

Abstract
A deeper understanding of the role of specific genes, proteins, pathways and networks in health and disease, coupled with the development of technologies to assay these molecules and pathways in patients, promises to revolutionise the practice of clinical medicine. In particular, the discovery and development of novel drugs targeted to disease-specific alterations could benefit significantly from non-invasive imaging techniques assessing the dynamics of specific disease-related parameters. Here we review the application of imaging biomarkers in the management of patients with brain tumours, especially malignant glioma. This first part of the review focuses on imaging biomarkers of general biochemical and physiological processes related to tumour growth such as energy, protein, DNA and membrane metabolism, vascular function, hypoxia and cell death. These imaging biomarkers are an integral part of current clinical practice in the management of primary brain tumours. The second article of the review discusses the use of imaging biomarkers of specific disease-related molecular genetic alterations such as apoptosis, angiogenesis, cell membrane receptors and signalling pathways. Current applications of these biomarkers are mostly confined to experimental small animal research to develop and validate these novel imaging strategies with future extrapolation in the clinical setting as the primary objective.
During the past decade, the molecular and genetic causes underlying many chronic illnesses, such as cancer, metabolic disorders and neurological and psychiatric disorders, have been characterised through major advances in genomics, proteomics and metabolomics. Knowledge of the underlying genetic and molecular defects in specific receptors, cytokines and growth factors and understanding of related pathophysiological changes in several signalling pathways associated with cancer have been adopted for the development of novel efficient therapeutic approaches targeting these disease-causing molecular defects. Owing to this multitude of possible drug targets, the discovery and development of successful novel drugs is very challenging. As a consequence, biomarkers are playing an increasing role in the evaluation and validation of drug actions [1].
Biomarkers
Biomarkers are anatomical, physiological, biochemical or molecular parameters associated with the presence and severity of specific disease states [2]. Biomarkers are detectable and measurable by a variety of methods, including physical examination, laboratory assays and medical imaging. Imaging biomarkers are the subset of biomarkers that manifest themselves via imaging means, including optical, ultrasound, radiographs, CT, positron emission tomography (PET), single photon emission CT (SPECT) and MRI. Imaging biomarkers include monitoring key morphological (tumour location, size, invasiveness), biochemical (metabolism, proliferation, angiogenesis, apoptosis) and physiological (vascular permeability, blood volume) processes, as well as disease-specific molecular genetic changes in receptors, cytokines, growth factors and related signalling pathways.
Evaluation of therapy response
Despite major developments in non-invasive clinical imaging, to date the evaluation of responses to cancer therapy are still based largely on volumetric and morphological criteria, in particular relative tumour sizes before and after treatment [1]. These approaches, which were introduced more than 25 years ago [3], and revised in 2000 [4], are known as the WHO (World Health Organization) and RECIST (Response Evaluation Criteria In Solid Tumors) criteria, respectively. According to the earlier (WHO) criteria, the size of a tumour should be estimated based on two perpendicular diameters, and positive tumour response to therapy should be defined as a reduction of at least 50% in the product of these two diameters [3]. In 1990, Macdonald et al [5] suggested that the two-dimensional (2D) WHO criteria should also be applied to brain tumours. For solid tumours, these WHO criteria were replaced in 2000 by unidimensional (1D) measurements (RECIST), which defined therapy-induced response as a 30% decrease in the largest dimension of the tumour [4], whereas for brain tumours the Mcdonald criteria are still the standard for many clinical brain tumour trials, although a recent study reported high concordance between the 1D and the 2D methods in assessing enhancing tumour progression and in estimating mean progression-free survival (mPFS) and 6 month progression-free survival in adult brain tumour patients [6]. These criteria, however, are very limited in their ability to assess the early effects of therapy, as was shown recently by the results of clinical trials using growth factor receptor inhibitors, in which RECIST-based X-ray CT was unable to predict patient survival benefit [7].
Molecular imaging
The development of novel targeted cancer therapies could benefit significantly from the introduction of approaches to provide disease-specific molecular information through (targeted) non-invasive imaging measurements. This would not only allow patient stratification and selection but also an early assessment of treatment response long before any reduction in tumour volume. This will in turn lead to a predictive individualised patient care.
Targeted or molecular imaging (MI) involves the coupling of conventional imaging technologies with the use of specific molecular probes and aims at the non-invasive characterisation of the dynamics of disease-specific molecular changes in vivo. To image molecular and cellular processes with exquisite spatial, temporal and biochemical resolution in vivo, several imaging modalities have been developed and implemented. Molecular imaging modalities that are currently being used for clinical application can be roughly divided into two groups: those primarily providing structural information like CT, MRI or ultrasound; and those primarily aiming at functional or molecular information, like PET, SPECT and optical imaging. Molecular imaging usually exploits specific molecular probes as the source of imaging contrast to report on the underlying biochemistry and cell biology associated with disease progression and response to therapy. It should be pointed out that it is the combination of different imaging technologies merging structural and functional information that provide complementary types of information enabling accurate, repetitive, non-invasive disease phenotyping and the measurement of therapeutic outcomes. Furthermore, combining two or more imaging modalities in a so-called multimodal imaging approach has the potential to exploit the strengths and overcome the shortcomings of each modality alone. The nuclear imaging techniques, PET and SPECT, have a very high sensitivity where very low levels (10−11 to 10−12 mol l–1) of specific radiotracer accumulation can be detected, have unlimited depth penetration, excellent signal-to-background ratios and a broad range of clinically applicable probes. The major drawbacks of radionuclide imaging are their inherently limited spatial resolution (2–6 mm), the exposure of patients to radiation, which lead to the need of dosimetry and may limit the number of examinations that can be performed, and, finally, the need of access to a cyclotron and radiochemistry facility. MRI techniques have good depth penetration and a spectacular spatial resolution which is not limited by detector geometry, as with nuclear imaging, or by tissue scattering properties, as by optical imaging; however, temporal resolution is limited and molecular probe detection is several orders of magnitude less sensitive than nuclear imaging techniques (10−3 to 10−5 mol l–1). However, this low sensitivity can be overcome to some extent by the use of targeted contrast agents that give large signal amplification.
Optical imaging techniques are cost-effective and time-efficient, do not involve ionising radiation, require less resources and space than PET and MRI and have excellent temporal resolution. However, the common disadvantages of these techniques are the lack of optimal quantitative or tomographic information and the limited spatial resolution and depth penetration, the main reason for the lack of clinical application to date. However, optical imaging techniques may enable the development of a new generation of MI agents to be used in the future in humans at specific sites, such as tissues and lesions close to the skin surface or tissues accessible by endoscopy and during intra-operative visualisation [8].
Research showing that molecular imaging can provide supporting data for clinical outcomes and perhaps endpoints that in themselves reflect and are recognised as meaningful clinical surrogates is in its early days [9]. However, molecular imaging in oncology clearly has the potential to guide patient management and drug selection through response monitoring and tumour-specific target identification. Several molecular imaging probes are being developed and validated as early response markers to assess the impact of novel therapeutics on various universal characteristics of tumour morphology, physiology and biochemistry such as tumour metabolism, proliferation, hypoxia and apoptosis as well as on specific molecular genetic tumour characteristics. Prominent examples of targets for drug development include specific kinases, cell adhesion molecules, cellular receptors and signalling. Probes to image such targets have included small molecules, peptides, antibodies labelled with radionuclides (e.g. 11C, 18F, 99mTc and 123I), fluorochromes or magnetic ligands [10].
