the choroid may indicate focal spread of the tumour (Figure 8). Normal choroid has fine uniform linear enhancement. Choroidal invasion is usually associated with a higher mortality rate: slight choroidal invasion increases the mortality to 24%, and significant invasion raises the mortality to 65% [19-21].
Figure 8.
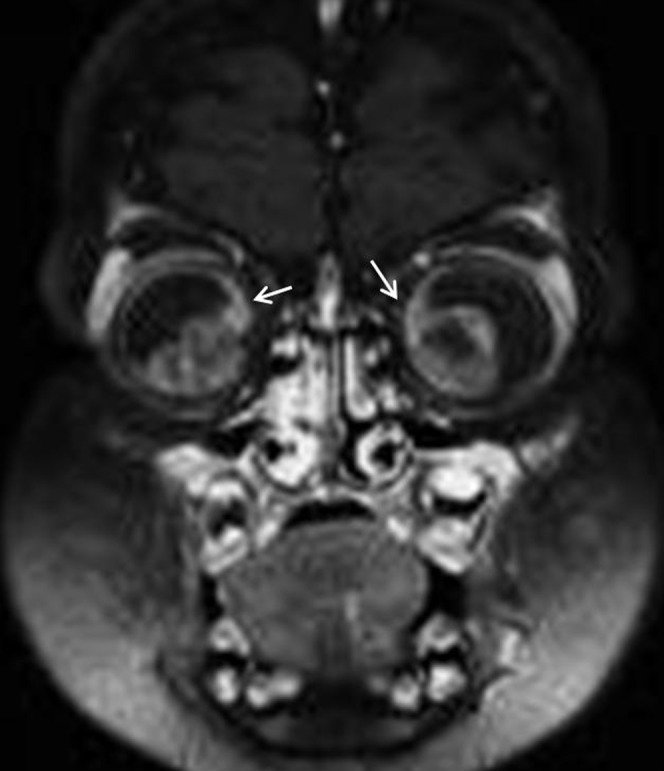
Retinoblastoma (RB) with choroidal invasion. Coronal contrast T1 weighted image shows focal choroidal thickening and enhancement along the superior aspect of RB on both sides (arrows).
Extraocular extension
Pre-operative detection of optic nerve extension of RB permits the physician to alter surgical management strategies. If the optic nerve is not invaded, mortality is less than 10%. If invasion of the optic nerve passes through the lamina cribrosa, mortality rises to 15%. If there is optic nerve involvement posterior to the lamina cribrosa, mortality rises to 44%. Optic nerve thickening and enhancement are indicators of tumour invasion. The length of optic nerve enhancement is a useful criterion to increase the specificity of MRI, as the enhancing segment of post-laminar invasion is longer (≥2 mm) than those associated with posterior bulging of the lamina cribrosa (<2 mm). The optic nerve appears thickened with an irregular outline. Abnormally high enhancement is seen within and around the affected nerve (Figure 9) [22-24].
Figure 9.

Retinoblastoma (RB) with optic nerve invasion. (a) Axial contrast T1 weighted image shows right RB with infiltration of a small part of the right optic nerve (arrowhead). (b) Axial contrast T1 weighted image in another patient shows left ocular mass infiltrating most of the left optic nerve (arrow).
Orbital extension of RB develops in fewer than 10% of patients and is associated with a higher mortality rate. Orbital extension of RB may be through the choroidal vasculature or the optic nerve (Figure 10) [19-21].
Figure 10.
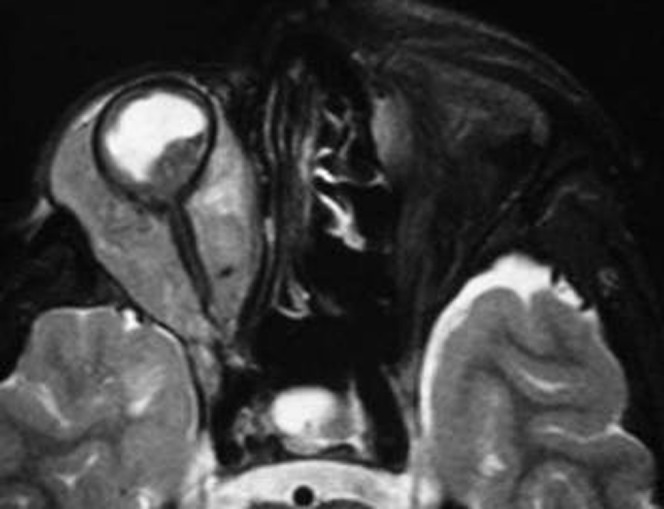
Retinoblastoma with orbital extension. Axial T2 weighted image shows right ocular mass with diffuse retro-orbital extension.
