

Abstract
Despite advances in therapy, gliomas remain associated with poor prognosis. Clinical advances will be achieved through molecularly targeted biological therapies, for which knowledge of molecular genetic and gene expression characteristics in relation to histopathology and in vivo imaging are essential. Recent research supports the molecular classification of gliomas based on genetic alterations or gene expression profiles, and imaging data supports the concept that molecular subtypes of glioma may be distinguished through non-invasive anatomical, physiological and metabolic imaging techniques, suggesting differences in the baseline biology of genetic subtypes of infiltrating glioma. Furthermore, MRI signatures are now being associated with complex gene expression profiles and cellular signalling pathways through genome-wide microarray studies using samples obtained by image guidance which may be co-registered with clinical imaging. In this review we describe the pathobiology, molecular pathogenesis, stem cells and imaging characteristics of gliomas with emphasis on astrocytomas and oligodendroglial neoplasms.
Gliomas are the most common primary central nervous system tumour and, although they represent only 2% of cancers, their treatment presents one of the greatest challenges to oncologists. Despite advances in therapy these tumours remain associated with high morbidity and mortality and, compared with other cancers, account for the greatest number of average years of life lost to cancer [1]. Infiltrative gliomas are incurable and the prognostic outlook remains poor, with low-grade tumours likely to progress and short survival, frequently less than a year, for many patients with high-grade glioma (HGG). Median survival with currently available therapies remains much less than 2 years for the most aggressive form, glioblastoma multiforme (GBM), which accounts for 60–70% of gliomas [2].
The causes of treatment failure, disease progression and recurrence arise from the fundamental biology of gliomas, challenging the biologist to advance our understanding of the genetic aberrations and cellular mechanisms that dictate tumour behaviour and provide better diagnostic tools and new biologically targeted therapies [3-7]. In the same way, development of advanced multimodal magnetic resonance (MR) and positron emission tomography (PET) imaging has allowed non-invasive characterisation of the physiology and metabolism of these tumours, leading to improved diagnosis and prognostication and better discrimination between treatment effects and tumour recurrence [8-11]. In many recent research studies, attempts are being made to correlate features of imaging with the genetics and biology of the tumour tissues giving rise to these imaging signatures [12-14]. This review provides an overview of the pathobiology and molecular genetics of gliomas, with focus on diffusely infiltrating astrocytomas and oligodendroglial neoplasms and describes imaging characteristics in relation to biology and genetics in these tumours.
Pathological features of gliomas
Gliomas are histologically heterogeneous tumours often diagnosed from small tissue specimens, which may be prone to sampling error unless obtained using imaging techniques to guide biopsy or sampling during resection. Early tumour classifications relied on comparing tumour features with those of normal tissue. Thus brain tumours with cells resembling astrocytes were termed astrocytomas and those with cells resembling oligodendrocytes were called oligodendrogliomas. Although historically a number of classification schemes have been used [15], currently the most widely used classification and grading system is that of the World Health Organization (WHO) [16]. The WHO classification is based on histological features including cellularity, mitotic activity, nuclear atypia, vascularity and necrosis. It also recognises four prognostic grades and a variety of histological subtypes of which astrocytomas (60–70%), oligodendrogliomas (10–30%) and ependymomas (<10%) are the most common (Figure 1). A variety of other histopathology types, e.g. gangliogliomas and gangliocytomas, are less commonly encountered. However, irrespective of the classification system used, the small sample size and subjective criteria means that for many gliomas diagnosis may be difficult and subject to inter-and intraobserver variation [17,18].
Figure 1.

World Health Organization (WHO) histopathology classification of gliomas. (a) Pilocytic astrocytoma WHO grade I, with compact bundles of "piloid"/elongated cells containing nuclei with minimal atypia; (b) astrocytoma WHO grade II, showing increased cellularity with occasional atypical nuclei and some cells with enlarged cytoplasm; (c) astrocytoma WHO grade III, containing darkly stained nuclei with increased cytoplasm and occasional mitoses; (d) glioblastoma WHO grade IV, illustrating tumour necrosis without pseudopalisading as is commonly seen in glioblastomas; (e) oligodendroglioma WHO grade II, showing perinuclear haloes, "chickenwire" vasculature and microcalcification; (f) oligodendroglioma WHO grade III, with increased cellularity retaining roundness of nuclei associated with cell necrosis, note the mild infiltration by neutrophils.
Pilocytic astrocytomas correspond to WHO grade I and are relatively circumscribed, slow growing, often cystic astrocytomas comprising 5–6% of all gliomas and show contrast enhancement on MRI [19,20]. They are most common in children and young adults, in whom the majority arise in the cerebellum, hypothalamus and third ventricular region and are usually curable by surgical excision. The common gliomas that arise in the cerebral hemispheres are called "diffuse" gliomas because of their marked propensity to infiltrate the surrounding brain parenchyma, irrespective of grade. These migratory cells are refractory to conventional therapy and may contribute to treatment failure [21]. Diffuse astrocytomas include astrocytomas of WHO grades II, III and IV. Astrocytomas WHO grade II represent 10–15% of all astrocytic tumours, have a peak incidence in adults between 30 and 40 years, show a predominance in males, are more common supratentorially and rare in the cerebellum. These tumours consist of well-differentiated fibrillary or gemistocytic astrocytes on a background of loosely structured, often microcystic, matrix. Compared with normal brain, there is moderately increased cellularity and occasional nuclear atypia, but mitotic activity is generally absent [22]. Diffuse astrocytomas WHO grade II show hypointensity on T1 weighted and hyperintensity on T2 weighted MRI and rarely show gadolinium enhancement [9]. Gliomas of grades I and II are termed low grade and often considered "benign", although this may be misleading as some show rapid clinical evolution. HGGs include grades III and IV, are more aggressive and are often referred to as "malignant", although metastasis outside the central nervous system is exceedingly rare. Low-grade astrocytomas have an inherent tendency to undergo transformation to more aggressive high-grade tumours, associated with loss of cell cycle control and increased angiogenesis. WHO grade III astrocytomas show increased cellularity, mitotic activity and nuclear atypia, becoming more atypical with progressive anaplasia [23]. Anaplastic astrocytomas are likely to show patchy contrast enhancement on MRI or CT due to blood–brain barrier breakdown associated with vascular proliferation, and bright T2 signal due to oedema [9,24]. Glioblastoma, WHO grade IV, is the most common form of astrocytic tumour and accounts for 60–75% of astrocytic tumours, affecting predominantly adults with a peak incidence between 45 and 75 years of age and occurring most commonly in the subcortical white matter of the cerebral hemispheres. Glioblastoma is an anaplastic cellular glioma with pleomorphic astrocytic cells with marked nuclear atypia and high mitotic rates. Glioblastomas are rapidly evolving tumours typically with neoplastic infiltration of adjacent normal brain tissue and solid proliferating tumour at the periphery. The rapid tumour growth results in spontaneous necrosis with pseudopalisading of tumour cells, quickly developing a necrotic tumour core. The essential diagnostic features that distinguish glioblastomas from lower grade gliomas are prominent endothelial proliferation forming multilayered vessels and/or necrosis [25,26]. On neuroimaging, glioblastomas commonly show extensive peritumoural oedema and marked peripheral gadolinium enhancement, indicating viable tumour surrounding extensive central necrosis and marked mass effect secondary to vasogenic oedema [9,24]. Variants of glioblastoma include giant cell glioblastoma displaying numerous multinucleated tumour cells, gliosarcoma with regions of glial and mesenchymal differentiation, small cell glioblastomas with highly monomorphic small densely packed round cells and glioblastomas with an oligodendroglial component that contains foci that resemble oligodendrogliomas [25,27]. Although primary and secondary glioblastomas are morphologically identical, primary glioblastomas arise de novo with a short clinical history (usually <3 months) without evidence of an earlier precursor lesion, typically in an older population. In contrast, secondary glioblastomas are less frequent (<10% of glioblastomas), progress over a period of years from an earlier lower grade astrocytoma and are more common in younger patients [28,29].
Oligodendroglial tumours include oligodendrogliomas and oligoastrocytomas and the WHO classification recognises two malignancy grades, namely grades II and III. Compared with astrocytomas, oligodendroglial tumours are more likely to have an indolent clinical course and to be chemosensitive [30-32]. Historically, oligodendroglial tumours account for 5–6% of all gliomas, but their reported incidence has risen in recent years, possibly owing to less stringent diagnostic criteria and the recognition of the clinical need to distinguish oligodendrogliomas from astrocytomas [33-36]. Oligodendrogliomas arise, with a peak incidence between 40 and 45 years of age, commonly in the cortex and white matter of the cerebral hemispheres, with 50–60% in the frontal lobes [37]. The distinguishing feature of these tumours is their uniform round nuclei with clear perinuclear halos, which result as an artefact of fixation, and the delicate branching patterns of their vasculature that resemble "chicken wire" [38,39]. Oligodendroglioma WHO grade II are diffusely infiltrating, well-differentiated tumours with moderate cellularity, occasional mitoses and nuclear atypia [39]. On neuroimaging, grade II oligodendrogliomas show hypointensity on T1 weighted and hyperintensity on T2 weighted MRI and frequently display calcification on CT, although this is not diagnostic [40]. Approximately 50% of grade II oligodendrogliomas show diffuse contrast enhancement [41,42]. In addition to rounded hyperchromatic nuclei, perinuclear halos and a network of branching vasculature, anaplastic oligodendrogliomas (WHO grade III) exhibit increased cellularity, mitotic activity and prominent microvascular proliferation and/or necrosis [43]. On imaging, they show heterogeneous patterns, owing to variable extents of cystic degeneration, haemorrhage, necrosis and calcification; contrast enhancement is usual but peripheral enhancement is uncommon [40]. Oligoastrocytomas are mixed gliomas with neoplastic cells that phenotypically resemble tumour cells in oligodendrogliomas or astrocytomas present either in discrete regions or diffusely admixed throughout the tumour [44] (Figure 2). Compared with WHO grade II oligoastrocytomas, anaplastic oligoastrocytomas (WHO grade III), have increased cellularity, mitotic activity, nuclear atypia and microvascular proliferation, but since the revision of the WHO classification system in 2007, those with necrosis are considered glioblastomas with an oligodendroglial component [26,45]. Anaplastic oligodendrogliomas and oligoastrocytomas may arise either de novo or through progression from a lower grade tumour [46]. Gliomas display a spectrum of morphological appearances ranging from those with classic features of pure oligodendroglioma at one extreme to typical astrocytomas at the other, resulting in a diagnostic challenge for all but the classic oligodendrogliomas and astrocytomas and considerable variation between pathologists; diagnosis of these gliomas remains controversial [18,38]. Therefore, in order to minimise errors and maximise treatment effects, a multidisciplinary approach is needed integrating clinical, neuroradiological, histological and molecular genetic data on individual patient's biopsies to reach a final diagnosis.
Figure 2.

Oligoastrocytomas. (a) Oligoastrocytoma World Health Organization (WHO) grade II showing a moderately cellular glioma composed of an oligodendroglial (left aspect) and an astrocytic component (right aspect) with calcification. (b) Oligoastrocytoma WHO grade III showing densely packed diffuse oligodendroglial and astrocytic cells with scattered mitoses.