Here we will review the application of imaging biomarkers for the management of patients with brain tumours, especially for the assessment of molecular targeted therapies in patients with malignant gliomas. After a short preface on primary brain tumours, the first part of this review will discuss imaging biomarkers of fundamental biochemical and physiological glioma properties. The second part of this review will describe imaging biomarkers of specific receptors and signalling pathways, which are in pre-clinical development.
Primary brain tumours
Diffuse infiltrative gliomas are the most common primary intracranial neoplasms, accounting for 40% of all primary and 78% of all malignant central nervous system (CNS) tumours. The term glioma includes several heterogeneous entities that have in common a presumed glial cell of origin. Subcategories of gliomas include astrocytomas, oligodendrogliomas, oligo-astrocytomas, ependymomas and glioblastomas. According to the WHO classification, gliomas receive a histopathological grade on the basis of the presence of nuclear changes, mitotic activity, and the presence of endothelial proliferation and necrosis [11]. Low-grade gliomas (WHO II) are defined as diffusely infiltrative astrocytotic tumours with cytological atypia; tumours presenting anaplastic cell types and high mitotic activity are classified as WHO III; gliomas with additional microvascular proliferation and/or necrosis as WHO grade IV. The term malignant glioma comprises WHO grade IV tumours, such as glioblastomas with its variants, and WHO grade III tumours, anaplastic forms of astrocytoma, oligodendroglioma and oligastrocytoma. The histological grading is highly relevant to predict the patients’ prognosis. The median survival of patients with WHO grade II glioma is usually more than 5 years, whereas the median survival of patients with high-grade gliomas remains modest (<15 months) despite recent advances in microsurgical techniques, radiation and chemotherapy [12]. Surgical resection and radiotherapy have been the mainstay of treatment [13] and only recently have the benefits of chemotherapy been unequivocally shown in a randomised trial [14]. Yet, even under such therapies, recurrence is the norm, and disease will follow a fatal course in virtually all patients with malignant glioma. This inability to successfully treat brain tumours is mostly due to the lack of understanding the underlying complex brain tumour biology, and treatment outcome is hardly predictable because of individually different molecular tumour phenotypes that are responsible for tumour growth.
Glioblastoma may arise through two distinct pathways of neoplastic progression. Tumours that progress from lower grade (II or III) astrocytic tumours are termed secondary or type 1 glioblastoma multiforme (GBM); tumours that arise de novo without any evidence of a lower-grade precursor are termed primary or type 2 GBM. Secondary GBM develop in younger patients (fifth to sixth decade) with time to progression from low-grade lesions ranging from months to decades. Primary GBM develop in older individuals (sixth to seventh decade) and have short clinical histories (less than 3 months).
A complex series of molecular changes leads to glioma development that results in deregulation of the cell cycle, alterations of apoptosis and cell differentiation, in neovascularization as well as tumour cell migration and invasion into brain parenchyma. During progression from low-grade astrocytoma (WHO grade II) to anaplastic astrocytoma (WHO grade III) and to GBM (WHO grade IV) a stepwise accumulation of these genetic alterations occurs. Primary and secondary GBM harbour distinct molecular genetic alterations [15,16]. Most importantly, molecular alterations have been identified that indicate therapeutic response of patients and, thus, have prognostic relevance [17-20].
An improved understanding of the biology of glial tumourigenesis is highly important for the development of molecular therapeutic targets in order to overcome current therapeutic limitations. Over the past years several drugs targeting cancer-associated molecular changes have been developed and first clinical trials have been conducted.
A major challenge in clinical studies to date remains the identification and validation of optimal endpoints for targeted therapy. For the time being, progression-free survival (PFS) (typically 6 months PFS) remains the most frequently recommended primary endpoint for glioma trials because it is assumed to be a surrogate marker of preservation of neurological and functional status as well as a predictor of overall survival (OS) [13]. However, it is of note that such assumptions lack objective validation and cannot be universally applied. Because assessment of PFS depends on radiographic evaluation (McDonald criteria), it is possible that mismatches between biological activity and radiographic effects as seen in certain types of treatment associated with a cytostatic rather than a cytotoxic effect (i.e. local therapies and anti-angiogenic drugs) may limit the value of PFS as a predictor of clinical benefit or survival. Therefore, other endpoints as surrogate markers for treatment response to targeted therapies need to be identified and validated. Imaging biomarkers are gaining increased attention as they can provide valuable information on fundamental biochemical and physiological processes as well as specific molecular changes associated with targeted therapies.
Imaging fundamental properties of gliomas
Tumour cell energy metabolism
Energy metabolism is a central function of all tissues, including tumours. It has long been known that tumours contain large quantities of lactic acid, because they rely on anaerobic glycolysis for a major part of their energy consumption [21]. However, this is a relatively inefficient process, since anaerobic glycolysis generates only a limited amount of energy (adenosine triphosphate) per molecule of glucose. This inefficient use of "fuel" results in a strongly increased demand for glucose in cancer cells. Generally, tumour cells have upregulated all the mechanisms that are necessary to obtain glucose, such as the insulin-dependent GLUT1 transmembrane transporter and intracellular enzymes such as hexokinase. These two families of proteins are frequently overexpressed in tumours [22,23].
FDG PET
18F-FDG PET is based on the uptake principle of the 18F-FDG tracer that is a direct analogue of glucose. The strongly increased uptake of 18F-FDG in cancer cells contrasts with most normal cells and organs. In addition, once 18F-FDG has entered the cell, it is phosphorylated but, unlike glucose itself, it is not further metabolised. This intracellular trapping, in combination with the relatively high resolution of PET cameras, leads to adequate detection properties of metabolically active tumours and metastases.
18F-FDG PET has been approved by the Food and Drug Administration (FDA) for assessment of abnormal glucose metabolism to assist in the evaluation of malignancy in patients with known or suspected abnormalities found by other testing modalities, or in patients with an existing diagnosis of cancer [21]. The European Organization for Research and Treatment of Cancer PET Group has established response assessment guidelines for PET [24]. However, the diagnostic value of 18F-FDG PET is limited because the uptake is variable in some types of cancer and 18F-FDG uptake is not tumour specific. Brain shows high 18F-FDG uptake because the brain exclusively uses glucose for energy metabolism [25] and physiological 18F-FDG uptake is also seen in tonsils, salivary glands, vocal cords, reactive lymph nodes, liver, gastrointestinal tract and testes, as well as muscles [26]. In addition, many pathological processes other than malignant neoplasms (benign tumours, hyperplastic processes, epileptic foci and infected and inflamed tissue [27]) show increased 18F-FDG uptake. Even though 18F-FDG is not a tumour-specific tracer, 18F-FDG PET has become a powerful and quantitative tool in clinical oncology, which allows the detection and staging of disease by identifying and localising tumours with high glucose metabolism. In neuro-oncology, 18F-FDG has been used to detect the metabolic differences between normal brain tissue, low-grade and high-grade gliomas, and radionecrosis [28-31] (Figure 1). Moreover, increased intratumoural glucose consumption correlates with tumour grade, cell density, biological aggressiveness and patient survival in both primary and recurrent gliomas [30,32,33]. However, because of the relatively high glucose metabolism in normal grey matter, clear tumour location and delineation with 18F-FDG PET is difficult [34,35].
Figure 1.