Intracranial extension
RB may extend into the suprasellar region via the optic nerve [24]. Cerebrospinal fluid seeding by RB commonly presents with diffuse leptomeningeal enhancement in the subarachnoid and intrathecal spaces [25]. Brain metastasis may occur in late stages of the disease and may be haemorrhagic [26].
Distant metastasis
The risk of distant metastasis markedly increases with extraocular extension. Tumours in the orbit, conjunctiva or eyelid may gain access to blood and lymphatic vessels. Haematogenous metastases are found in the lungs, skull, distal bones and brain, while lymphatic metastases may be found in regional lymph nodes. Most cases of metastatic RB develop within 2 years following the original diagnosis, and late metastasis are rare [2,9,10].
Prognostic value
The risk factors for poor prognosis and metastasis of RB include invasion to the post-laminar optic nerve, massive invasion of ocular coats (choroid and sclera), vitreous seeds and AES enhancement, or bilaterally of the disease. If the tumour is confined to the globe, 5 year survival is over 90%, whereas if the tumour extends outside the globe, the mortality is over 90% [19-24].
Bilateral, trilateral and quadrilateral retinoblastoma
Bilateral RB represents the hereditary form of RB and occurs in 30% of patients. Trilateral RB refers to the occurrence of bilateral ocular RBs and primitive midline neuroectodermal tumour arising in the pineal region or the suprasellar cistern (Figure 11); trilateral RBs represent 1.5–5% of patients with RB. Tetralateral RBs present bilateral ocular RB and tumour in both suprasellar and pineal regions [1,4,26].
Figure 11.

Trilateral retinoblastoma. (a) Axial T2 weighted image showing bilateral intraocular masses. (b) Axial contrast T1 weighted image of brain showing associated pineal mass.
Associated brain abnormalities
Pineoblastoma (5.5%) is associated with hereditary RB. Structural brain abnormalities, such as corpus callosum agenesis and Dandy–Walker variant are seen in patients with the 13q deletion syndrome. The incidence of pineal cysts (2.2%) in patients with RBs is similar to that in healthy children and is not associated with hereditary RB [27].
Post-treatment
The treatment of RB depends on several parameters: tumour volume, tumour position, intraocular tumour extension, extraocular stage of disease and laterality of the tumour. Conservative treatment with preservation of the useful vision of the eye applied in the early stages of small RB. Most children with unilateral RB are treated with eye enucleation followed with adjuvant therapy, while children with bilateral RB are treated with chemoreduction and thermotherapy [9,10].
Recurrence
MRI has an important tool for monitoring focal response to therapy and development of recurrent tumours. The growth patterns of recurrent RB may be intraocular tumour (Type A), intraorbital tumour with local spread into the optic nerve (Type B) or tumour extension to the lateral aspect of orbit and invading brain via sphenoidal bone (Type C) (Figure 12) [28].
Figure 12.
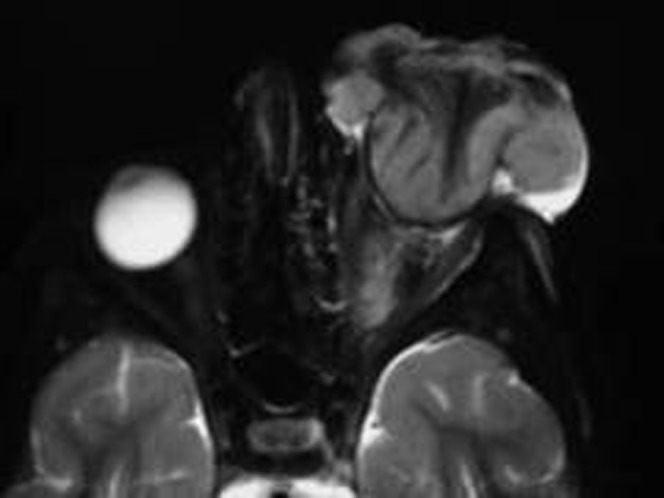
Recurrent retinoblastoma (RB). Axial T2 weighted image of patient after therapy shows large fungating recurrent left RB.