Molecular pathogenesis
It is now recognised the classification of gliomas based on genetic alterations or gene expression profiles may act as an adjunct to histopathological diagnosis and in some instances more closely correlates with prognosis than histopathology assessment [33,47-50]. Indeed a single histological subtype may encompass more than one molecular subtype, each with different prognoses. Development of gliomas and their malignant transformation results from sequential accumulation of genetic alterations [4,7,48,51] (Figure 3). Genetic changes found in gliomas include amplification and/or overexpression of oncogenes and loss of tumour suppressor genes and DNA repair genes through mutation, loss of heterozygosity (LOH) or by epigenetic mechanisms such as promoter hypermethylation. These genetic changes result progressively in uncontrolled proliferation and loss of normal cell cycle control mechanisms, diminished ability to undergo apoptosis in response to genotoxic agents, failure of DNA repair mechanisms, increasing genetic instability and deregulation of growth factor signalling pathways. The molecular pathogenesis differs between adult and paediatric gliomas, as does the incidence of the various forms of glioma.
Figure 3.

Molecular pathogenesis of adult astrocytic and oligodendroglial neoplasms. The illustration shows the progression of low-grade astrocytomas and oligodendrogliomas to higher grade with sequential accumulation of genetic alterations and impact on the biological properties of these tumours. Genetic alterations seen in lower grade tumours are retained on progression. Anaplastic oligodendrogliomas may arise through progression or de novo, but irrespective of route have similar clinical behaviour and molecular genetic characteristics with 1p/19q loss as their genetic hallmark. Glioblastomas arise de novo or progress from lower grade astrocytomas. Although indistinguishable clinically, they may be separated by their spectrum of genetic alterations, but these genetic alterations are not mutually exclusive to either lineage. The most common genetic alterations used to distinguish molecular subtypes of glioma are shown in red. AII, astrocytoma WHO grade II; AIII, astrocytoma WHO grade III; amp, amplification; del, deletion; GBM, glioblastoma WHO grade IV; meth, methylation; mut, mutation; OII, oligodendroglioma WHO grade II; OIII, oligodendroglioma WHO grade III. OE, overexpression.
Adult gliomas
TP53 mutation is by far the most common genetic alteration in adult diffuse astrocytomas WHO grade II, accompanied by LOH of the other allele at 17p13 in over 60% of these tumours [48,52], affecting the G1 cell cycle checkpoint. Low-grade astrocytomas show overexpression of platelet-derived growth factor (PDGF) ligands and receptors, which may stimulate cell proliferation [48,53]. Additional genetic changes seen in a proportion of these tumours include LOH of 22q and gain of chromosome 7 and amplification of 8q [48,54]. Progression to anaplasia is accompanied by a variety of genetic changes in genes that regulate the G1 cell cycle checkpoint, such that this control point is aberrant in all high-grade astrocytomas. These genetic changes include TP53 mutation, retinoblastoma gene (Rb1) mutation, deletion of CDKN2A or CDKN2B, which code for the negative regulators of G1 to S phase cell progression p16INK4A and p15INK4b, and cyclin-dependent kinase (CDK) 4 and CDK6 amplification. Other genetic deletions in addition to 17p and 22q may include deletions on chromosomes 6, 9p, 11p and 19q [48]. LOH 10q and phosphatase and tensin homologue on chromosome 10 (PTEN) mutation, both of which are associated with poor prognosis [55,56] are seen in 35–60% and 18–23% of anaplastic astrocytomas respectively [55,57-59]. Glioblastomas exhibit the greatest variety of genetic alterations and these can be used to distinguish between glioblastomas that have progressed from an earlier astrocytoma (secondary glioblastomas) and those that have arisen de novo [25,29,48]. Secondary glioblastomas are likely to have TP53 mutation (65%), loss of 17p, overexpression of PDGF receptor (PDGFR), abnormalities in the p16 and Rb1 pathways and LOH 10q. Glioblastomas that arise de novo are characterised by epidermal growth factor receptor (EGFR) amplification, genetic alterations that disrupt cell cycle control, such as p14ARF or p16INK4a deletions or MDM2 and MDM4 amplification or overexpression and infrequently TP53 mutation. LOH of 10q and 10p resulting in complete loss of one copy of chromosome 10 and deletions or mutations in the PTEN gene that negatively regulates the PI3kinase pathway are also likely [25,48,60]. Although distinct genetic alterations may be used to distinguish primary and secondary glioblastomas, these changes are not mutually exclusive and similar molecular pathways are affected.
EGFR is a transmembrane protein responsible for transmission of signal from its extracellular ligands, epidermal growth factor (EGF) and tumour necrosis factor alpha (TNF-α) through tyrosine kinase activity and signal transduction pathways to regulate cell proliferation. EGFR amplification, which is rare in secondary glioblastomas, occurs in 40% of primary glioblastomas and results in overexpression of the EGFR protein [60,61]. 20–50% of glioblastomas with EGFR amplification have a mutated form, EGFRvIII, which codes for a tumour-specific truncated and constitutively active autophosphorylated receptor with tyrosine kinase activity and may confer an unfavourable prognosis [25,62]. EGFR and EGFRvIII are potentially important targets for biological therapy in glioblastomas.
The most common genetic change seen in oligodendroglial tumours is a combined loss of chromosome arms 1p and 19q observed in the majority of oligodendrogliomas (50–80%) irrespective of grade [33,51], suggesting this is an early event in the development of these tumours. This genetic alteration arises through an unbalanced centromeric translocation between chromosomes 1p and 19q, resulting in complete loss of the 1p and 19q arms [63]. Loss of 1p and 19q is uncommon in other gliomas and is now considered the genetic hallmark of oligodendrogliomas [64]. However, it is important to distinguish tumours with the 1p/19q co-deletion from those with an oligodendroglial phenotype and 1p partial deletions, typically 1p36, which occur in astrocytic tumours and are associated with poor prognosis [65]. Complete loss of 1p and 19q is associated with good prognosis, prolonged survival [46,66,67] and sensitivity to chemotherapy, and radiotherapy has been shown to be a better predictor of response to therapy and overall survival than conventional histopathology [31,68-71]. Genetic testing for combined losses of 1p and 19q is now being used clinically to distinguish tumours of oligodendroglial and astrocytic genetic lineages, as a predictive and prognostic marker, and in the stratification of clinical trials. Similar to astrocytomas, progression to anaplasia in oligodendrogliomas is associated with genetic alterations that regulate cell cycle control, including Rb1, CDKN2A, CDKN2B and p14ARF, but TP53 mutations and gene amplifications are rare [33,72]. These tumour suppressor genes are frequently silenced in oligodendroglial tumours by promoter hypermethylation [73]. Loss of 10q occurs in approximately 10–20% associated with poor prognosis [31,33,46,74], and occasionally PTEN mutation is observed [74]. Additional genetic alterations include gains on chromosomes 7 and 15q, and losses on 4q, 6, 9p, 11, 13q, 18 and 22q [33,75]. Oligoastrocytomas can be divided into two groups, those with loss of 1p and 19q resembling oligodendrogliomas (∼50%), or those with loss of 17p and TP53 mutation typical of astrocytomas (∼33%) [33,76], suggesting a clonal origin of these tumours [77,78]. Anaplastic oligoastrocytomas show similar genetic alterations to anaplastic tumours of the oligodendroglial or astrocytic lineages [33,51].
O6-methylguanine-DNA methyltransferase (MGMT) is a DNA repair enzyme that removes alkyl groups from the O6 position of guanine in DNA following alkylation and methylation by agents such as nitrosoureas and temozolomide. The MGMT gene, at 10q26, is silenced by epigenetic hypermethylation of the gene promoter in approximately 50% of glioblastomas [79], whereas more frequent and extensive methylation is found in oligodendroglial tumours with 1p/19q loss [80]. The consequent reduction in MGMT expression is now believed to contribute to chemosensitivity. Glioblastomas with MGMT promoter hypermethylation are more likely to have longer survival when treated with concurrent temozolomide and radiotherapy [79], while recent research suggests the extent of methylation may influence survival [81]. Tumours with MGMT promoter hypermethylation are more likely to show radiation or treatment-induced pseudoprogression immediately after treatment, which is difficult to distinguish from disease progression by conventional MRI [82,83].
More recently, through whole-genome sequencing, heterozygous point mutations were found in the IDH1 gene in 12% glioblastomas associated with younger age, secondary glioblastomas and increased survival [84]. IDH1 on chromosome 2q33, encodes isocitrate dehydrogenase 1, which catalyses the oxidative carboxylation of isocitrate to α-ketoglutarate, resulting in the production of cytosolic nicotinamide adenine dinucleotide phosphate (NADPH). All of the mutations were at codon 132, which lies within the active site, with the majority being a guanine to adenine transition resulting in the amino acid substitution of arginine to histidine (IDHR132H), which impairs the enzyme's affinity for its substrate through heterodimer formation and dominantly inhibits wild-type IDH1 activity. Mutations in IDH1 confer tumour suppressor activity and contribute to tumourigenesis through induction of the hypoxia-inducible factor 1 (HIF-1) pathway [85]. Point mutations in IDH1 and less commonly in the related IDH2 gene (at codon 172) are frequently found in diffusely infiltrating gliomas and are rare in all other central nervous system (CNS) and non-CNS tumours [86]. IDH1 mutations are found in ∼68, 74 and 77% of grades II and III astrocytomas, oligodendrogliomas and oligoastrocytomas, respectively and in secondary glioblastomas that develop from these lower grades tumours [87-90]. The gene mutation appears to be an early event in gliomagenesis, occurring before the acquisition of TP53 mutation or 1p/19q loss [90]. IDH1/IDH2 mutations are tightly correlated with co-deletion of 1p/19q and MGMT methylation, but are uncommon in tumours with EGFR amplification [91]. IDH1/IDH2 mutations are present in 78% secondary glioblastomas but only 5% of primary glioblastomas [87-90], and are associated with favourable prognosis in grade III astrocytomas and glioblastomas, and in WHO grades II, III and IV gliomas [89,91,92].
Classification of gliomas into molecular subtypes may be based not only on DNA-based genetic alterations, but also on transcription profiles through genome-wide gene expression microarrays using RNA extracted ideally from snap frozen tumour tissue. Gene expression profiles have identified clinically relevant subgroups of gliomas [49,50], distinguished oligodendroglial tumours with 1p/19q loss from those with intact 1p/19q [93,94], and between primary and secondary glioblastomas [95] and identified expression profiles associated with good prognosis [96-99].
Paediatric gliomas
Although gliomas account for around 12% of cancers in children under 15 years of age, relatively few studies have been performed on their molecular characteristics [100]. Low-grade gliomas (LGGs), most of which are juvenile pilocytic astrocytomas (JPA), constitute the majority and specific variants are associated with heritable diseases. Neurofibromatosis 1 (NF1) is caused by mutations within the neurofibromin gene located on chromosome 17q. In up to 15% of cases NF1 is associated with LGG of the optic tract and hypothalamus [101,102]. This suggests a role for NF1 or its signal transduction pathway (the mitogen-activated protein kinase (MAPK) pathway) in the development of sporadic JPA through ras inhibition, but this has not been proven. Chromosomal copy number changes are found in a proportion of pilocytic astrocytomas, but genetic alterations were more frequent and extensive in those older than 15 years [103]. Recently the MAPK pathway as been further implicated in the tumourigenesis of pilocytic astrocytomas. Jones et al [104] demonstrated a novel tandem duplication at locus 7q34 leading to the fusion of KIAA1549 and BRAF in 66% of pilocytic astrocytomas. This in combination with wild-type IDH1/2 has been shown to distinguish pilocytic astrocytomas from grade II astrocytomas at a molecular level [105]. Tandem duplication at 3p25 leading to MAPK activation through a fusion between SRGAP3 and RAF1 has been reported in approximately 2% of pilocytic astrocytomas [106]. Low-grade astrocytoma is also a feature of Li–Fraumeni syndrome, which is triggered by a TP53 germline mutation. TP53 mutations are found in up to 19% of sporadic grade II diffuse paediatric astrocytomas [107]. Few other molecular genetic abnormalities have been identified in paediatric LGG. In particular EGFR gene amplification and overexpression of the protein is rarely seen [107]; however Tabori et al [108] showed a high rate of EGFR amplification and overexpression in tumours from children with LGG disseminated within the CNS.