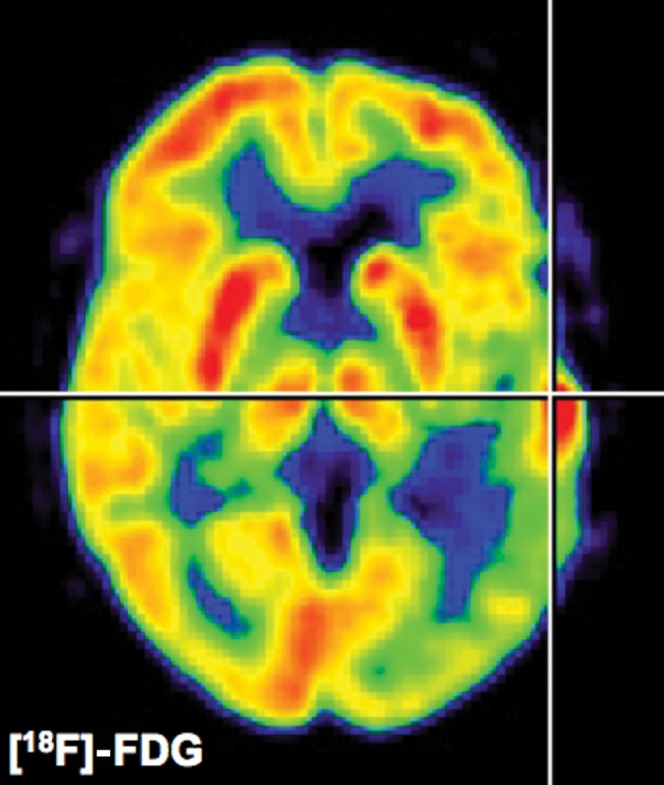
18F-fluorodeoxyglucose (FDG) positron emission tomography. Glucose metabolism can be determined quantitatively (μmol 100 g–1 min–1) in patients with gliomas. Low-grade gliomas will show a decreased glucose metabolism whereas high-grade gliomas as depicted in this figure will show an increased uptake of 18F-FDG which is proportional to the cellular density of the glioma. It should be pointed out that the extent of secondary inactivation of normal grey matter as depicted in this patient is of prognostic relevance. Because of high cortical background activity and glucose consumption in normal brain, improved glioma radiotracers with lower background activity in normal brain as shown in Figures 2 and 4 have been developed.
Furthermore, 18F-FDG PET can also be used to predict the outcome of therapy, since a decrease in 18F-FDG uptake can indicate a positive response to treatment. Over the last few years, several studies have demonstrated the ability of 18F-FDG PET to detect changes in glucose metabolism as early as 2 weeks after chemo- and radiotherapy in patients with various types of cancer [36-39]. Also molecular targeted therapies can decrease 18F-FDG uptake in the early stage. Preliminary observation of gefitinib therapy for patients with EGFR mutation-positive adenocarcinomas of the lung has demonstrated a rapid and significant decrease in the uptake of 18F-FDG in responding tumours after two doses of gefitinib therapy [26]. Also, in malignant gliomas and other CNS malignancies the response to chemotherapy has been assessed by PET [40]. In a pilot 18F-FDG PET study, 14 days after treatment with temozolomide a greater reduction in 18F-FDG uptake in foci of high metabolic activity was seen in patients with tumours that had an objective response to treatment 6 weeks later [41]. In a subsequent study, patients showing response to chemotherapy on PET survived longer than those who did not [42]. However, it should be pointed out that 18F-FDG uptake might also be due to migration of macrophages into the tumour, making the interpretation of 18F-FDG uptake difficult for the evaluation of therapy response [43]. Furthermore, changes in 18F-FDG uptake do not seem to be predictive of response to target-based therapy, time to progression or survival [44]. For these reasons, 18F-FDG PET does not play a major role in the assessment of novel therapies for gliomas [45].
Magnetic resonance spectroscopy
In contrast to the structural information provided by standard T1 and T2 weighted MRI with or without contrast, magnetic resonance spectroscopy (MRS) provides a qualitative analysis of a number of metabolites within the brain, and a quantitative analysis if a reference of known concentration is used [46]. These metabolites reflect aspects of neuronal integrity, energy metabolism, cell membrane proliferation or degradation, and necrotic transformation of brain or tumour tissue.
Major metabolites detected with 1H-MRS include N-acetyl aspartate (NAA), total cholines (tCho), total creatines (tCr), lactate (Lac), mobile lipids (Lip) and other macromolecules (MM), myo-inositol (mI) and glutamine and glutamate (Glx). The tCr signal arises from the adenosine diphosphate–adenosine triphosphate energy cycle metabolites creatine and phosphocreatine and is a marker for cellular energy metabolism. Compared with normal brain, tCr is generally reduced in astrocytomas and is almost absent in meningiomas, schwannomas and metastases [47]; however, the significance in relation to tumour metabolism is unclear. The lactate signal consists of two distinct resonant peaks called a doublet. Lactate is usually a product of anaerobic metabolism that occurs when the oxygen demand of a rapidly growing tumour exceeds that what is supplied by its neovasculature [48]. Tumours may also produce lactate because of their preference for aerobic glycolysis. Lactate is often more prominent in the highest grade tumours, but accumulation in cystic or necrotic regions can produce increased levels independent of grade.
31P-MRS can interrogate on intracellular energy stores by providing distinct signals from the three phosphorus atoms in adenosine triphosphate and phosphocreatine, which varies with intracellular pH, and from phosphomonoesters and phosphodiesters. These signals can show changes in malignant tumours [49].
Tumour cell protein metabolism and related alterations
Synthesis of proteins is a fundamental process for all cellular functions. The process of protein synthesis in tumours is increased because of the uncontrolled and accelerated growth of cancer [21]. As a consequence, the demand for amino acids, the building blocks of proteins, is increased. Protein metabolism can be divided into amino acid transport (from plasma into the cell) by specific amino acid transporters (A- and L-amino acid transporters) and protein synthesis (incorporation into protein). Therefore, it is expected that radiolabelled amino acid tracers will also accumulate in tumours, and subsequent imaging of the amino acid metabolism could provide useful information with regard to tumour metabolism.
PET/SPECT
Nearly all amino acids and slightly modified variants have been radiolabelled, but only a few have clinical use. Among these few are 11C-methionine (MET), 18F-fluoroethyl-tyrosine (FET) and 11C-tyrosine (TYR) for PET imaging, and 123I-iodomethyl-tyrosine (IMT) for SPECT imaging [50]. From these tracers, only 11C-TYR uptake represents both transport and protein synthesis, whereas the remaining tracers represent increased transport into tumour cells. In comparison with 18F-FDG PET, uptake of amino acids is less influenced by inflammation, although for amino acid tracers tumour specificity is not perfect [51].
In neuro-oncology, radiolabelled methionine and thymidine compounds have been shown to be more specific tracers in tumour detection, delineation and staging owing to their relatively low uptake in normal brain (Figure 2). The increased 11C-MET uptake (factor of 1.3–3.5 compared with a contralateral control region) is related to increased transport mediated by type L-amino acid transporters located at the blood–brain barrier (BBB) and this increased transport seems to be directly regulated by tumour growth factors [52,53]. 11C-MET PET detects parts of brain tumours as well as infiltrating areas with high sensitivity (87%) and specificity (89%) by an uptake threshold of 1.3-fold [54]. Furthermore, 11C-MET uptake correlates with microvessel density [55] and proliferative activity [56] and enables differentiating between WHO II and WHO grade III/IV gliomas [57]. By using a threshold of 1.5 11C-MET PET permits the differentiation between non-tumoural lesions and gliomas with a sensitivity of 79% [58]. However, substantial differences in uptake between tumour types limit the specificity of amino acid uptake imaging for non-invasive grading of tumours of unknown histological type [40]. 11C-MET is well suited to follow the effects of radiotherapy [59] and brachytherapy, as there is a reduction in relative methionine uptake [35]; 11C-MET also enables differentiating recurrent tumour from radiation necrosis while the tracer accumulation is nearly independent from disruption of the BBB or macrophage activity within the necrosis [60]. Most importantly, 11C-MET and 18F-FET are useful tools to evaluate responses of gliomas to chemotherapy with PCV (procarbuzine, bis-2-chloroethyl-3-cyclohexyl-1-nitrosourea and vincristine) [61,62], temozolomide (TMZ) [63], revealing tumour response after three cycles of TMZ chemotherapy (Figure 3), and convection-enhanced delivery (CED) of placlitaxel [64].