There are difficulties in the evaluation for post-treatment recurrence, such as post-operative changes/scar, artefacts from prostheses, metallic hardware and atrophy of the optic nerve on the side of globectomy.
Second primary malignancy
Second primary malignancy is commonly seen in heritable forms of RB. It occurs in association with radiation treatment or can be an independent process. The risk of developing a second primary tumour increases from 20% within 10 years to over 90% at 30 years. The secondary tumours may be mesenchymal tumours, including osteogenic sarcoma (Figure 13), chondrogenic sarcoma, fibrosarcoma and malignant fibrous histiocytoma. Periodic imaging surveillance of the orbit for local recurrence and for detection of second malignancies is very useful [29].
Figure 13.
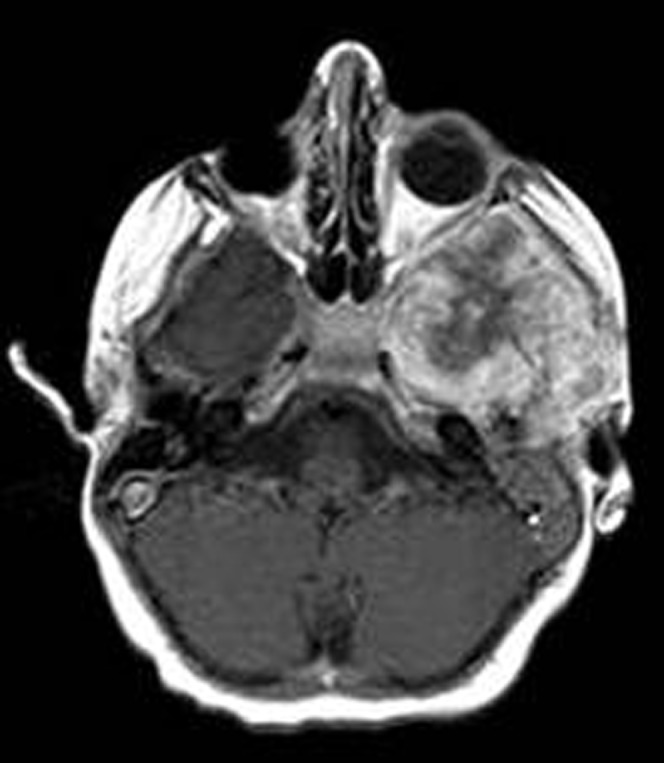
Second primary osteosarcoma. Axial contrast T1 weighted image shows intense enhanced osteosarcoma in left sphenoid bone in a patient previously treated with radiation and surgery for a right retinoblastoma (image courtesy of Dr Mauricio Castillo).
Hour-glass deformity
Characteristic facial deformities following irradiation during infancy occurs in patients with bilateral RBs. This includes hypotelorism, enophthalmos, depressed temporal bones, atrophy of the temporalis muscles and a depressed nasion. These distinctive features are termed the hour-glass facial deformity [30].
Differentiation from simulating lesions
RB must be differentiated from other intraocular lesions presenting with leukocoria. The commonest cause of leukocoria is RB (56–72%) but other causes include persistent hyperplastic primary vitreous (19–28%), coloboma (11.5%), sclerosing endophthalmitis (6.5–16%), Coats' disease (4–16%), retinal astrocytoma (3%), medulloepithelioma, retinal dysplasia in the form of Walker–Warburg syndrome and Noirre syndrome as well as retrolental fibroplasias of prematurity [8-10].
Coats' disease
Coats' disease is a primary congenital, non-familial idiopathic vascular anomaly of the retina. It is characterised by telangiectatic, leaky retinal vessels that lead to progressive retinal exudates. It usually occurs in young males (70%) with an incidence peak at age 6–8 years. It is mostly unilateral (90%). Patients present with leukocoria, strabismus, failed school screening or painful glaucoma. The MRI findings are retinal detachment without intraocular mass. The lipoproteinaceous sub-retinal exudation is usually seen as a mild to moderate hyperintense signal on T1 and T2 weighted MRI. There is enhancement along the leaves of the detached retina denoting presence of abnormal vessels. Proton MR spectroscopy of the lipoproteinaceous exudates shows a large peak at 1–1.6 ppm [31-34].