In contrast to adults, little is known regarding the progression from low to high grade, as malignant transformation is rare (<10%). A recent study by Broniscer et al [109] investigated 11 patients with progression from a previous LGG. Small numbers of assessable patients precluded any statistically significant conclusions but p53 overexpression was observed more frequently after transformation and PTEN deletion was found after transformation in over 50%. De novo high-grade astrocytomas are much less common in children, and their molecular abnormalities are reminiscent of secondary glioblastoma in adults [110]. The mechanisms of tumourigenesis in children are distinct from those in adults as evidenced by the presence of other genetic differences, the rarity of progression in the paediatric group and recent studies using comparative genomic hybridisation [111]. Various genetic pathways have been implicated including p53, EGFR, Rb, PI3K, MGMT and mismatch repair (MMR). Overexpression of p53 and TP53 mutations are associated with adverse outcomes [110]. The former is found in one-third of paediatric patients and increases with tumour grade [110]. TP53 mutations are significantly less common in children under 3 years of age than in older children [112]. Paediatric HGGs infrequently show EGFR amplification, a common feature of adult de novo glioblastomas [113-115]. However, EGFR overexpression is present in up to 85% of paediatric supratentorial HGGs [113,115]. RNA expression studies have shown overexpression of EGFR and other genes associated with angiogenesis [116]. The role of the Rb1 pathway in paediatric HGGs is not well characterised despite disruption being observed in the majority of adult HGGs. In particular p16 inactivation is only observed in 9% of paediatric tumours (50–70% adults) and CDK4 amplification in 6% of paediatric HGGs (15% in adults) [114]. PTEN mutations have been observed in 6% of paediatric anaplastic astrocytomas and 20% of glioblastomas and have been associated with reduced survival [114]. Donson et al [117] recently indicated that children with glioblastomas with methylated MGMT may also benefit from temozolomide, and this marker may be a prognostic factor for survival. Microsatellite instability is used as a surrogate marker for defects in MMR genes and has been found to be markedly more common in paediatric HGGs while being rarely exhibited in adult tumours [118]. Paediatric oligodendrogliomas and oligoastrocytomas are uncommon and their genetics has not been characterised extensively; however, 1p/19q loss is rarely seen [119].
Origins of glioma and stem cells
To date, neither histopathological classification nor molecular genetic characterisation has been able to adequately predict or account for the resistance displayed by the majority of gliomas to radiotherapy and chemotherapy and the early recurrence of those that do respond to therapy. The cellular origin of gliomas remains a topic of debate. One hypothesis is that gliomas arise from neoplastic transformation and dedifferentiation of mature glial cells, astrocytes, oligodendrocytes and ependymal cells, giving rise to tumours resembling these cells [15,120]. However, evidence to support this mechanism of gliomagenesis is limited and does not account for mixed gliomas. Alternatively, the cancer stem cell hypothesis suggests tumour cells with stem-cell like properties are present within tumours and are responsible for their initiation and continued repopulation. It was previously thought the only dividing cells within the adult brain were glial cells. It is now known there are both neural stem cells and glial progenitor cells in multiple regions of the adult brain, including the subventricular zone (SVZ), the dentate gyrus, hippocampus and subcortical white matter [121-125]. Self-renewing neural stem cells are able to divide asymmetrically [126] and their progenitors are capable of differentiating along either neuronal or glial lineages to produce mature differentiated cells [121,123]. Neural stem cells and glial progenitor cells have migratory potential and there is increasing evidence that neoplastic transformation of these cells gives rise to the various types of glioma [121,127] (Figure 4). Gene expression research supports the concept that histologically similar tumours may be molecularly distinct because they arise from distinct populations of site-restricted progenitors [128]. In addition, MRI studies suggest the spatial relationship of glioblastomas with the SVZ may influence invasiveness and patterns of recurrence [129]. Brain tumour stem cells, which are also referred to as brain tumour-initiating cells or brain tumour stem-like cells (BTSCs), have been identified in glioblastomas [130,131], low-grade astrocytomas [132], anaplastic oligoastrocytomas [133], ependymomas [134] and established cell lines [135] on the basis of their ability to grow as neurospheres in vitro and/or expression of stem cell markers, such as CD133 (promin1), nestin and musashi-1. These BTSCs, present as only a minor subset of the total tumour population, are capable of self-renewal, i.e. generate new stem cells by either asymmetric or symmetric cell division, and generate new tumours when orthoptically transplanted into immune-compromised mice [132,136]. BTSCs are particularly chemo- and radioresistant [137-139], possibly owing to their increased capacity to repair DNA damage [140], and expression of drug transporters [141,142]. BTSCs contribute to tumour angiogenesis [143], while the tumour vascular microenvironment regulates BTSC behaviour [144]. There is increasing evidence that therapeutic resistance is due to the presence of stem cells within gliomas and these cells should be a key therapeutic target for eradication of the tumour [145]. Gene expression profiling has been used to identify cell signalling pathways active in stem cells derived from glioblastomas [146] and ependymomas [134]. Stem cell-related "self-renewal" gene expression signatures have been associated with resistance to concomitant chemoradiotherapy also in glioblastomas [97]. Such research is essential for the identification of therapeutic targets to eliminate cancer stem cells. By the same context, future imaging research should be directed to the development of imaging biomarkers associated with the presence of stem cells within gliomas.
Figure 4.
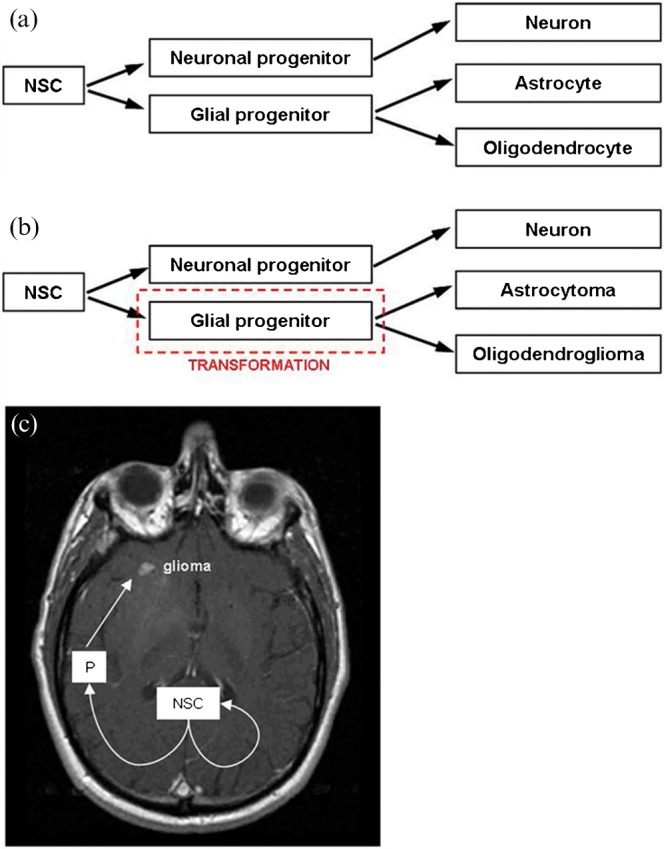
Neural stem cell and progenitor cell theory of glioma development. (a) normal cell development. (b) astrocytoma, oligodendroglioma and other glioma subtype development may follow neoplastic transformation of a glial progenitor cell resulting in neoplastic cells that phenotypically resemble normal cells. (c) asymmetrical division of neural stem cell results in a cell population, which remains at the germinal origin, and a progenitor cell population, which migrates and proliferates causing distant glioma (adapted from Sanai et al [121] and Berger et al [127]). NSC, neural stem cell; P, progenitor cell.
Glioma biology and imaging
Histological grade
The histological features used to classify gliomas are reflected macroscopically in their imaging characteristics, which may be used as a non-invasive tool to distinguish tumour from normal brain and to predict grade and thereby prognosis. The proliferation rate of gliomas, reflected in their mitotic index and frequently measured immunohistochemically by labelling S-phase cells via the Ki-67 or MIB-1 antigens, increases progressively from low to high grade. This increased cell division is associated with elevated choline signal in proton magnetic resonance spectroscopy (MRS), reflecting cell membrane phospholipid turnover [147] and increased uptake of amino acids and glucose in PET [148]. Increased cellularity in HGGs results in fewer neurones within tumour tissues and a decreased N-acetylaspartate signal in MRS [147,149], as well as decreased apparent diffusion coefficient (ADC) in diffusion-weighted imaging, reflecting more solid tissue with decreased water mobility [9,150]. High growth rates in HGGs necessitate increased metabolism, which becomes anaerobic as growth outstrips oxygen supply, and hypoxia and necrosis result, associated with elevated lipid and lactate signals in MRS [9,10].
The vasculature is a key feature in the histopathology diagnosis of gliomas and permits imaging associations with grade through perfusion-weighted imaging. The vessels within tumour tissue in LGGs may be somewhat more numerous than in normal brain and may adopt "chicken-wire" patterns in oligodendrogliomas, but vessel structure is unaltered and the blood–brain barrier remains intact. Neovascularisation in gliomas is associated with malignant transformation and biological aggressiveness, resulting through angiogenesis, intussusception and vessel co-option regulated by secretion of endothelial growth factors in response to hypoxia and degradation and remodelling of extracellular matrix macromolecules, leading to loss of blood–brain barrier integrity [151,152]. Vessels in glioblastomas are tortuous, disorganised and highly permeable, with larger lumens and abnormalities in the vessel wall, pericyte coverage and basement membrane. Aberrant microvasculature typically appears as "glomeruloid tufts" consisting of multilayered, mitotically active endothelial cells and perivascular cells [26,152]. However, these changes are not uniform, leading to considerable spatial and temporal phenotypic heterogeneity in blood vessel architecture within HGGs, which is reflected in perfusion-weighted MRI. The distinction of tumour tissue from normal brain and between gliomas of different grade has been demonstrated by a variety of perfusion parameters. These include relative cerebral blood volume (rCBV), relative cerebral blood flow (rCBF) and contrast transfer coefficient (Ktrans), which measures vascular permeability [10,11,153,154].
Molecular subtypes of glioma
The application of molecular genetic markers to classify gliomas has led to investigations comparing the imaging characteristics of molecular subgroups.