Figure 2.
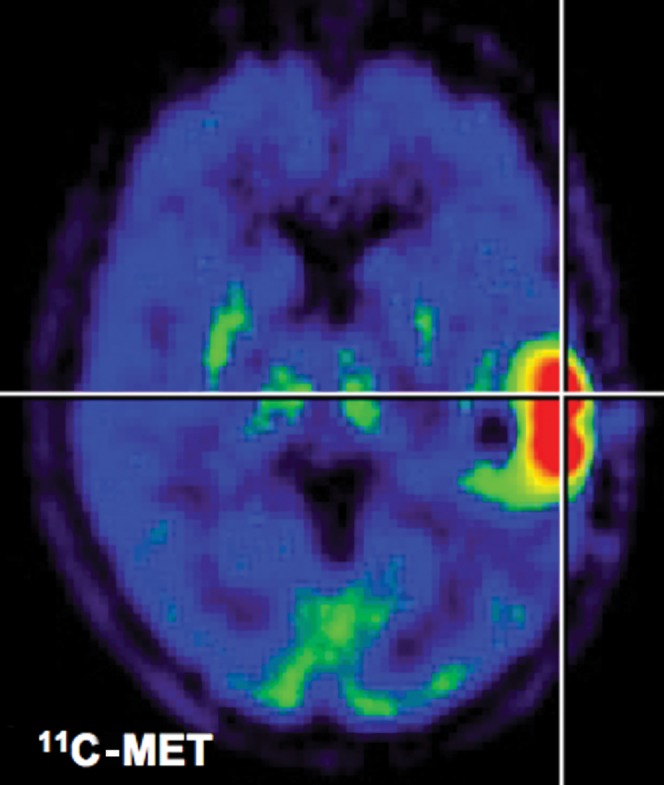
11C-methionine positron emission tomography (PET). After application of radiolabelled amino acids, they are transported through endothelial cells into the tumour. Background uptake in brain is limited, enabling the delineation of the glioma. This is a clear advantage over 18F-fluorodeoxyglucose PET. Owing to metabolisation of methionine, quantification of uptake is only possible in a semi-quantitative manner (tumour-to-background ratios).
Figure 3.

11C-methionine positron emission tomography (MET-PET) for imaging-guided chemotherapy. A patient with an anaplastic oligoastrocytoma after resection and radiation therapy presented with increased seizure activity as sign of tumour recurrence. Based on MRI (upper left) a clear indication of active tumour could not be found. 11C-MET-PET identified recurrent tumour (lower left). The patient was put on temozolomide treatment. Follow-up 11C-MET-PET could identify positive temozolomide (TMZ) response (lower row) whereas MRI did not show a clear change over time (upper right) (modified from Galldiks et al [63] with permission). CTX, chemotherapy; OA, oligoastrocytoma; OP, operation; RTX, radiotherapy.
11C-MET metabolism remains a rather indirect marker for proliferation, although correlations with in vitro proliferation markers such as the proliferating cell nuclear antigen (PCNA) have been described recently [56] and because of the short half-life of 11C (20 min) the use of 11C-MET remains restricted to PET centres with a cyclotron. A critical review on the role of 11C-MET in the clinical management of cerebral gliomas has been published recently [65].
Tumour cell DNA metabolism
As a high rate of cellular proliferation is a key feature of malignant tumours, in recent years much attention has been attracted to the search for cellular proliferation markers, the uptake of which would accurately reflect DNA synthesis. It is important to realise that there are two main pathways involved in DNA synthesis: (i) the exogenous or "salvage" pathway, which uses DNA precursors, nucleic acids, from outside the cell that are phosphorylated by thymidine kinase 1 (TK1); and (ii) the endogenous pathway, in which intracellular molecules, such as uridine monophosphate, enter DNA synthesis after phosphorylation by thymidylate synthase. TK1 is a cytosolic enzyme that is expressed during the S phase of the cell cycle, and TK1 expression is specifically increased in dividing cells and decreased in non-dividing cells [66].
PET
A variety of nucleic acids have been radiolabelled as PET tracers for cellular proliferation. For more than 40 years, 3H-thymidine incorporation has been the gold standard for assessing proliferation in vitro [10]. Thymidine labelled with positron-emitting nuclides can enable in vivo imaging using PET. Like the tritiated derivative, thymidine labelled with 11C in the pyrimidine ring provides the authentic substrate for the salvage pathway transporters, thymidine kinase and DNA polymerase and is incorporated into DNA. However, the 11C derivatives are also substrates for the catabolic pathway, yielding 11C-CO2 and other labelled derivatives [67]. Validated kinetic models of 11C-thymidine that account for this metabolism have been used to characterise human tumours, including response to chemotherapy directed at the de novo DNA synthesis pathway [68].
The deoxyribose group of thymidine can also be labelled with 18F at the 3′-deoxy position, resulting in 3′-deoxy-3′-fluorothymidine (FLT) [69]. Also 18F-FLT is transported across the cell membrane by nucleoside transporter proteins and once intracellular FLT is phosphorylated by TK1. Similar to 18F-FDG , 18F-FLT is trapped intracellularly, because after initial phosphorylation by TK1 the resulting monophosphate cannot be incorporated into DNA due to lack of a hydroxyl group. Thus, uptake of 18F-FLT appears to be a measure of the activity of the salvage pathway and therefore depends on the activity of TK1. In general, TK1 is approximately 10-fold increased in cancer cells, especially during the S phase of the cell cycle [70]. It has now been demonstrated in many types of cancer that 18F-FLT uptake in vivo is a measure of tumour proliferation activity and that there exists a significant correlation between 18F-FLT uptake and the in vitro proliferation marker Ki-67 in various tumour type [71-76]. In addition, in vitro and animal studies comparing 18F-FDG and 18F-FLT have repeatedly confirmed that 18F-FLT uptake in inflammatory tissue is considerably less than 18F-FDG. However, uptake in tumour tissue itself appears to be lower than 18F-FDG , as has been demonstrated in many types of cancer [70]. Since high uptake is among the principle requirements of any successful imaging method (especially when applied in cancer staging), this is an important drawback. 18F-FLT PET, therefore, seems less suitable for staging of cancer and currently most research focuses on response evaluation, as 18F-FLT has great potential utility in following response to therapy. A responding tumour cell may continue to metabolise 18F-FDG to maintain ion gradients or to provide energy for the P-glycoprotein (P-gp) pump function or protein biosynthesis; however, it will not synthesise new DNA and will not accumulate 18F-FLT. A decrease in DNA synthesis is likely following either cytostatic or cytotoxic therapy, highlighting the general use of 18F-FLT PET for detecting response [10]. Clinical studies to quantify response will benefit from using rigorously quantitative methods to distinguish thymidine delivery and transport from thymidine kinase enzyme activity [77]. In a recent study, we demonstrated that 18F-FLT uptake (i) enables differentiating between low-grade and high-grade tumours and (ii) is mainly due to increased transport and to a lower extent to phosphorylation by TK1 in gliomas (Figure 4) [78]. Furthermore, we demonstrated that kinetic analysis of 18F-FLT tracer uptake is essential for the in vivo assessment of tumour proliferation. A significant correlation was observed between the metabolic rate constant Ki and the proliferation index as measured by Ki-67 immunostaining (Ki, r=0.79; p=0.004) in patients first diagnosed with high-grade gliomas, whereas 18F-FLT uptake ratios did not correlate with the proliferation index (Figure 5) [79]. Thus, parametric images determined by kinetic analysis provide an accurate method for the assessment of tumour proliferation in vivo.