Persistent hyperplastic primary vitreous
Persistent hyperplastic primary vitreous is caused by the failure of the embryonic hyaloid vascular system to regress normally and extensive proliferation of embryonic connective tissue. It is commonly unilateral (90%) but may be bilateral in patients with retinal dysplasia. It is characterised by a leukocoria in a microphthalmic eye. MR shows microphthalmia; however, the eye can be normal in size or even buphthalmos in a patient with glaucoma. An enhanced triangular or tubular retrolental tissue that represents the persistent foetal tissue in the hyaloid canal (also known as the Cloquet's canal) is a characteristic finding. The tissue may extend into the hyaloid canal, giving a triangular shape. The vitreous is hyperintense with fluid–fluid levels owing to a sedimentation effect within the subretinal and subhyaloid haemorrhagic exudates. Other findings include an anterior displaced lens, small irregular lens and shallow anterior chamber (Figure 14) [35-37].
Figure 14.

Persistent hyperplastic primary vitreous. (a) Axial T1 weighted image shows retrolental opacity with linear vessel along the hyaloid canal. (b) Sagittal T1 weighted image shows fluid level in vitreous (image courtesy of Dr Mauricio Castillo).
Norrie disease
Norrie disease is a rare X-linked recessive syndrome consisting of retinal malformation, deafness and mental retardation. Female carriers are completely healthy. The ocular changes in male patients include retinal detachments and vitreo-retinal haemorrhage. MRI shows bilateral microphthalmia with hyperintense vitreous, caused by chronic vitreous or subretinal haemorrhage. It may be associated with bilateral persistence hyperplastic primary vitreous, hypoplastic optic nerves, abnormal lenses and developmental anomalies of the brain (Figure 15) [38-39].
Figure 15.

Norrie disease. (a) Axial T2 weighted image shows bilateral microphthalmia with hyperintense subretinal fluid. (b) Axial T2 weighted image shows a fluid level with associated persistent hyperplastic primary vitreous (PHPV) (arrows) (image courtesy of Dr Mauricio Castillo).
Walker–Warburg syndrome
Walker–Warburg syndrome is an autosomal recessive disorder caused by an abnormality of chromosome 9q34 that is characterised by profound hypotonia and ocular lesions in the form of microphthalmia and retinal non-attachment. MRI shows bilateral retinal detachment, subretinal or vitreous haemorrhage and gravitational intravitreal fluid. The congenital non-attached retina or the totally detached retina exhibits a characteristic narrow funnel shape, or a triangular intravitreal mass adjacent to hyaloid canal. Brain anomalies include diffuse cobblestone lissencephaly and unmyelinated white matter with hydrocephalus [39-40].
Sclerosing endophthalmitis (toxocariasis)
Sclerosing endophthalmitis is a granulomatous chorioretinitis uveitis that develops secondary to a Toxocara canis infestation. It affects patients over the age of 5 years bilaterally in 85% of cases. On MRI, a central vitreous mass appears isointense to vitreous on T1 weighted images and iso- or hypointense relative to vitreous according to fibrosis. There is a moderate to marked enhancement of the granuloma. Associated exudative subretinal fluid can be present with variable hyperintensity on T1 weighted and T2 weighted scans. It differs from RB by its central position, the fact that it is hyperintense to vitreous on T2 weighted images, the patient age and a positive serologic enzyme-linked immunosorbent assay (ELISA) [10-11].
Tuberculous endophathalmitis
Ocular tuberculosis results from endogenous spread from systemic foci. Chorioretinitis and uveitis are the most common type. Most patients have a chronic course of visual disturbance. On imaging, ocular tuberculosis usually manifests as a unilateral choroidal mass that shows contrast enhancement. The mass can fill the entire vitreous cavity and extend to the extraocular space in advanced cases [41].