1p/19q genotype
As described above, oligodendroglial tumours are typified by 1p/19q loss, whereas astrocytomas are more likely to have TP53 mutation and loss of 17p, providing a more robust means to distinguish oligodendrogliomas from astrocytomas than conventional histopathological classification. This has prompted numerous studies to investigate whether imaging might provide a non-invasive means to distinguish these genetic subtypes. A preferential distribution of oligodendroglial tumours with TP53 mutation to the temporal lobe and those with 1p/19q to the frontal, parietal and occipital [155,156] or frontal [157] lobes or non-temporal locations [76] has been reported. However, other studies found no significant differences between genotype and tumour location [46,67,158]. In conventional MR studies oligodendrogliomas with 1p/19q loss were more likely to have indistinct borders on T1 weighted images, mixed signal intensities on T1 and T2, paramagnetic susceptibility effect and calcification [14]. However, T1 and T2 signal inhomogeneity was associated with intact 1p/19q in oligoastrocytomas [76]. In a similar study of oligodendroglial tumours diagnosed by frame-based serial stereotactic biopsy, assessment of in vivo histological growth patterns in relation to co-registered clinical imaging characteristics showed tumours with intact 1p/19q were more likely to have an infiltrative growth pattern [12]. Transition in cellularity across the tumour margin was similar irrespective of genotype; however, those with a sharp/smooth border on T2 weighted imaging (Figure 5a,b) and homogeneous signal intensity on T1 and T2 were more likely to have intact 1p/19q [12]. To provide a more objective method to asses differences in imaging characteristics, Brown et al [159] have used a quantitative method of MR analysis based on an S-transform to measure image texture in a series of low-grade oligodendroglial tumours in relation to genotype (Figure 6). Differences in texture were seen between tumours with 1p/19q loss and those with intact 1p/19q on contrast-enhanced T1 and T2 weighted MRI, enabling non-invasive discrimination of genotype with high sensitivity and specificity. The biological basis of these differences in imaging characteristics on anatomical MRI is as yet unknown, although Megyesi et al [14] postulated that differences in signal intensity and tumour borders may be due to increased "invasiveness" in tumours with loss of 1p/19q.
Figure 5.

Multimodal imaging characteristics of oligodendroglial tumours classified by genotype. (a, b) T2 weighted MRI. (c, d) Negative enhancement integral colour maps used to derive relative cerebral blood volume (rCBV). (e, f) Apparent diffusion coefficient (ADC) maps. The left panel shows 1p/19q loss tumours exhibiting indistinct, (a) irregular T2 border, (c) high rCBV and (e) low histogram ADC. The right panel shows 1p/19q intact tumours displaying sharp, (b) smooth T2 border, (d) low rCBV and (f) high histogram ADC.
Figure 6.

Analysis of image texture in relation to genotype. (a, b) T2 and fluid-attenuated inversion-recovery weighted images of an oligodendroglioma with loss of 1p and 19q and (c, d) an oligodendroglioma with intact 1p and 19q. The similar-looking images were correctly classified with regard to genotype using texture analysis. Images are cropped to 128 × 128 pixels (green) before being transformed; ROIs are as indicated (pink). Reproduced with permission from Brown et al. [159].
Distinction between oligodendroglial genotypes has also been observed using multimodal advanced MRI. Using dynamic susceptibility contrast MRI (DSC-MRI) in a study of 37 oligodendroglial neoplasms, tumours with 1p/19q loss were more likely to have high rCBV than those with intact 1p/19q (Figure 5c,d) with receiver operating characteristic analysis revealing a cut-off of 1.59 for identifying genotype with sensitivity 92% and specificity 76% [160]. In this study rCBV was determined by placing regions of interest over areas of highest blood volume to derive rCBVmax. Subsequently elevated rCBV in association with 1p/19q loss has been supported in other small series of oligodendroglial tumours measuring rCBV [161-163] or using histogram analysis [164]. These data warrant further study in larger series to determine the potential of rCBV measurements for the non-invasive prediction of genotype. In histopathology comparisons, rCBV was higher in low-grade oligodendrogliomas than astrocytomas [165], but similar in some, but not all studies, of low and high grade oligodendrogliomas [166,167], rendering DSC-MRI-based tumour grading potentially inaccurate [160,166,168]. Therefore, unlike astrocytomas, high rCBV measurements may not be indicative of aggressive biology in oligodendroglial tumours with 1p/19q loss, suggesting differences in the baseline biology of the two genetic subtypes of glioma. These differences may be attributable to the elevated microvessel density seen in both low- and high-grade oligodendrogliomas and may reflect their "chicken wire" vasculature [169,170]. In diffusion MRI, ADC has been shown to vary according to 1p/19q status [171]. Oligodendroglial tumours with intact 1p/19q were more likely to have a higher maximum and histogram ADC and a greater ADC transition coefficient (ATC) (Figure 5e,f). Similarly, Tozer et al [172] have reported that low-grade oligodendrogliomas had a lower ADC histogram than astrocytomas. These differences may reflect the cellularity of these tumours, as astrocytomas have a tendency towards lower cell density and to be more cystic than oligodendrogliomas, whereas tumours with intact 1p/19q are more likely to show infiltrative growth patterns [12]. Alternatively differences in ADC may reflect the extracellular matrix composition of these tumours; indeed, a recently published study of oligodendroglial genotype failed to demonstrate any relationship between a quantitative assessment of cellularity and ADC [173].
In contrast, to perfusion and diffusion MRI, oligodendroglial tumours with 1p/19q loss could not be distinguished from those with intact 1p/19q using routine clinical MRS and measurement of N-acetylaspartate, choline, myoinositol or lipid and lactate metabolite ratios [174]. However, in another study, lipid, lactate, glutamine and glutamate metabolite ratios distinguished low-grade oligodendrogliomas from astrocytomas [175].
Metabolic imaging with radiolabelled tracers has been used in a number of glioma studies to yield diagnostic or prognostic information [148], but few studies have so far related metabolism to genotype. Brain tumour metabolism was assessed by single-photon emission CT (SPECT) in oligodendroglial neoplasms characterised by genotype using thallium-201 (201Tl) and [18F]-fluorodeoxyglucose (FDG) [42]. 201Tl has a low uptake in normal cerebral tissue because of restricted passive diffusion across the blood–brain barrier. Its uptake in gliomas is thought to depend on disruption of the blood–brain barrier and activity of the Na/K ATPase pump, indicating cell viability and to a lesser extent blood flow. FDG, an analogue of glucose, is transported into the cell by facilitated diffusion, where it is subsequently phosphorylated by hexokinase to fluoroglucose-6-phosphate, but unlike glucose, this is not a substrate for further metabolism and becomes trapped intracellularly. There was a positive correlation between 201Tl and FDG uptake, and between metabolic rate and both histological grade and contrast enhancement, although some low-grade oligodendroglial tumours showed high metabolism. There was a clear association between metabolism and genotype. Oligodendroglial tumours with 1p/19q loss were more likely to show increased 201Tl uptake and, to a lesser extent, increased FDG uptake than those with intact 1p/19q [42]. In support of these observations, other studies report high FDG metabolic activity in low-grade oligodendrogliomas [176,177]. Stockhammer et al [178] have recently investigated FDG uptake by PET and shown raised glucose utilisation in six of eight grade II gliomas with 1p/19q loss compared with zero of eight with intact 1p/19q. In a case study, a spinal oligoastrocytoma with 1p/19q loss had high glucose uptake [179]. These data suggest increased metabolic activity in oligodendrogliomas with 1p/19q loss. In summary, imaging data have revealed differences between gliomas of astrocytic and oligodendroglial genetic lineages that must reflect differences in their biology, but which are as yet poorly understood. In addition non-invasive imaging molecular diagnosis of oligodendrogliomas may potentially have future clinical utility.
Primary and secondary glioblastomas
Evidence that molecular subtypes of glioblastoma may be distinguished by MRI is emerging. Aghi et al [180] categorised glioblastomas according to EGFR amplification (primary glioblastomas) and mutations in exons 5–8 of the TP53 gene (secondary glioblastomas) and compared this to four imaging variables: (a) T2/T1, the ratio of T2 bright volume to enclosed T1 enhancing volume; (b) percentage of tumour volume that was necrosis; (c) and (d) T1 and T2 border sharpness coefficients (BSCs). The percentage necrosis and T1 BSC did not differ between glioblastoma subtypes, but EGFR-amplified tumours had increased T2/T1 ratio and decreased T2 BSC, suggesting increased angiogenesis, oedema and/or invasion through alterations in EGFR signalling pathways in EGFR-overexpressing tumours (Figure 7). EGFR-amplified tumours exhibit elevated oedema, possibly through EGFR activation of hypoxia-inducible transcription factor, potentially resulting in increased angiogenesis, due to increased secretion of vascular endothelial growth factor (VEGF) and increased invasion of surrounding normal brain, possibly due to matrix metalloproteinase activity [116,181-183]. In a similar study, imaging characteristics of glioblastomas were correlated with expression of the p53 protein measured immunohistochemically. Tumours with greater than 50% of cells showing p53 immunopositivity were more likely to show ring enhancement and well-defined borders on T1 weighted images with contrast than tumours with lower proportions of p53-positive cells [184]. Glioblastomas with unmethylated MGMT were more likely to show ring enhancement and differed from MGMT methylated tumours in texture analysis using S-transform [185]. These data support the distinction of molecular subtypes of glioblastoma through conventional MRI.
Figure 7.

Border sharpness coefficient (BSC) in glioblastomas. The tumour shown in (a) had the largest T2 BSC, signifying a sharp T2 bright border, and proved to be non- epidermal growth factor receptor (EGFR)-amplified, whereas the tumour in (b) had the smallest T2 BSC, signifying a fuzzy T2 bright border, and proved to be EGFR-amplified Reproduced with permission from Aghi et al. [180].
Gene expression and imaging
Many imaging studies have sought to explain the imaging findings in terms of the histopathological and biological characteristics of the tumour tissue. For example [11C]-methione uptake measured by PET correlated with the proliferative activity measured immunohistochemically in LGGs [186] and rCBV correlated with tumour grade and the expression of VEGF in non-enhancing gliomas [187]. However, most studies suffer the limitation that imaging and histology are likely to be heterogeneous and the sample analysed may not be from the tumour region from which imaging signatures are derived. Additionally, most studies have used a candidate approach, looking only at specific aspects of histology, biology or expression of single gene markers. These limitations are now being addressed through image-guided sample acquisition and expression profiling studies. Using computer-assisted frameless navigation to obtain samples from tumour regions with defined imaging characteristics, Hobbs et al [188] demonstrated differences in the protein expression profiles from enhancing and non-enhancing tumour regions. Similarly van Meter et al [189] obtained image-guided biopsy specimens from the tumour core and enhancing periphery of untreated glioblastomas and used microarray gene expression profiling to demonstrate intratumoural regional differences in molecular phenotype, with many of the genes identified as differentially expressed being potential therapeutic targets, e.g. EGFR, VEGF and matrix metalloproteinases. More recently, upregulation of genes associated with hypoxia, angiogenesis and oedema was observed in biopsies from completely enhancing compared with incompletely enhancing glioblastomas [190]. In another similar study, Barajas et al [191] showed differences in rCBV and ADC between enhancing and non-enhancing peritumoural regions in glioblastomas and in the gene expression profiles of biopsies obtained from these regions, with upregulation of biological processes associated with mitosis, angiogenesis and apoptosis within contrast-enhancing regions. Diehn et al [13] have combined neuroimaging and microarray analysis to create a multidimensional map of gene expression patterns in glioblastoma that provided clinically relevant insights into tumour biology. Tumour contrast enhancement and mass effect predicted activation of specific hypoxia and proliferation gene-expression programmes, respectively (Figure 8), and an infiltrative imaging phenotype was identified that predicted shorter survival. These studies demonstrate the association of complex molecular signatures with readily identifiable imaging characteristics.
Figure 8.