Figure 4.
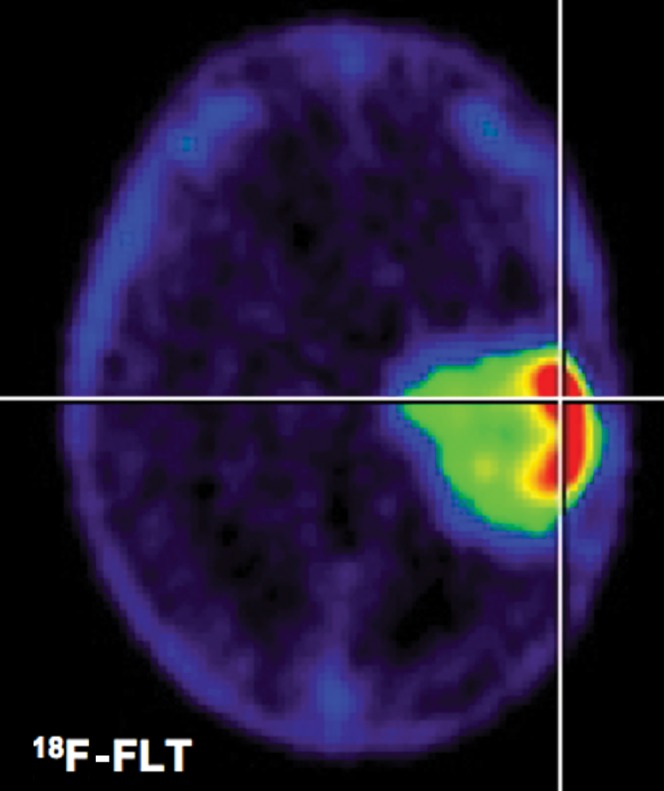
3′-deoxy-3′-fluorothymidine positron emission tomography (FLT-PET). After systemic administration of 18F-FLT, it accumulates in brain tumours according to World Health Organization grade. Background uptake in normal brain is lowest compared with 18F-Fluorodeoxyglucose (FDG)- and 11C-methionine (MET)-PET. Kinetic analysis seems necessary to determine proliferative activity and to distinguish 18F-FLT uptake due to leakage of the blood–brain barrier (see Figure 5).
Figure 5.

Parametric mapping of 18F-3′-deoxy-3′-fluorothymidine (FLT) Ki (a) and correlation to Ki-67 (b). (a) Coregistered 18F-FLT positron emission tomography (PET), 11C-methionine (MET)-PET, parametric map of 18F-FLT Ki and MRI (T1+Gd) in a 63-year-old patient with a first diagnosed glioblastoma multiforme. 18F-FLT- and 11C-MET-PET images show high tracer uptake ratios (18F-FLT, 6.7-fold; 11C-MET, 3.2-fold) than the contralateral tissue, parametric 18F-FLT mapping shows an increased kinetic metabolic rate constant Ki (0.0161 ml g–1 min–1) which is related to a high percentage Ki-67 expression (45%). (b) The relationship between 18F-FLT Ki and Ki-67 expression in 11 patients with newly diagnosed glioma (modified from Ullrich et al [79] with permission).
In the field of imaging antiproliferative treatment effects, recent studies demonstrated that changes in 18F-FLT PET may allow an early assessment of treatment response [80,81]. In a recent small prospective study in patients with recurrent malignant gliomas, changes in 18F-FLT uptake predicted overall survival already 1–2 weeks after start of treatment [80]. Patients were treated with the anti-VEGF antibody bevacizumab in combination with irinotecan, and 18F-FLT PET scans were performed at baseline, 1–2 weeks and after 6 weeks from start of therapy. A greater than 25% reduction in tumour 18F-FLT uptake as measured by the standardised uptake value (SUV) was defined as a metabolic response. All 18F-FLT responses were compared with radiological response as shown by MRI and with patient survival. Metabolic responders had a longer OS than non-responders (10.8 vs 3.4 months, p=0.003) and a trend for improved PFS was also seen (p=0.061). Both early and late 18F-FLT responses were more significant predictors of OS (1–2 weeks; p=0.006; 6 weeks; p=0.002) compared with MRI response (p=0.060 for both 6 weeks and best response). However, 18F-FLT accumulation within the tumour might not only be due to proliferation but also be due to leakage of the BBB, as 18F-FLT uptake depends on BBB damage. Thus, changes in intracerebral 18F-FLT uptake after therapy have to be considered with caution. We believe that kinetic analysis provides a more accurate measure to evaluate changes in FLT uptake after therapy since it allows to distinguish tracer uptake that is due to transport from uptake that is due to DNA synthesis/proliferation (see Figure 5).
In an experimental setting, our group performed a comparison of 18F-FLT and 18F-FDG PET for their potential to identify response to EGFR inhibitors early after treatment in a model of EGFR-dependent lung cancer [82]. While tumours sensitive to the EGFR tyrosine kinase inhibitor (TKI) erlotinib exhibited a striking and reproducible decrease in 18F-FLT uptake after only 2 days of treatment, 18F-FDG PET revealed no consistent reduction in tumour glucose consumption (Figure 6). The specificity of these results was confirmed by the complete lack of 18F-FLT-PET response of tumours expressing the T790M erlotinib resistance mutation of EGFR. This study clearly demonstrates the ability of 18F-FLT-PET to identify an erlotinib response in EGFR-dependent tumours at a very early stage and indicates that 18F-FLT-PET imaging may represent an appropriate method for early prediction of response to EGFR TKI treatment in patients with non-small cell lung cancer.
Figure 6.

Multitracer positron emission tomography (PET) evaluation of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor (TKI) treatment. 18F-3′-deoxy-3′-fluorothymidine (FLT) PET indicates response to therapy after 2 days of erlotinib treatment (50 mg kg–1) in the EGFR TKI-sensitive non-small cell lung cancer xenografts PC9 (n=8; vehicle, n=2) and HCC827 (n=7; vehicle, n=2) (a). The PET signal remains reduced also after 4 days of erlotinib treatment. No significant decrease in 18F-FLT uptake was observed in the EGFR TKI-resistant H1975 xenografts (n=8; vehicle, n=2). Statistical analysis revealed a significant reduction in 18F-FLT uptake in HCC827 and PC9 xenografts (b) and no significant changes in the H1975 xenografts during therapy. 18F-FDG metabolism only slightly decreased after 4 days of erlotinib treatment in the HCC827 xenografts. However, even after 4 days of erlotinib treatment the reduction in 18F-FDG uptake was not significant. The decrease in 18F-FLT uptake was accompanied by inhibition of cell growth as assessed by Ki-67 staining (c). The strong correlation between 18F-FLT uptake and the in vitro proliferation marker Ki-67 highlights the ability of 18F-FLT to assess cell proliferation in vivo (r=0.87, p<0.001). In contrast, the correlation of Ki-67 expression to 18F-FDG uptake was much lower (r=0.38, p=0.0037) (d) (modified from Ullrich et al 2008 with permission [82]).