Medulloepithelioma
Medulloepithelioma is a rare primary malignant non-hereditary embyrogenic intraocular tumour that commonly arises from the ciliary body and rarely from retina or optic nerve. The mean age at diagnosis is 5 years. There is no known gender or racial predilection. The most common signs are leukocoria and a mass of the iris or ciliary body. This tumour is divided into non-teratoid (60%) and teratoid (40%) histological subtypes. The tumour frequently extends locally to involve the iris or the adjacent anterior retina and may grow into the vitreous and invade through the cornea or sclera. MRI shows a mass localised to the ciliary body that is slightly to moderately hyperintense to vitreous on T1 weighted images and hypointense on T2 weighted images. The tumour shows marked homogeneous or heterogeneous enhancement [42].
Retinal astrocytic hamartoma
Retinal astrocytoma is a rare, benign retinal low grade tumour or hamartoma that can arise from the nerve fibre of the retina or optic nerve. It can present as an isolated lesion or in association with tuberous sclerosis or neurofibromatosis. It develops in 15% of patients who have tuberous sclerosis. It is associated with tuberous sclerosis in 50% and neurofibromatosis Type I in 14%. It may be associated with an exudative retinal detachment, haemorrhage and calcification. Unlike RB, it rarely grows in size and if it does, only modestly [2,10,11].
Retinopathy of prematurity
Retinopathy of prematurity is seen in premature, low birthweight infants. It used to be related to excessive oxygen therapy that was previously used to treat hyaline membrane disease, but it is now uncommon owing to the advent of exogenous surfactant therapy. It is usually bilateral and fairly symmetric. In early stages, the eyes may be microphthalmic. Hyperintense vitreous on T1 and T2 weighted images is common, which is a result of chronic haemorrhage. Phthisis bulbi may be the end result for the most severely affected. Calcifications may occur. Patient history, clinical findings, bilaterality and associated periventricular leukomalacia may be seen in the brain and are usually suggestive of a diagnosis [9-10].
Conclusion
MRI is essential for initial diagnosis, extension, staging and treatment planning of RB. It has been used to follow-up and monitor patients after treatment as well as for differentiating RB from simulating lesions in children with leukocoria.
Acknowledgments
We thank Professor Mauricio Castillo at University of North Carolina for his help with some of the figures in this article. This review was first presented as an educational exhibit at the Assembly and Annual Meeting of the Radiological Society of North America (RSNA) in 2009.
References
- 1.Kadom N, Sze R. Radiological reasoning: leukocoria in a child. AJR Am J Roentgenol 2008;191:S40–4 [DOI] [PubMed] [Google Scholar]
- 2.Chung E, Specht C, Schroeder J. Pediatric orbit tumors and tumor-like lesions: Neuroepithelial lesions of the ocular globe and optic nerve. RadioGraphics 2007;27:1159–86 [DOI] [PubMed] [Google Scholar]
- 3.Mafee MF, Karimi A, Shah J, Rapoport M, Ansari SA. Anatomy and pathology of the eye: role of MR imaging and CT. Magn Reson Imaging Clin North Am 2006;14:249–70 [DOI] [PubMed] [Google Scholar]
- 4.Belden C. MR imaging of the globe and optic nerve. Neuroimaging Clin North Am 2004;14:809–25 [DOI] [PubMed] [Google Scholar]
- 5.Mafee MF, Mafee RF, Malik M, Pierce J. Medical imaging in pediatric ophthalmology. Pediatr Clin North Am 2003;50:259–86 [DOI] [PubMed] [Google Scholar]
- 6.Gorospe L, Royo A, Berrocal T, Garcia-Raya P, Moreno P, Abelairas J. Imaging of orbital disorders in pediatric patients. Eur Radiol 2003;13:2012–16 [DOI] [PubMed] [Google Scholar]