Gene expression surrogates for MRI traits. (a) Association between the hypoxia gene expression module and contrast enhancement. Tumour arrays were clustered by using only cDNA clones contained within the gene module. The value of the imaging trait for each tumour is indicated by the coloured box above the expression map. Representative MRI are depicted on the left and a subset of named genes is labelled. (b) expanded view of the association between the proliferation gene-expression module and mass effect. Reproduced with permission from Diehn et al [13].
Conclusion
In summary, molecular pathology research and advances in non-invasive multimodal imaging techniques have greatly increased our understanding of the relationship between genotype and histological and imaging phenotypes in diffusely infiltrating gliomas. Further research is important to develop the clinical potential for non-invasive multimodal MRI techniques to discriminate histological and molecular subtypes of glioma, and to decipher the molecular processes giving rise to macroscopic imaging characteristics, which are essential for the development of new imaging biomarkers and discovery of new clinically relevant therapeutic targets.
References
- 1.Burnet NG, Jefferies SJ, Benson RJ, Hunt DP, Treasure FP. Years of life lost (YLL) from cancer is an important measure of population burden—and should be considered when allocating research funds. Br J Cancer 2005;92:241–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, van denBent MJ, Mason WP, Weller M, Mirimanoff RO, et al. Changing paradigms—an update on the multidisciplinary management of malignant glioma. Oncologist 2006;11:165–80 [DOI] [PubMed] [Google Scholar]
- 3.Colman H, Aldape K. Molecular predictors in glioblastoma: toward personalized therapy. Arch Neurol 2008;65:877–83 [DOI] [PubMed] [Google Scholar]
- 4.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol 2006;1:97–117 [DOI] [PubMed] [Google Scholar]
- 5.Collins VP. Mechanisms of disease: genetic predictors of response to treatment in brain tumors. Nat Clin Pract Oncol 2007;4:362–74 [DOI] [PubMed] [Google Scholar]
- 6.Mason WP, Cairncross JG. Invited article: the expanding impact of molecular biology on the diagnosis and treatment of gliomas. Neurology 2008;71:365–73 [DOI] [PubMed] [Google Scholar]
- 7.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 2007;21:2683–710 [DOI] [PubMed] [Google Scholar]
- 8.Cao Y, Sundgren PC, Tsien CI, Chenevert TT, Junck L. Physiologic and metabolic magnetic resonance imaging in gliomas. J Clin Oncol 2006;24:1228–35 [DOI] [PubMed] [Google Scholar]
- 9.Jenkinson MD, Du Plessis DG, Walker C, Smith TS. Advanced MRI in the management of adult gliomas. Br J Neurosurg 2007;21:550–61 [DOI] [PubMed] [Google Scholar]
- 10.Price SJ. The role of advanced MR imaging in understanding brain tumour pathology. Br J Neurosurg 2007;21:562–75 [DOI] [PubMed] [Google Scholar]
- 11.Jackson A, O'Connor JP, Parker GJ, Jayson GC. Imaging tumor vascular heterogeneity and angiogenesis using dynamic contrast-enhanced magnetic resonance imaging. Clin Cancer Res 2007;13:3449–59 [DOI] [PubMed] [Google Scholar]
- 12.Jenkinson MD, du Plessis DG, Smith TS, Joyce KA, Warnke PC, Walker C. Histological growth patterns and genotype in oligodendroglial tumours: correlation with MRI features. Brain 2006;129:1884–91 [DOI] [PubMed] [Google Scholar]
- 13.Diehn M, Nardini C, Wang DS, McGovern S, Jayaraman M, Liang Y, et al. Identification of noninvasive imaging surrogates for brain tumor gene-expression modules. Proc Natl Acad Sci USA 2008;105:5213–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Megyesi JF, Kachur E, Lee DH, Zlatescu MC, Betensky RA, Forsyth PA, et al. Imaging correlates of molecular signatures in oligodendrogliomas. Clin Cancer Res 2004;10:4303–6 [DOI] [PubMed] [Google Scholar]
- 15.Martin-Villalba A, Okuducu AF, von Deimling A. The evolution of our understanding on glioma. Brain Pathol 2008;18:455–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleihues P, Louis DN, Wiestler OD, Burger PC, Scheithauer BW. WHO grading of tumours of the central nervous system. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO Classification of tumours of the central nervous system. Lyon, France: IARC Press, 2007: 10–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giannini C, Scheithauer BW, Weaver AL, Burger PC, Kros JM, Mork S, et al. Oligodendrogliomas: reproducibility and prognostic value of histologic diagnosis and grading. J Neuropathol Exp Neurol 2001;60:248–62 [DOI] [PubMed] [Google Scholar]
- 18.Kros JM, Gorlia T, Kouwenhoven MC, Zheng PP, Collins VP, Figarella-Branger D, et al. Panel review of anaplastic oligodendroglioma from European Organization For Research and Treatment of Cancer Trial 26951: assessment of consensus in diagnosis, influence of 1p/19q loss, and correlations with outcome. J Neuropathol Exp Neurol 2007;66:545–51 [DOI] [PubMed] [Google Scholar]
- 19.Murakami R, Hirai T, Kitajima M, Fukuoka H, Toya R, Nakamura H, et al. Magnetic resonance imaging of pilocytic astrocytomas: usefulness of the minimum apparent diffusion coefficient (ADC) value for differentiation from high-grade gliomas. Acta Radiol 2008;49:462–7 [DOI] [PubMed] [Google Scholar]
- 20.Scheithauer BW, Hawkins C, Tihan T, VandenBerg SR, Burger PC. Pilocytic astrocytoma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous system. Lyon, France: IARC Press, 2007: 14–21 [Google Scholar]
- 21.Lefranc F, Brotchi J, Kiss R. Possible future issues in the treatment of glioblastomas: special emphasis on cell migration and the resistance of migrating glioblastoma cells to apoptosis. J Clin Oncol 2005;23:2411–22 [DOI] [PubMed] [Google Scholar]
- 22.von Deimling A, Burger PC, Nakazato Y, Ohgaki H, Kleihues P. Diffuse astrocytoma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous system. Lyon, France: IACR Press, 2007: 25–39 [Google Scholar]
- 23.Kleihues P, Burger PC, Rosenblaum MK, Paulus W, Scheithauer BW. Anaplastic astrocytoma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous system. Lyon, France: IARC Press, 2007: 30–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rees J. Advances in magnetic resonance imaging of brain tumours. Curr Opin Neurol 2003;16:643–50 [DOI] [PubMed] [Google Scholar]
- 25.Kleihues P, Burger PC, Aldape K, Brat DJ, Biernat W, Bigner DD. Glioblastoma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous system. Lyon, France: IARC Press, 2007: 33–49 [Google Scholar]
- 26.Scheithauer BW, Fuller GN, VandenBerg SR. The 2007 WHO classification of tumors of the nervous system: controversies in surgical neuropathology. Brain Pathol 2008;18:307–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller CR, Perry A. Glioblastoma. Arch Pathol Lab Med 2007;131:397–406 [DOI] [PubMed] [Google Scholar]
- 28.Kleihues P, Ohgaki H. Primary and secondary glioblastomas: from concept to clinical diagnosis. Neuro-oncol 1999;1:44–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohgaki H, Dessen P, Jourde B, Horstmann S, Nishikawa T, Di Patre PL, et al. Genetic pathways to glioblastoma: a population-based study. Cancer Res 2004;64:6892–9 [DOI] [PubMed] [Google Scholar]
- 30.Cairncross JG, Macdonald DR. Successful chemotherapy for recurrent malignant oligodendroglioma. Ann Neurol 1988;23:360–4 [DOI] [PubMed] [Google Scholar]
- 31.Ino Y, Betensky RA, Zlatescu MC, Sasaki H, Macdonald DR, Stemmer-Rachamimov AO, et al. Molecular subtypes of anaplastic oligodendroglioma: implications for patient management at diagnosis. Clin Cancer Res 2001;7:839–45 [PubMed] [Google Scholar]
- 32.van denBent MJ. Diagnosis and management of oligodendroglioma. Semin Oncol 2004;31:645–52 [DOI] [PubMed] [Google Scholar]
- 33.Reifenberger G, Louis DN. Oligodendroglioma: toward molecular definitions in diagnostic neuro-oncology. J Neuropathol Exp Neurol 2003;62:111–26 [DOI] [PubMed] [Google Scholar]
- 34.Perry A. Oligodendroglial neoplasms: current concepts, misconceptions, and folklore. Adv Anat Pathol 2001;8:183–99 [DOI] [PubMed] [Google Scholar]
- 35.Burger PC. What is an oligodendroglioma? Brain Pathol 2002;12:257–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cairncross JG. Imaging molecular signatures in oligodendroglioma. Clin Cancer Res 2004;10:7109–11 [DOI] [PubMed] [Google Scholar]
- 37.Kros JM, Pieterman H, van Eden CG, Avezaat CJ. Oligodendroglioma: the Rotterdam-Dijkzigt experience. Neurosurgery 1994;34:959–66, discussion 66 [DOI] [PubMed] [Google Scholar]
- 38.Aldape K, Burger PC, Perry A. Clinicopathologic aspects of 1p/19q loss and the diagnosis of oligodendroglioma. Arch Pathol Lab Med 2007;131:242–51 [DOI] [PubMed] [Google Scholar]
- 39.Reifenberger G, Kros JM, Louis DN, Collins VP. Oligodendroglioma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous system. Lyon, France: IARC Press, 2007: 54–59 [Google Scholar]
- 40.Engelhard HH, Stelea A, Mundt A. Oligodendroglioma and anaplastic oligodendroglioma: clinical features, treatment, and prognosis. Surg Neurol 2003;60:443–56 [DOI] [PubMed] [Google Scholar]
- 41.Reiche W, Grunwald I, Hermann K, Deinzer M, Reith W. Oligodendrogliomas. Acta Radiol 2002;43:474–82 [PubMed] [Google Scholar]
- 42.Walker C, du Plessis DG, Fildes D, Haylock B, Husband D, Jenkinson MD, et al. Correlation of molecular genetics with molecular and morphological imaging in gliomas with an oligodendroglial component. Clin Cancer Res 2004;10:7182–91 [DOI] [PubMed] [Google Scholar]
- 43.Reifenberger G, Kros JM, Louis DN, Collins VP. Anaplastic oligodendroglioma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous System. Lyon, France: IARC Press, 2007: 60–2 [Google Scholar]
- 44.von Deimling A, Reifenberger G, Kros JM, Louis DN, Collins VP. Oligoastrocytoma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous system. Lyon, France: IARC Press, 2007: 63–5 [Google Scholar]
- 45.von Deimling A, Reifenberger G, Kros JM, louis DN, Collins VP. Anaplastic oligoastrocytoma. Louis DN, Ohgaki H, Wiestler OD, Cavanee WK, WHO classification of tumours of the central nervous system. Lyon, France: IARC Press, 2007: 66–7 [Google Scholar]
- 46.Walker C, du Plessis DG, Joyce KA, Fildes D, Gee A, Haylock B, et al. Molecular pathology and clinical characteristics of oligodendroglial neoplasms. Ann Neurol 2005;57:855–65 [DOI] [PubMed] [Google Scholar]