MRI
MRI with and without contrast media is widely used for primary diagnosis of brain tumours. A set of various MRI acquisition parameters, such as T1, T2, proton-, diffusion- and perfusion-weighted images as well as fluid-attenuated inversion-recovery (FLAIR) sequences give a characteristic pattern of each tumour depending on tumour type and grade. Highly proliferative active tumours, such as glioblastomas, lead to a destruction of the BBB with subsequent leakage of contrast media, which is being used for diagnostic purposes in T1 weighted MRI. In contrast, low-grade tumours usually have no or minimal contrast enhancement. However, Scott et al [83] recently reported that 9% of malignant gliomas did not show contrast enhancement whereas 48% of low-grade gliomas enhanced, indicating that contrast enhancement in T1 may not allow for exact differentiation between low- and high-grade gliomas.
The contrast-enhancing lesion (T1+Gd) corresponds histologically to a hypercellular region with neovascularisation inducing high vascular permeability, whereas central hypointense areas (T1) are mainly caused by central necrosis. To monitor therapeutic response, conventional contrast-enhanced MRI has the major disadvantage that residual tumour and post-therapy changes can both result in abnormal enhancement due to therapy-induced leakage of the BBB and these techniques usually fail to detect early effects of radio- and chemotherapy, because individual treatment effects on tumour size are only visible after more than 12 months [84] with a substantial interobserver variability in the assessment of treatment response [85]. Especially after the application of biologically active agents the detection of changes in tumour viability is limited [86].
Diffusion-weighted MRI (DWI) may serve surrogate information on the cellular density of gliomas. In areas with high cell density, diffusion of water molecules is reduced because of the limited intercellular space. Therefore, highly proliferating tumour regions with high cell densities may display a decrease in the apparent diffusion coefficient (ADC) [87]. Although this might suggest that ADC can be used to distinguish between tumour and peritumoural lesions, Pauleit et al [88] reported a strong overlap between ADC values in tumour and peritumoural tissue. Further studies are required to investigate the correlations between biological tumour characteristics—as determined by histology—and ADC values.
Tumour cell membrane metabolism
Choline is an important precursor in the synthesis of cell membranes. The uptake of choline appears to be driven by the activity of choline kinase, which is generally increased in tumour cells due to increased cell membrane turnover. Choline is intracellularly phosphorylated and subsequently enters the phosphatidylcholine biosynthesis pathway. High levels of choline and its metabolites have long been known from MRS imaging. However, radiolabelled choline variants can also be used for PET imaging of cancer.
PET
The most used variant, 11C-choline (11C-ch), is chemically identical to choline. However, 18F variants with a more favourable half-life, such as 18F-Fluorocholine (18F-FCh) and 18F-Fluoroethylcholine (18F-FECh), also exist [21]. Similarly to 18F-FDG and 18F-FLT, the phosphorylated versions of these choline-based tracers accumulate in cells at a rate proportional to the rate of cell proliferation. Although choline can be considered a general tumour PET tracer, it appears to be taken up by prostate tissue [89] and brain [90] in particular. However, as the application of this technique is still in its infancy, it is too early to define its precise role.
MRS
MRS has been used for many years to study the metabolism of tumour cells in vitro and in vivo [46]. A particular focus has been the levels of the phospholipid metabolites, phosphocholine (PC) and phosphoethanolamine (PE), which are frequently elevated in proliferating tumour cells and decreased in concentration in tumours responding to treatment [1]. The increases in phosphocholine levels are thought to be due to increases in choline kinase activity and phospholipase C-mediated catabolism [91].
In most brain tumours, the choline-to-NAA ratios are high. NAA is a metabolic intermediary product that is mostly present in neurons and some non-neuronal cell types, such as oligodendrocytes. High lipid and choline peaks are associated with histological indicators of malignancy and glioma grade, such as expression of the proliferation markers Ki-67 and MIB-1 labelling, cellular anaplastic features, necrosis and nuclear density [92,93]. Moreover, MRS can distinguish between tumour recurrence and radiation necrosis such that a tCho/tCr ratio of greater than 3 in an enlarging lesion suggests viable tumour cells and thus tumour recurrence, whereas a ratio of less than 2 suggests that radiation necrosis is the predominant feature [94]. MRS can also provide predictive information on tumour progression during therapy, even before changes on conventional MRI (changes in tumour volume or contrast enhancement) are visible [95] and can produce biochemical information on apoptosis of cancer cells in vivo. It has been shown that accumulation of lipids can be used as a marker for growth arrest and can be detected by MRS, reflecting cell damage as early as 2–4 days after ganciclovir treatment in gene therapy of experimental gliomas [96]. Also other studies in pre-clinical model systems have demonstrated the potential of spectroscopic measurements to detect the action of new therapeutic agents non-invasively [1]. An anticancer drug that inhibits heat shock protein-90(Hsp90), 17-allylamino, 17-demethoxygeldanamycin (17AAG), was shown in 31P-MRS studies of cultured tumour cells and tumour xenografts to increase the concentration of phosphocholine [97]. It was suggested that these measurements have the potential to act as non-invasive pharmacodynamic markers for analysing tumour response to treatment with 17AAG or other Hsp90 inhibitors. Similar studies have been performed with other anticancer drugs, including a choline kinase inhibitor and a mitogen-activated protein kinase (MAPK) signalling inhibitor [98,99]. Again, it was the level of phosphocholine that provided a pharmacodynamic marker for assessing tumour response. However, a problem with MRS measurements, particularly the 31P-MRS measurements, is their lack of sensitivity, which leads to long data acquisition times and poor spatial resolution limiting its ability to fully address the anatomical and biological heterogeneity of brain tumours.
Angiogenesis and vascular functions
Vasculogenesis, angiogenesis and arteriogenesis are the three mechanisms of vascular network development. Vasculogenesis occurs mainly at the embryonic stage by angioblast differentiation into endothelial cells to form blood vessels. Angiogenesis, the development of new vessels from pre-existing ones, occurs later in embryogenesis, and after birth during normal processes such as female reproductive cycle and wound healing. Arteriogenesis refers to the enlargement of the arteriolar network to sustain higher metabolic demands, such as when vascular stenosis or occlusion occurs [100]. Vascular development in tumours is thought to occur mainly by angiogenesis [101]. However, the role of post-natal vasculogenesis by recruitment of bone marrow-derived endothelial progenitor cells (EPCs) into the tumour vasculature has been recently discussed [102].
Angiogenesis is a key oncological process essential for tumour growth, tumour invasion and initiation of metastasis [103]. For a tumour to grow beyond 1–2 mm in diameter there is a requirement for the growth of new blood vessels to supply essential nutrients, in particular oxygen.
As increased tumour angiogenesis is normally associated with poor disease prognosis the development of therapeutic agents that can reduce or inhibit angiogenesis (anti-angiogenic drugs) or destroy the neovasculature of tumours (antivascular drugs) has received considerable interest [1]. One possible advantage of these drugs, compared with conventional chemotherapy, is their potential for reduced susceptibility to acquired drug resistance (ADR). This is a major clinical problem observed in approximately 30% of cancer patients undergoing chemotherapy and results from the intrinsic genetic instability and heterogeneity of tumour cells [104]. Endothelial cells, on the other hand, on which antivascular and anti-angiogenic drugs act, are genetically more stable and more homogeneous and are thus presumed to be not so susceptible to ADR. Our growing understanding of the molecular mechanisms underlying angiogenesis, including the role of several growth factors that regulate the process, has enabled the development of a large number of drug candidates against vascular-specific targets that can inhibit angiogenesis (see Part II of the review).