- 7.Smirniotopoulos JG, Bargallo N, Mafee MF. Differential diagnosis of leukokoria: radiologic-pathologic correlation. RadioGraphics 1994;14:1059–79 [DOI] [PubMed] [Google Scholar]
- 8.O'Brien J. Retinoblastoma: clinical presentation and the role of neuroimaging. AJNR Am J Neuroradiol 2001;22:426–8 [PubMed] [Google Scholar]
- 9.Apushkin MA, Apushkin MA, Shapiro MJ, Mafee MF. Retinoblastoma and simulating lesions: role of imaging. Neuroimaging Clin North Am 2005;15:49–67 [DOI] [PubMed] [Google Scholar]
- 10.Kaufman LM, Mafee MF, Song CD. Retinoblastoma and simulating lesions. Role of CT, MR imaging and use of Gd-DTPA contrast enhancement. Radiol Clin North Am 1998;36:1101–7 [DOI] [PubMed] [Google Scholar]
- 11.Lemke A, Kazi I, Mergner U, Foerster P, Heimann H, Bechrakis N, et al. Retinoblastoma—MR appearance using a surface coil in comparison with histopathological results. Eur Radiol 2007;17:49–60 [DOI] [PubMed] [Google Scholar]
- 12.Schueler AO, Hosten N, Bechrakis NE, Lemke AJ, Foerster P, Felix R, et al. High resolution magnetic resonance imaging of retinoblastoma. Br J Ophthalmol 2003;87:330–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Galluzzi P, Hadjistilianou T, Cerase A, De Francesco S, Toti P, Venturi C. Is CT still useful in the study protocol of retinoblastoma? AJNR Am J Neuroradiol 2009;30:1760–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Graaf P, Barkhof F, Moll AC, Imhof SM, Knol DL, van derValk P, et al. Retinoblastoma: MR imaging parameters in detection of tumor extent. Radiology 2005;235:197–207 [DOI] [PubMed] [Google Scholar]
- 15.Gizewski E, Wanke I, Jurklies C, Gungor A, Forsting A. T1 Gd-enhanced compared with CISS sequences in retinoblastoma: superiority of T1 sequences in evaluation of tumour extension. Neuroradiology 2005;47:56–61 [DOI] [PubMed] [Google Scholar]
- 16.Mafee MF, Rapoport M, Karimi A, Ansari SA, Shah J. Orbital and ocular imaging using 3- and 1.5-T MR imaging systems. Neuroimaging Clin North Am 2005;15:1–21 [DOI] [PubMed] [Google Scholar]
- 17.Saket RR, Mafee MF. Anterior-segment retinoblastoma mimicking pseudoinflammatory angle-closure glaucoma: review of the literature and the important role of imaging. AJNR Am J Neuroradiol 2009;30:1607–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Graaf P, van derValk P, Moll A, Imhof S, Schoutenvan A, Meeteren V, et al. Contrast-enhancement of the anterior eye segment in patients with retinoblastoma: correlation between clinical, MR imaging, and histopathologic findings. AJNR Am J Neuroradiol 2010;31:237–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galluzzi P, Cerase A, Hadjistilianou T, De Francesco S, Toti P, Vallone I, et al. Retinoblastoma: abnormal gadolinium enhancement of anterior segment of eyes at MR imaging with clinical and histopathologic correlation. Radiology 2003;228:683–90 [DOI] [PubMed] [Google Scholar]
- 20.de Graaf P, Knol D, Moll A, Imhof S, Schouten-van Meeteren A, Castelijns J. Eye size in retinoblastoma: MR imaging measurements in normal and affected eyes. Radiology 2007;244:273–80 [DOI] [PubMed] [Google Scholar]
- 21.Brisse HJ, Lumbroso L, Freneaux PC, Validire P, Doz F, Quintana E, et al. Sonographic, CT, and MR imaging findings in diffuse infiltrative retinoblastoma: report of two cases with histologic comparison. AJNR Am J Neuroradiol 2001;22:499–504 [PMC free article] [PubMed] [Google Scholar]
- 22.Brisse H, Guesmi M, Aerts I, Sastre-Garau X, Savignoni A, Lumbrosole Rouic L, et al. Relevance of CT and MRI in retinoblastoma for the diagnosis of postlaminar invasion with normal-size optic nerve: A retrospective study of 150 patients with histologic comparison. Pediatr Radiol 2007;37:649–56 [DOI] [PubMed] [Google Scholar]
- 23.Ainbinder D, Haik B, Frei D, Gupta K, Mafee M. Gadolinium enhancement: improved MRI detection of retinoblastoma extension into the optic nerve. Neuroradiology 1996;38:778–81 [DOI] [PubMed] [Google Scholar]