- 47.Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, et al. Gene expression profiling of gliomas strongly predicts survival. Cancer Res 2004;64:6503–10 [DOI] [PubMed] [Google Scholar]
- 48.Reifenberger G, Collins VP. Pathology and molecular genetics of astrocytic gliomas. J Mol Med 2004;82:656–70 [DOI] [PubMed] [Google Scholar]
- 49.Nutt CL, Mani DR, Betensky RA, Tamayo P, Cairncross JG, Ladd C, et al. Gene expression-based classification of malignant gliomas correlates better with survival than histological classification. Cancer Res 2003;63:1602–7 [PubMed] [Google Scholar]
- 50.Yamanaka R, Arao T, Yajima N, Tsuchiya N, Homma J, Tanaka R, et al. Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 2006;25:5994–6002 [DOI] [PubMed] [Google Scholar]
- 51.Hartmann C, Mueller W, von Deimling A. Pathology and molecular genetics of oligodendroglial tumors. J Mol Med 2004;82:638–55 [DOI] [PubMed] [Google Scholar]
- 52.Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res 2000;60:417–24 [PubMed] [Google Scholar]
- 53.Hermanson M, Funa K, Koopmann J, Maintz D, Waha A, Westermark B, et al. Association of loss of heterozygosity on chromosome 17p with high platelet-derived growth factor alpha receptor expression in human malignant gliomas. Cancer Res 1996;56:164–71 [PubMed] [Google Scholar]
- 54.Nishizaki T, Kubota H, Harada K, Harada K, Ito H, Suzuki M, et al. Clinical evidence of distinct subgroups of astrocytic tumors defined by comparative genomic hybridization. Hum Pathol 2000;31:608–14 [DOI] [PubMed] [Google Scholar]
- 55.Balesaria S, Brock C, Bower M, Clark J, Nicholson SK, Lewis P, et al. Loss of chromosome 10 is an independent prognostic factor in high-grade gliomas. Br J Cancer 1999;81:1371–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, et al. PTEN mutation, EGFR amplification, and outcome in patients with anaplastic astrocytoma and glioblastoma multiforme. J Natl Cancer Inst 2001;93:1246–56 [DOI] [PubMed] [Google Scholar]
- 57.Ichimura K, Schmidt EE, Miyakawa A, Goike HM, Collins VP. Distinct patterns of deletion on 10p and 10q suggest involvement of multiple tumor suppressor genes in the development of astrocytic gliomas of different malignancy grades. Genes Chromosomes Cancer 1998;22:9–15 [DOI] [PubMed] [Google Scholar]
- 58.Davies MP, Gibbs FE, Halliwell N, Joyce KA, Roebuck MM, Rossi ML, et al. Mutation in the PTEN/MMAC1 gene in archival low grade and high grade gliomas. Br J Cancer 1999;79:1542–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Watanabe K, Peraud A, Gratas C, Wakai S, Kleihues P, Ohgaki H. p53 and PTEN gene mutations in gemistocytic astrocytomas. Acta Neuropathol (Berl) 1998;95:559–64 [DOI] [PubMed] [Google Scholar]
- 60.Ohgaki H. Genetic pathways to glioblastomas. Neuropathology 2005;25:1–7 [DOI] [PubMed] [Google Scholar]
- 61.Biernat W, Huang H, Yokoo H, Kleihues P, Ohgaki H. Predominant expression of mutant EGFR (EGFRvIII) is rare in primary glioblastomas. Brain Pathol 2004;14:131–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin Cancer Res 2005;11:1462–6 [DOI] [PubMed] [Google Scholar]
- 63.Jenkins RB, Blair H, Ballman KV, Giannini C, Arusell RM, Law M, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer Res 2006;66:9852–61 [DOI] [PubMed] [Google Scholar]
- 64.Fuller CE, Perry A. Molecular diagnostics in central nervous system tumors. Adv Anat Pathol 2005;12:180–94 [DOI] [PubMed] [Google Scholar]
- 65.Idbaih A, Marie Y, Pierron G, Brennetot C, Hoang-Xuan K, Kujas M, et al. Two types of chromosome 1p losses with opposite significance in gliomas. Ann Neurol 2005;58:483–7 [DOI] [PubMed] [Google Scholar]
- 66.Smith JS, Perry A, Borell TJ, Lee HK, O'Fallon J, Hosek SM, et al. Alterations of chromosome arms 1p and 19q as predictors of survival in oligodendrogliomas, astrocytomas, and mixed oligoastrocytomas. J Clin Oncol 2000;18:636–45 [DOI] [PubMed] [Google Scholar]
- 67.Van DenBent MJ, Looijenga LH, Langenberg K, Dinjens W, Graveland W, Uytdewilligen L, et al. Chromosomal anomalies in oligodendroglial tumors are correlated with clinical features. Cancer 2003;97:1276–84 [DOI] [PubMed] [Google Scholar]
- 68.Walker C, Haylock B, Husband D, Joyce KA, Fildes D, Jenkinson MD, et al. Clinical use of genotype to predict chemosensitivity in oligodendroglial tumors. Neurology 2006;66:1661–7 [DOI] [PubMed] [Google Scholar]
- 69.Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 1998;90:1473–9 [DOI] [PubMed] [Google Scholar]
- 70.van denBent MJ, Carpentier AF, Brandes AA, Sanson M, Taphoorn MJ, Bernsen HJ, et al. Adjuvant procarbazine, lomustine, and vincristine improves progression-free survival but not overall survival in newly diagnosed anaplastic oligodendrogliomas and oligoastrocytomas: a randomized European Organisation for Research and Treatment of Cancer phase III trial. J Clin Oncol 2006;24:2715–22 [DOI] [PubMed] [Google Scholar]
- 71.Giannini C, Burger PC, Berkey BA, Cairncross JG, Jenkins RB, Mehta M, et al. Anaplastic oligodendroglial tumors: refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol 2008;18:360–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hartmann C, Mueller W, Lass U, Kamel-Reid S, von Deimling A. Molecular genetic analysis of oligodendroglial tumors. J Neuropathol Exp Neurol 2005;64:10–14 [DOI] [PubMed] [Google Scholar]
- 73.Wolter M, Reifenberger J, Blaschke B, Ichimura K, Schmidt EE, Collins VP, et al. Oligodendroglial tumors frequently demonstrate hypermethylation of the CDKN2A (MTS1, p16INK4a), p14ARF, and CDKN2B (MTS2, p15INK4b) tumor suppressor genes. J Neuropathol Exp Neurol 2001;60:1170–80 [DOI] [PubMed] [Google Scholar]
- 74.Sasaki H, Zlatescu MC, Betensky RA, Ino Y, Cairncross JG, Louis DN. PTEN is a target of chromosome 10q loss in anaplastic oligodendrogliomas and PTEN alterations are associated with poor prognosis. Am J Pathol 2001;159:359–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jeuken JW, von Deimling A, Wesseling P. Molecular pathogenesis of oligodendroglial tumors. J Neurooncol 2004;70:161–81 [DOI] [PubMed] [Google Scholar]
- 76.Eoli M, Bissola L, Bruzzone MG, Pollo B, Maccagnano C, De Simone T, et al. Reclassification of oligoastrocytomas by loss of heterozygosity studies. Int J Cancer 2006;119:84–90 [DOI] [PubMed] [Google Scholar]
- 77.Dong ZQ, Pang JC, Tong CY, Zhou LF, Ng HK. Clonality of oligoastrocytomas. Hum Pathol 2002;33:528–35 [DOI] [PubMed] [Google Scholar]
- 78.Walker C, du Plessis DG, Joyce KA, Machell Y, Thomson-Hehir J, Al Haddad SA, et al. Phenotype versus genotype in gliomas displaying inter- or intratumoral histological heterogeneity. Clin Cancer Res 2003;9:4841–51 [PubMed] [Google Scholar]
- 79.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med 2005;352:997–1003 [DOI] [PubMed] [Google Scholar]
- 80.Mollemann M, Wolter M, Felsberg J, Collins VP, Reifenberger G. Frequent promoter hypermethylation and low expression of the MGMT gene in oligodendroglial tumors. Int J Cancer 2005;113:379–85 [DOI] [PubMed] [Google Scholar]
- 81.Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R, et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer 2009;101:124–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brandes AA, Franceschi E, Tosoni A, Blatt V, Pession A, Tallini G, et al. MGMT promoter methylation status can predict the incidence and outcome of pseudoprogression after concomitant radiochemotherapy in newly diagnosed glioblastoma patients. J Clin Oncol 2008;26:2192–7 [DOI] [PubMed] [Google Scholar]
- 83.Brandsma D, Stalpers L, Taal W, Sminia P, van denBent MJ. Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol 2008;9:453–61 [DOI] [PubMed] [Google Scholar]
- 84.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321:1807–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science 2009;324:261–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ducray F, El Hallani S, Idbaih A. Diagnostic and prognostic markers in gliomas. Curr Opin Oncol 2009;21:537–42 [DOI] [PubMed] [Google Scholar]
- 87.Ichimura K, Pearson DM, Kocialkowski S, Backlund LM, Chan R, Jones DT, et al. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro Oncol 2009;11:341–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Balss J, Meyer J, Mueller W, Korshunov A, Hartmann C, von Deimling A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol 2008;116:597–602 [DOI] [PubMed] [Google Scholar]
- 89.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med 2009;360:765–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Watanabe T, Nobusawa S, Kleihues P, Ohgaki H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am J Pathol 2009;174:1149–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sanson M, Marie Y, Paris S, Idbaih A, Laffaire J, Ducray F, et al. Isocitrate dehydrogenase 1 codon 132 mutation is an important prognostic biomarker in gliomas. J Clin Oncol 2009;27:4150–4 [DOI] [PubMed] [Google Scholar]
- 92.Weller M, Felsberg J, Hartmann C, Berger H, Steinbach JP, Schramm J, et al. Molecular predictors of progression-free and overall survival in patients with newly diagnosed glioblastoma: a prospective translational study of the German Glioma Network. J Clin Oncol 2009;27:5743–50 [DOI] [PubMed] [Google Scholar]
- 93.Tews B, Felsberg J, Hartmann C, Kunitz A, Hahn M, Toedt G, et al. Identification of novel oligodendroglioma-associated candidate tumor suppressor genes in 1p36 and 19q13 using microarray-based expression profiling. Int J Cancer 2006;119:792–800 [DOI] [PubMed] [Google Scholar]
- 94.Huang H, Okamoto Y, Yokoo H, Heppner FL, Vital A, Fevre-Montange M, et al. Gene expression profiling and subgroup identification of oligodendrogliomas. Oncogene 2004;23:6012–22 [DOI] [PubMed] [Google Scholar]
- 95.Tso CL, Freije WA, Day A, Chen Z, Merriman B, Perlina A, et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res 2006;66:159–67 [DOI] [PubMed] [Google Scholar]
- 96.Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006;9:157–73 [DOI] [PubMed] [Google Scholar]
- 97.Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. J Clin Oncol 2008;26:3015–24 [DOI] [PubMed] [Google Scholar]