Several imaging modalities have been used to evaluate the morphological and physiological effects of anti-angiogenic or antivascular drugs. Conventional morphological imaging techniques (e.g. CT) are not normally used, since these drugs usually only arrest tumour growth and stabilise disease rather than effect tumour regression [105-107]. Therefore, imaging modalities that are specifically sensitive to vascular function have been developed and include methods to assess blood volume and flow or vascular permeability. In clinical neuro-oncology, magnetic resonance [perfusion or dynamic contrast-enhanced (DCE) MRI] and nuclear techniques (H2O or CO imaging) are the main tools to monitor these events [100].
MRI
A critical parameter in many brain disorders is the tissue perfusion status, and several MR techniques are able to measure brain haemodynamics, such as vascular permeability, cerebral blood volume (CBV) and cerebral blood flow (CBF) [108]. Most pharmacokinetic modelling of the tissue response curve after bolus injection of a paramagnetic contrast agent is based upon DCE T1 weighted MR measurements and allows calculation and quantification of vascular permeability [109], whereas single-shot T2* weighted DCE is commonly used for whole-brain mapping of relative CBV (rCBV) and CBF, especially in clinical settings [110]. In recent years, several approaches have been developed to estimate vascular permeability and CBF and volume from a single dynamic imaging acquisition, either T1 [111] or T2* weighted [110] or by dual-gradient echo dynamic imaging [112].
Vascular permeability is considered to be a surrogate marker of tumour vascularity and angiogenesis [113,114]. The rCBV provides information about tumour angiogenesis [115] and several studies have demonstrated a correlation between rCBV and glioma grade [116,117] or survival [118]. A decrease in Ki (influx constant) and KTrans (volume transfer coefficient), representative of blood flow and vascular permeability, in patients treated with an angiogenesis inhibitor (an anti-VEGF antibody or a tyrosine kinase inhibitor) can be detected with DCE MRI as early as 2 days after the start of therapy [119,120]. However, no significant difference in any of these parameters was correlated to clinical response rate. In two glioma studies [121,122], a correlation between a decrease in the rCBV and a favourable outcome was observed in patients treated with thalidomide or cilengitide (anti-integrins), whereas no correlation was identified with the use of conventional contrast enhancement. Although tumour enhancement on gadolinium-enhanced T1 weighted MRI has an important value in prediction of the malignancy of gliomas, this enhancement only reflects disruption of the BBB and not tumour angiogenesis.
PET
PET imaging can be used clinically to assess tumour perfusion and vascular volume [1]. H215O, a biologically and metabolically inert molecule, is normally used to quantify CBF (in ml 100 g–1 min–1) as the tracer diffuses freely into and out of the tumour interstitium, and is frequently used to assess malignant angiogenesis and response to treatment [123]. The half-life of 15O, however, is very short (ca 2 min), and therefore the procedure must be repeated at short intervals. Spatial resolution is another major limitation, and spill-over phenomenon of the large blood vessels adjacent to the tumour may render difficulties into the analysis of small capillaries in tumours. Measurement of circulatory parameters, including blood volume, can also be performed by inhaling trace amounts of carbon monoxide (CO), labelled with either 11C or 15O, which binds tightly to haemoglobin in red blood cells, and is thus partially distributed in relation to blood volume [124]. However, CO is also distributed according to haematocrit, which can be highly variable in tumours [125].
Tumour hypoxia
In solid tumours, hypoxia may result from unregulated cellular growth, but it is also a common attribute of the tumour phenotype and may even be a factor in tumourigenesis [10]. Hypoxia-induces tissue changes that result in a selection of cells with mutant p53 expression [126]. After DNA damage, hypoxic cells do not readily undergo apoptosis. Hypoxia enhances expression of endothelial cytokines, such as VEGF, interleukin-1, tumour necrosis factor-α and transforming growth factor-β, and a cellular O2-sensing mechanism triggers production of hypoxia-inducible factor-1α (HIF-1α) [127]. HIF-1α induction initiates a cascade of events (HIF-1 pathway) culminating in angiogenesis. Tumour hypoxia and HIF-1α activation may also contribute to the metabolic switch to glycolysis that characterises tumour cells [128]. However, many cancer cells use glycolysis for energy production regardless of the availability of oxygen, suggesting that the two processes are independent [129]. Direct imaging of these hypoxia-related molecular genetic changes are described in Part II of this review.
Hypoxia is prevalent in nearly all tumours; however, hypoxia in tumours does not depend on tumour size, grade and extent of necrosis or blood haemoglobin status and seems to be an independent predictor of outcome [10]. In addition to inducing radioresistance, hypoxia promotes resistance to several chemotherapeutic agents potentially through three mechanisms: (i) hypoxia impedes drugs from reaching the cells from blood vessels, (ii) slows proliferation and (iii) promotes gene expression changes that enable cellular rescue from severe damage [130].
PET
PET imaging is an ideal modality for evaluating hypoxia. The PET imaging agent fluoromisonidazole (FMISO) is a 18F fluorinated derivative of misonidazole, an azomycin hypoxic cell sensitizer introduced two decades ago, which binds covalently to intracellular molecules at a rate inversely proportional to intracellular O2 concentration [10]. In 3 studies of 34 patients with glioma, only glioblastomas demonstrated constant uptake of 18F-FMISO, whereas neither low-grade gliomas nor anaplastic gliomas showed this effect [131-133]. Moreover, uptake of such a marker was related only to tumour VEGFR immunostaining, but not to microvessel density or HIF-1 expression [131]. Because hypoxia is associated with poor response to both radiation and chemotherapy, identifying hypoxia should have prognostic value and might enable advantageous treatment modifications during treatment [10].
MRI
Tissue oxygenation levels can be directly addressed by blood oxygenation level-dependent contrast (BOLD) MRI. This technique is based upon the magnetic property of blood which is dependent on the oxygenation state of haemoglobin [108]. Deoxygenated haemoglobin is paramagnetic, whereas oxyhaemoglobin is diamagnetic. As blood oxygen level decreases, deoxyhaemoglobin concentration increases, thereby resulting in a decrease in signal intensity on T2 and T2* weighted sequences. Decreased signal in T2* imaging has been correlated with hypoxic areas, and this has been confirmed in histological studies [134]. It should be pointed out, that blood oxygenation level-dependent contrast MRI has technical limitations, and routine use is difficult.
Tumour cell death, apoptosis and necrosis
Programmed cell death, or apoptosis, is an essential component of normal human growth and development, immunoregulation and tissue homeostasis. This highly regulated and genetically defined cellular process constitutes the main mechanism by which cells die, both in healthy and diseased tissues. A deficiency or excess in apoptosis is an integral component of autoimmune disorders, transplant rejection, neurodegenerative disorders and cancer. The other well-recognised form of cell death, necrosis, is an accidental and unregulated form of cell death that normally occurs as a result of a physicochemical insult [1]. Cell death is now recognised as a complex continuum of mechanisms, which include the apoptotic pathways, necrosis and autophagy. The last one being an alternative form of cell death that occurs normally in some cell types following nutrient deprivation. Pre-clinical and clinical studies have shown that the detection of apoptosis, or cell death in general, can potentially be used to provide an early indication of the success of therapy, providing prognostic information and guiding the course of subsequent treatment [135,136].