- 24.Meli F, Boccaleri C, Manzitti J, Lylyk P. Meningeal dissemination of retinoblastoma: CT findings in eight patients. AJNR Am J Neuroradiol 1990;11:983–6 [PMC free article] [PubMed] [Google Scholar]
- 25.Atlas SW, Kemp SS, Rorke L, Grossman RI. Hemorrhagic intracranial retinoblastoma metastases: MR pathology correlation. J Comput Assist Tomogr 1988;12:286–9 [DOI] [PubMed] [Google Scholar]
- 26.Provenzale J, Gururangan S, Klintworth G. Trilateral retinoblastoma: clinical and radiologic progression. AJR Am J Roentgenol 2004;83:505–11 [DOI] [PubMed] [Google Scholar]
- 27.Rodjan F, de Graaf P, Moll A, Imhof S, Verbeke J, Sanchez E, et al. Brain Abnormalities on MR imaging in patients with retinoblastoma. AJNR Am J Neuroradiol 2010;31:1385–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tateishi U, Hasegawa T, Miyakawa K, Sumi M, Moriyama N. CT and MRI features of recurrent tumors and second primary neoplasms in pediatric patients with retinoblastoma. AJR Am J Roentgenol 2003;181:879–84 [DOI] [PubMed] [Google Scholar]
- 29.Chan LL, Czerniak BA, Ginsberg LE. Radiation induced osteosarcoma after bilateral childhood retinoblastoma. AJR Am J Roentgenol 2000;174:1288. [DOI] [PubMed] [Google Scholar]
- 30.Yue N, Benson M. The hourglass facial deformity as a consequence of orbital irradiation for bilateral retinoblastoma. Pediatr Radiol 1996;26:421–3 [DOI] [PubMed] [Google Scholar]
- 31.Förl B, Schmack I, Grossniklaus HE, Rohrschneider K. Coats' disease. Important differential diagnosis for retinoblastoma. Ophthalmologe 2008;105:761–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edward DP. Mafee MF. Garcia-Valenzuela E. Weiss RA. Coats' disease and persistent hyperplastic primary vitreous. Role of MR imaging and CT. Radiol Clin North Am 1998;36:1119–31 [DOI] [PubMed] [Google Scholar]
- 33.Dhoot D, Weissman J, Landon R, Evans M, Stout T. Optic nerve enhancement in Coats disease with secondary glaucoma. J AAPOS 2009;13:301–2 [DOI] [PubMed] [Google Scholar]
- 34.Eisenberg L, Castillo M, Kwock L, Mukherji SK, Wallace DK. Proton MR spectroscopy in Coats disease. AJNR Am J Neuroradiol 1997;18:727–9 [PMC free article] [PubMed] [Google Scholar]
- 35.Kaste SC, Jenkins JJ, 3rd, Meyer D, Fontanesi J, Pratt CB. Persistent hyperplastic primary vitreous of the eye: imaging findings with pathologic correlation. AJR Am J Roentgenol 1994;162:437–40 [DOI] [PubMed] [Google Scholar]
- 36.Castillo M, Wallace DK, Mukherji SK. Persistent hyperplastic primary vitreous involving the anterior eye. AJNR Am J Neuroradiol 1997;18:1526–8 [PMC free article] [PubMed] [Google Scholar]
- 37.Kker W, Ramaekers V. Persistent hyperplastic primary vitreous: MRI. Neuroradiology 1999;41:520–2 [DOI] [PubMed] [Google Scholar]
- 38.Hatsukawa Y, Nakao T, Yamagishi T, Okamoto N, Isashiki Y. Novel nonsense mutation (Tyr44stop) of the Norrie disease gene in a Japanese family. Br J Ophthalmol 2002;86:1453–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.de Graaf P, van derValk A, Moll A, Imhof S, Schouten-van Meeteren A, Castelijns J. Retinal dysplasia mimicking intraocular tumor: MR imaging findings with histopathologic correlation. AJNR Am J Neuroradiol 2007;28:1731–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Khalaf S, Tareef R. Walker–Warburg Syndrome. JAAPOS 2006;10:486–8 [DOI] [PubMed] [Google Scholar]
- 41.Raina U, Tuli D, Arora R, Mehta D, Taneja M. Tubercular endophthalmitis simulating retinoblastoma. Am J Ophthalmol 2000;130:843–5 [DOI] [PubMed] [Google Scholar]
- 42.Vajaranant T, Mafee M, Kapur R, Rapoport M, Edward D. Medulloepithelioma of the ciliary body and optic nerve: clinicopathologic, CT, and MR imaging features. Neuroimaging Clin North Am 2005;15:69–83 [DOI] [PubMed] [Google Scholar]