- 98.Colman H, Zhang L, Sulman EP, McDonald JM, Shooshtari NL, Rivera A, et al. A multigene predictor of outcome in glioblastoma. Neuro Oncol 2010;12:49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Shirahata M, Oba S, Iwao-Koizumi K, Saito S, Ueno N, Oda M, et al. Using gene expression profiling to identify a prognostic molecular spectrum in gliomas. Cancer Sci 2009;100:165–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Korshunov A, Sycheva R, Gorelyshev S, Golanov A. Clinical utility of fluorescence in situ hybridization (FISH) in nonbrainstem glioblastomas of childhood. Mod Pathol 2005;18:1258–63 [DOI] [PubMed] [Google Scholar]
- 101.Vinchon M, Soto-Ares G, Ruchoux MM, Dhellemmes P. Cerebellar gliomas in children with NF1: pathology and surgery. Childs Nerv Syst 2000;16:417–20 [DOI] [PubMed] [Google Scholar]
- 102.Listernick R, Charrow J, Gutmann DH. Intracranial gliomas in neurofibromatosis type 1. Am J Med Genet 1999;89:38–44 [DOI] [PubMed] [Google Scholar]
- 103.Jones DT, Ichimura K, Liu L, Pearson DM, Plant K, Collins VP. Genomic analysis of pilocytic astrocytomas at 0.97 Mb resolution shows an increasing tendency toward chromosomal copy number change with age. J Neuropathol Exp Neurol 2006;65:1049–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008;68:8673–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Korshunov A, Meyer J, Capper D, Christians A, Remke M, Witt H, et al. Combined molecular analysis of BRAF and IDH1 distinguishes pilocytic astrocytoma from diffuse astrocytoma. Acta Neuropathol 2009;118:401–5 [DOI] [PubMed] [Google Scholar]
- 106.Jones DT, Kocialkowski S, Liu L, Pearson DM, Ichimura K, Collins VP. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 2009;28:2119–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Nakamura M, Shimada K, Ishida E, Higuchi T, Nakase H, Sakaki T, et al. Molecular pathogenesis of pediatric astrocytic tumors. Neuro Oncol 2007;9:113–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Tabori U, Rienstein S, Dromi Y, Leider-Trejo L, Constantini S, Burstein Y, et al. Epidermal growth factor receptor gene amplification and expression in disseminated pediatric low-grade gliomas. J Neurosurg 2005;103:357–61 [DOI] [PubMed] [Google Scholar]
- 109.Broniscer A, Baker SJ, West AN, Fraser MM, Proko E, Kocak M, et al. Clinical and molecular characteristics of malignant transformation of low-grade glioma in children. J Clin Oncol 2007;25:682–9 [DOI] [PubMed] [Google Scholar]
- 110.Pollack IF, Finkelstein SD, Woods J, Burnham J, Holmes EJ, Hamilton RL, et al. Expression of p53 and prognosis in children with malignant gliomas. N Engl J Med 2002;346:420–7 [DOI] [PubMed] [Google Scholar]
- 111.Rickert CH, Strater R, Kaatsch P, Wassmann H, Jurgens H, Dockhorn-Dworniczak B, et al. Pediatric high-grade astrocytomas show chromosomal imbalances distinct from adult cases. Am J Pathol 2001;158:1525–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pollack IF, Finkelstein SD, Burnham J, Holmes EJ, Hamilton RL, Yates AJ, et al. Age and TP53 mutation frequency in childhood malignant gliomas: results in a multi-institutional cohort. Cancer Res 2001;61:7404–7 [PubMed] [Google Scholar]
- 113.Bredel M, Pollack IF, Hamilton RL, James CD. Epidermal growth factor receptor expression and gene amplification in high-grade non-brainstem gliomas of childhood. Clin Cancer Res 1999;5:1786–92 [PubMed] [Google Scholar]
- 114.Raffel C, Frederick L, O'Fallon JR, Atherton-Skaff P, Perry A, Jenkins RB, et al. Analysis of oncogene and tumor suppressor gene alterations in pediatric malignant astrocytomas reveals reduced survival for patients with PTEN mutations. Clin Cancer Res 1999;5:4085–90 [PubMed] [Google Scholar]
- 115.Sung T, Miller DC, Hayes RL, Alonso M, Yee H, Newcomb EW. Preferential inactivation of the p53 tumor suppressor pathway and lack of EGFR amplification distinguish de novo high grade pediatric astrocytomas from de novo adult astrocytomas. Brain Pathol 2000;10:249–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Khatua S, Peterson KM, Brown KM, Lawlor C, Santi MR, LaFleur B, et al. Overexpression of the EGFR/FKBP12/HIF-2alpha pathway identified in childhood astrocytomas by angiogenesis gene profiling. Cancer Res 2003;63:1865–70 [PubMed] [Google Scholar]
- 117.Donson AM, Addo-Yobo SO, Handler MH, Gore L, Foreman NK. MGMT promoter methylation correlates with survival benefit and sensitivity to temozolomide in pediatric glioblastoma. Pediatr Blood Cancer 2007;48:403–7 [DOI] [PubMed] [Google Scholar]
- 118.Alonso M, Hamelin R, Kim M, Porwancher K, Sung T, Parhar P, et al. Microsatellite instability occurs in distinct subtypes of pediatric but not adult central nervous system tumors. Cancer Res 2001;61:2124–8 [PubMed] [Google Scholar]
- 119.Kreiger PA, Okada Y, Simon S, Rorke LB, Louis DN, Golden JA. Losses of chromosomes 1p and 19q are rare in pediatric oligodendrogliomas. Acta Neuropathol 2005;109:387–92 [DOI] [PubMed] [Google Scholar]
- 120.Sehgal A. Molecular changes during the genesis of human gliomas. Semin Surg Oncol 1998;14:3–12 [DOI] [PubMed] [Google Scholar]
- 121.Sanai N, Alvarez-Buylla A, Berger MS. Neural stem cells and the origin of gliomas. N Engl J Med 2005;353:811–22 [DOI] [PubMed] [Google Scholar]
- 122.Vescovi AL, Galli R, Reynolds BA. Brain tumour stem cells. Nat Rev Cancer 2006;6:425–36 [DOI] [PubMed] [Google Scholar]
- 123.Das S, Srikanth M, Kessler JA. Cancer stem cells and glioma. Nat Clin Pract Neurol 2008;4:427–35 [DOI] [PubMed] [Google Scholar]
- 124.Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G, 2nd, Jiang L, et al. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med 2003;9:439–47 [DOI] [PubMed] [Google Scholar]
- 125.Germano I, Swiss V, Casaccia P. Primary brain tumors, neural stem cell, and brain tumor cancer cells: where is the link? Neuropharmacology 2010;58:903–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sommer L, Rao M. Neural stem cells and regulation of cell number. Prog Neurobiol 2002;66:1–18 [DOI] [PubMed] [Google Scholar]
- 127.Berger F, Gay E, Pelletier L, Tropel P, Wion D. Development of gliomas: potential role of asymmetrical cell division of neural stem cells. Lancet Oncol 2004;5:511–14 [DOI] [PubMed] [Google Scholar]
- 128.Gilbertson RJ, Gutmann DH. Tumorigenesis in the brain: location, location, location. Cancer Res 2007;67:5579–82 [DOI] [PubMed] [Google Scholar]
- 129.Lim DA, Cha S, Mayo MC, Chen MH, Keles E, VandenBerg S, et al. Relationship of glioblastoma multiforme to neural stem cell regions predicts invasive and multifocal tumor phenotype. Neuro Oncol 2007;9:424–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396–401 [DOI] [PubMed] [Google Scholar]
- 131.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, et al. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004;64:7011–21 [DOI] [PubMed] [Google Scholar]
- 132.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, et al. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003;63:5821–8 [PubMed] [Google Scholar]
- 133.Yi L, Zhou ZH, Ping YF, Chen JH, Yao XH, Feng H, et al. Isolation and characterization of stem cell-like precursor cells from primary human anaplastic oligoastrocytoma. Mod Pathol 2007;20:1061–8 [DOI] [PubMed] [Google Scholar]
- 134.Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 2005;8:323–35 [DOI] [PubMed] [Google Scholar]
- 135.Kondo T, Setoguchi T, Taga T. Persistence of a small subpopulation of cancer stem-like cells in the C6 glioma cell line. Proc Natl Acad Sci U S A 2004;101:781–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, et al. Cancerous stem cells can arise from pediatric brain tumors. Proc Natl Acad Sci USA 2003;100:15178–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Liu G, Yuan X, Zeng Z, Tunici P, Ng H, Abdulkadir IR, et al. Analysis of gene expression and chemoresistance of CD133+ cancer stem cells in glioblastoma. Mol Cancer 2006;5:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Eramo A, Ricci-Vitiani L, Zeuner A, Pallini R, Lotti F, Sette G, et al. Chemotherapy resistance of glioblastoma stem cells. Cell Death Differ 2006;13:1238–41 [DOI] [PubMed] [Google Scholar]
- 139.Kang MK, Kang SK. Tumorigenesis of chemotherapeutic drug-resistant cancer stem-like cells in brain glioma. Stem Cells Dev 2007;16:837–47 [DOI] [PubMed] [Google Scholar]
- 140.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444:756–60 [DOI] [PubMed] [Google Scholar]
- 141.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005;5:275–84 [DOI] [PubMed] [Google Scholar]
- 142.Salmaggi A, Boiardi A, Gelati M, Russo A, Calatozzolo C, Ciusani E, et al. Glioblastoma-derived tumorospheres identify a population of tumor stem-like cells with angiogenic potential and enhanced multidrug resistance phenotype. Glia 2006;54:850–60 [DOI] [PubMed] [Google Scholar]
- 143.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J Clin Oncol 2008;26:2839–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Charles N, Holland EC. The perivascular niche microenvironment in brain tumor progression. Cell Cycle 2010;9:3012–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheng L, Bao S, Rich JN. Potential therapeutic implications of cancer stem cells in glioblastoma. Biochem Pharmacol 2010;80:654–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Gunther HS, Schmidt NO, Phillips HS, Kemming D, Kharbanda S, Soriano R, et al. Glioblastoma-derived stem cell-enriched cultures form distinct subgroups according to molecular and phenotypic criteria. Oncogene 2008;27:2897–909 [DOI] [PubMed] [Google Scholar]
- 147.Cha S. Perfusion MR imaging: basic principles and clinical applications. Magn Reson Imaging Clin N Am 2003;11:403–13 [DOI] [PubMed] [Google Scholar]
- 148.Herholz K, Coope D, Jackson A. Metabolic and molecular imaging in neuro-oncology. Lancet Neurol 2007;6:711–24 [DOI] [PubMed] [Google Scholar]
- 149.Law M, Yang S, Wang H, Babb JS, Johnson G, Cha S, et al. Glioma grading: sensitivity, specificity, and predictive values of perfusion MR imaging and proton MR spectroscopic imaging compared with conventional MR imaging. AJNR Am J Neuroradiol 2003;24:1989–98 [PMC free article] [PubMed] [Google Scholar]
- 150.Kono K, Inoue Y, Nakayama K, Shakudo M, Morino M, Ohata K, et al. The role of diffusion-weighted imaging in patients with brain tumors. AJNR Am J Neuroradiol 2001;22:1081–8 [PMC free article] [PubMed] [Google Scholar]
- 151.Fischer I, Gagner JP, Law M, Newcomb EW, Zagzag D. Angiogenesis in gliomas: biology and molecular pathophysiology. Brain Pathol 2005;15:297–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Jain RK, di Tomaso E, Duda DG, Loeffler JS, Sorensen AG, Batchelor TT. Angiogenesis in brain tumours. Nat Rev Neurosci 2007;8:610–22 [DOI] [PubMed] [Google Scholar]
- 153.Patankar TF, Haroon HA, Mills SJ, Baleriaux D, Buckley DL, Parker GJ, et al. Is volume transfer coefficient (K(trans)) related to histologic grade in human gliomas? AJNR Am J Neuroradiol 2005;26:2455–65 [PMC free article] [PubMed] [Google Scholar]
- 154.Hakyemez B, Erdogan C, Ercan I, Ergin N, Uysal S, Atahan S. High-grade and low-grade gliomas: differentiation by using perfusion MR imaging. Clin Radiol 2005;60:493–502 [DOI] [PubMed] [Google Scholar]
- 155.Zlatescu MC, TehraniYazdi A, Sasaki H, Megyesi JF, Betensky RA, Louis DN, et al. Tumor location and growth pattern correlate with genetic signature in oligodendroglial neoplasms. Cancer Res 2001;61:6713–15 [PubMed] [Google Scholar]
- 156.Mueller W, Hartmann C, Hoffmann A, Lanksch W, Kiwit J, Tonn J, et al. Genetic signature of oligoastrocytomas correlates with tumor location and denotes distinct molecular subsets. Am J Pathol 2002;161:313–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Laigle-Donadey F, Martin-Duverneuil N, Lejeune J, Criniere E, Capelle L, Duffau H, et al. Correlations between molecular profile and radiologic pattern in oligodendroglial tumors. Neurology 2004;63:2360–2 [DOI] [PubMed] [Google Scholar]
- 158.Felsberg J, Erkwoh A, Sabel MC, Kirsch L, Fimmers R, Blaschke B, et al. Oligodendroglial tumors: refinement of candidate regions on chromosome arm 1p and correlation of 1p/19q status with survival. Brain Pathol 2004;14:121–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brown R, Zlatescu M, Sijben A, Roldan G, Easaw J, Forsyth P, et al. The use of magnetic resonance imaging to noninvasively detect genetic signatures in oligodendroglioma. Clin Cancer Res 2008;14:2357–62 [DOI] [PubMed] [Google Scholar]
- 160.Jenkinson MD, Smith TS, Joyce KA, Fildes D, Broome J, du Plessis DG, et al. Cerebral blood volume, genotype and chemosensitivity in oligodendroglial tumours. Neuroradiology 2006;48:703–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Law M, Brodsky JE, Babb J, Rosenblum M, Miller DC, Zagzag D, et al. High cerebral blood volume in human gliomas predicts deletion of chromosome 1p: Preliminary results of molecular studies in gliomas with elevated perfusion. J Magn Reson Imaging 2007;25:1113–19 [DOI] [PubMed] [Google Scholar]
- 162.Whitmore RG, Krejza J, Kapoor GS, Huse J, Woo JH, Bloom S, et al. Prediction of oligodendroglial tumor subtype and grade using perfusion weighted magnetic resonance imaging. J Neurosurg 2007;107:600–9 [DOI] [PubMed] [Google Scholar]
- 163.Kapoor GS, Gocke TA, Chawla S, Whitmore RG, Nabavizadeh A, Krejza J, et al. Magnetic resonance perfusion-weighted imaging defines angiogenic subtypes of oligodendroglioma according to 1p19q and EGFR status. J Neurooncol 2009;92:373–86 [DOI] [PubMed] [Google Scholar]
- 164.Emblem KE, Scheie D, Due-Tonnessen P, Nedregaard B, Nome T, Hald JK, et al. Histogram analysis of MR imaging-derived cerebral blood volume maps: combined glioma grading and identification of low-grade oligodendroglial subtypes. AJNR Am J Neuroradiol 2008;29:1664–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cha S, Tihan T, Crawford F, Fischbein NJ, Chang S, Bollen A, et al. Differentiation of low-grade oligodendrogliomas from low-grade astrocytomas by using quantitative blood-volume measurements derived from dynamic susceptibility contrast-enhanced MR imaging. AJNR Am J Neuroradiol 2005;26:266–73 [PMC free article] [PubMed] [Google Scholar]
- 166.Xu M, See SJ, Ng WH, Arul E, Back MF, Yeo TT, et al. Comparison of magnetic resonance spectroscopy and perfusion-weighted imaging in presurgical grading of oligodendroglial tumors. Neurosurgery 2005;56:919–26, discussion 19–26 [PubMed] [Google Scholar]
- 167.Spampinato MV, Smith JK, Kwock L, Ewend M, Grimme JD, Camacho DL, et al. Cerebral blood volume measurements and proton MR spectroscopy in grading of oligodendroglial tumors. AJR Am J Roentgenol 2007;188:204–12 [DOI] [PubMed] [Google Scholar]
- 168.Lev MH, Ozsunar Y, Henson JW, Rasheed AA, Barest GD, Harsh GR, 4th, et al. Glial tumor grading and outcome prediction using dynamic spin-echo MR susceptibility mapping compared with conventional contrast-enhanced MR: confounding effect of elevated rCBV of oligodendroglimoas. AJNR Am J Neuroradiol 2004;25:214–21 [PMC free article] [PubMed] [Google Scholar]
- 169.Birner P, Gatterbauer B, Oberhuber G, Schindl M, Rossler K, Prodinger A, et al. Expression of hypoxia-inducible factor-1 alpha in oligodendrogliomas: its impact on prognosis and on neoangiogenesis. Cancer 2001;92:165–71 [DOI] [PubMed] [Google Scholar]
- 170.Christov C, Adle-Biassette H, Le Guerinel C, Natchev S, Gherardi RK. Immunohistochemical detection of vascular endothelial growth factor (VEGF) in the vasculature of oligodendrogliomas. Neuropathol Appl Neurobiol 1998;24:29–35 [DOI] [PubMed] [Google Scholar]
- 171.Jenkinson MD, Smith TS, Brodbelt AR, Joyce KA, Warnke PC, Walker C. Apparent diffusion coefficients in oligodendroglial tumors characterized by genotype. J Magn Reson Imaging 2007;26:1405–12 [DOI] [PubMed] [Google Scholar]
- 172.Tozer DJ, Jager HR, Danchaivijitr N, Benton CE, Tofts PS, Rees JH, et al. Apparent diffusion coefficient histograms may predict low-grade glioma subtype. NMR Biomed 2007;20:49–57 [DOI] [PubMed] [Google Scholar]
- 173.Jenkinson MD, du Plessis DG, Smith TS, Brodbelt AR, Joyce KA, Walker C. Cellularity and apparent diffusion coefficient in oligodendroglial tumours characterized by genotype. J Neurooncol 2010;96:385–92 [DOI] [PubMed] [Google Scholar]
- 174.Jenkinson MD, Smith TS, Joyce K, Fildes D, du Plessis DG, Warnke PC, et al. MRS of oligodendroglial tumors: correlation with histopathology and genetic subtypes. Neurology 2005;64:2085–9 [DOI] [PubMed] [Google Scholar]
- 175.Rijpkema M, Schuuring J, Van DerMeulen Y, Van DerGraaf M, Bernsen H, Boerman R, et al. Characterization of oligodendrogliomas using short echo time 1H MR spectroscopic imaging. NMR Biomed 2003;16:12–18 [DOI] [PubMed] [Google Scholar]
- 176.Derlon JM, Chapon F, Noel MH, Khouri S, Benali K, Petit-Taboue MC, et al. Non-invasive grading of oligodendrogliomas: correlation between in vivo metabolic pattern and histopathology. Eur J Nucl Med 2000;27:778–87 [DOI] [PubMed] [Google Scholar]
- 177.Kaschten B, Stevenaert A, Sadzot B, Deprez M, Degueldre C, Del Fiore G, et al. Preoperative evaluation of 54 gliomas by PET with fluorine-18-fluorodeoxyglucose and/or carbon-11-methionine. J Nucl Med 1998;39:778–85 [PubMed] [Google Scholar]
- 178.Stockhammer F, Thomale UW, Plotkin M, Hartmann C, Von Deimling A. Association between fluorine-18-labeled fluorodeoxyglucose uptake and 1p and 19q loss of heterozygosity in World Health Organization Grade II gliomas. J Neurosurg 2007;106:633–7 [DOI] [PubMed] [Google Scholar]
- 179.Shimizu T, Saito N, Aihara M, Kurihara H, Nakazato Y, Ueki K, et al. Primary spinal oligoastrocytoma: a case report. Surg Neurol 2004;61:77–81, discussion 81 [DOI] [PubMed] [Google Scholar]
- 180.Aghi M, Gaviani P, Henson JW, Batchelor TT, Louis DN, Barker FG., 2nd Magnetic resonance imaging characteristics predict epidermal growth factor receptor amplification status in glioblastoma. Clin Cancer Res 2005;11:8600–5 [DOI] [PubMed] [Google Scholar]
- 181.Lal A, Glazer CA, Martinson HM, Friedman HS, Archer GE, Sampson JH, et al. Mutant epidermal growth factor receptor up-regulates molecular effectors of tumor invasion. Cancer Res 2002;62:3335–9 [PubMed] [Google Scholar]
- 182.Maity A, Pore N, Lee J, Solomon D, O'Rourke DM. Epidermal growth factor receptor transcriptionally up-regulates vascular endothelial growth factor expression in human glioblastoma cells via a pathway involving phosphatidylinositol 3′-kinase and distinct from that induced by hypoxia. Cancer Res 2000;60:5879–86 [PubMed] [Google Scholar]
- 183.Choe G, Park JK, Jouben-Steele L, Kremen TJ, Liau LM, Vinters HV, et al. Active matrix metalloproteinase 9 expression is associated with primary glioblastoma subtype. Clin Cancer Res 2002;8:2894–901 [PubMed] [Google Scholar]
- 184.Mut M, Turba UC, Botella AC, Baskurt E, Lopes MB, Shaffrey ME. Neuroimaging characteristics in subgroup of GBMs with p53 overexpression. J Neuroimaging 2007;17:168–74 [DOI] [PubMed] [Google Scholar]
- 185.Drabycz S, Roldan G, de Robles P, Adler D, McIntyre JB, Magliocco AM, et al. An analysis of image texture, tumor location, and MGMT promoter methylation in glioblastoma using magnetic resonance imaging. Neuroimage 2010;49:1398–405 [DOI] [PubMed] [Google Scholar]
- 186.Kato T, Shinoda J, Oka N, Miwa K, Nakayama N, Yano H, et al. Analysis of 11C-methionine Uptake in Low-Grade Gliomas and Correlation with Proliferative Activity. AJNR Am J Neuroradiol 2008;29:1867–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Maia AC, Jr, Malheiros SM, da Rocha AJ, da Silva CJ, Gabbai AA, Ferraz FA, et al. MR cerebral blood volume maps correlated with vascular endothelial growth factor expression and tumor grade in nonenhancing gliomas. AJNR Am J Neuroradiol 2005;26:777–83 [PMC free article] [PubMed] [Google Scholar]
- 188.Hobbs SK, Shi G, Homer R, Harsh G, Atlas SW, Bednarski MD. Magnetic resonance image-guided proteomics of human glioblastoma multiforme. J Magn Reson Imaging 2003;18:530–6 [DOI] [PubMed] [Google Scholar]
- 189.Van Meter T, Dumur C, Hafez N, Garrett C, Fillmore H, Broaddus WC. Microarray analysis of MRI-defined tissue samples in glioblastoma reveals differences in regional expression of therapeutic targets. Diagn Mol Pathol 2006;15:195–205 [DOI] [PubMed] [Google Scholar]
- 190.Pope WB, Chen JH, Dong J, Carlson MR, Perlina A, Cloughesy TF, et al. Relationship between gene expression and enhancement in glioblastoma multiforme: exploratory DNA microarray analysis. Radiology 2008;249:268–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Barajas RF, Jr, Hodgson JG, Chang JS, Vandenberg SR, Yeh RF, Parsa AT, et al. Glioblastoma multiforme regional genetic and cellular expression patterns: influence on anatomic and physiologic MR imaging. Radiology 2010;254:564–76 [DOI] [PMC free article] [PubMed] [Google Scholar]