MRI
In the field of magnetic resonance, early attempts to detect cell death/apoptosis focused on identifying metabolic markers of the process using spectroscopic techniques. Both 31P-MRS and 1H-MRS have been used to detect apoptosis by measuring the accumulation of choline [137] or cytoplasmic mobile lipid droplets [138] and were used to detect apoptosis in a gene therapy model of rat glioma [96]. Decreases in other chemical species, such as glutamine and glutamate, taurine, and reduced glutathione, can also be seen with apoptotic cell death. In contrast, necrosis in general is characterised by a completely different 1H nuclear magnetic resonance (NMR) profile in which there is a significant increase in all of the metabolites examined, with the exception of mobile lipids that remain unchanged [139].
Other MR parameters have also been proposed as surrogate markers for generalised tumour cell death following therapy (reviewed recently by Kettunen and Grohn [140]). Perhaps the most promising of these parameters is the apparent diffusion coefficient (ADC) of water, which increases markedly following induction of tumour cell death and is most likely the result of changes in tissue density [141]. DWI, which is sensitive to changes in water ADC, is considered a non-invasive indicator of cell density [142] and could be useful in the assessment of response to therapy, because the inverse relationship between ADC and cellular density suggests that the temporal evolution from viable tumour to treatment-induced necrotic tumour may be measurable by diffusion. Early increases in ADC values during therapy are hypothesised to relate to therapy-induced necrosis, whereas a drop in ADC values within the tumour compared with pre-treatment levels is thought to be an indicator of tumour regrowth [142]. Moffat et al [143] investigated if diffusion MRI could be used as a biomarker for early prediction of treatment response in brain cancer patients. 20 brain tumour patients (n=7 GBM, n=3 astrocytoma, n=2 anaplastic astrocytoma, n=4 anaplastic oligodendroglioma, n=2 germ cell or n=2 primitive neuroectodermal tumour) were examined by standard and diffusion MRI before initiation of treatment. Additional images were acquired 3 weeks after initiation of chemo- and/or radiotherapy. Images were co-registered to pre-treatment scans, and changes in tumour water diffusion values were calculated and displayed as a functional diffusion map (fDM) for correlation with clinical response. Of the 20 patients imaged during the course of therapy, 6 were classified as having a partial response, 6 as stable disease, and 8 as progressive disease. The fDMs were found to predict patient response at 3 weeks from the start of treatment, revealing that early changes in tumour diffusion values could be used as a prognostic indicator of subsequent volumetric tumour response. Similar results have been published by Hamstra et al [144], who examined 34 patients with malignant glioma by diffusion MRI before and after treatment. Functional diffusion maps were calculated from the differences in tumour–water diffusion values. Also in this study, MRI were found to be strongly correlated with patient radiographic response, time-to-progression and overall survival, providing a route to individualisation of treatment or evaluation of clinical response.
Direct targeted imaging of the molecular changes related to apoptosis are described in Part II of the review.
Conclusion
Over the past years various fields in clinical medicine and in particular oncology have undergone a rapid evolution from relatively broad histopathology-based diagnoses and non-specific therapies to a disease-specific molecular-targeted approach of disease diagnoses and treatment. Therefore, the development of novel targeted cancer therapies could benefit significantly from the introduction of approaches to provide disease-specific molecular information through (targeted) non-invasive imaging measurements. This would not only allow patient stratification and selection but also an early assessment of treatment response long before any reduction in tumour volume is visible and lead to a predictive individualised patient care. Imaging biomarkers of fundamental glioma properties are currently used in clinical practice and represent an important step towards such personalised medicine. However, the introduction of clinically applicable imaging agents for non-invasive evaluation of disease-related genes, proteins, signalling pathways and networks will substantially improve the management of such patients. Part II of this review describes current advances in pre-clinical development of specific imaging biomarkers of genes, proteins and signalling pathways and their application in molecular targeted therapies.
Acknowledgments
Our work is supported in part by the Deutsche Forschungsgemeinschaft (DFG-Ja98/1-2), by the 6th FW EU grants EMIL (LSHC-CT-2004-503569), DiMI (LSHB-CT-2005-512146) and CliniGene NoE (LSHB-CT-2006-018933).
References
- 1.Neves AA, Brindle KM. Assessing responses to cancer therapy using molecular imaging. Biochim Biophys Acta 2006;1766:242–61 [DOI] [PubMed] [Google Scholar]
- 2.Harrington DP. Imaging biomarkers and the future of radiology. J Am Coll Radiol 2006;3:317–18 [DOI] [PubMed] [Google Scholar]
- 3.Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer 1981;47:207–14 [DOI] [PubMed] [Google Scholar]
- 4.Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92:205–16 [DOI] [PubMed] [Google Scholar]
- 5.Macdonald DR, Cascino TL, Schold SC., Jr , Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. J Clin Oncol 1990;8:1277–80 [DOI] [PubMed] [Google Scholar]
- 6.Shah GD, Kesari S, Xu R, Batchelor TT, O’Neill AM, Hochberg FH, et al. Comparison of linear and volumetric criteria in assessing tumor response in adult high-grade gliomas. Neuro Oncol 2006;8:38–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hainsworth JD, Sosman JA, Spigel DR, Edwards DL, Baughman C, Greco A. Treatment of metastatic renal cell carcinoma with a combination of bevacizumab and erlotinib. J Clin Oncol 2005;23:7889–96 [DOI] [PubMed] [Google Scholar]
- 8.Cai W, Chen X. Multimodality molecular imaging of tumor angiogenesis. J Nucl Med 2008;49:113S–28S [DOI] [PubMed] [Google Scholar]
- 9.Hargreaves RJ. The role of molecular imaging in drug discovery and development. Clin Pharmacol Ther 2008;83:349–53 [DOI] [PubMed] [Google Scholar]
- 10.Kelloff GJ, Krohn KA, Larson SM, Weissleder R, Mankoff DA, Hoffman JM, et al. The progress and promise of molecular imaging probes in oncologic drug development. Clin Cancer Res 2005;11:7967–85 [DOI] [PubMed] [Google Scholar]
- 11.Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, et al. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 2002;61:215–225; discussion 226–9 [DOI] [PubMed] [Google Scholar]
- 12.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Omuro AM, Faivre S, Raymond E. Lessons learned in the development of targeted therapy for malignant gliomas. Mol Cancer Ther 2007;6:1909–19 [DOI] [PubMed] [Google Scholar]
- 14.Stupp R, Mason WP, van denBent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 2005;352:987–96 [DOI] [PubMed] [Google Scholar]
- 15.Miller CR, Perry A. Glioblastoma. Arch Pathol Lab Med 2007;131:397–406 [DOI] [PubMed] [Google Scholar]
- 16.Lang FF, Miller DC, Koslow M, Newcomb EW. Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg 1994;81:427–36 [DOI] [PubMed] [Google Scholar]
- 17.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, et al. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med 2005;353:2012–24 [DOI] [PubMed] [Google Scholar]
- 18.Cairncross JG, Ueki K, Zlatescu MC, Lisle DK, Finkelstein DM, Hammond RR, et al. Specific genetic predictors of chemotherapeutic response and survival in patients with anaplastic oligodendrogliomas. J Natl Cancer Inst 1998;90:1473–9 [DOI] [PubMed] [Google Scholar]
- 19.Cahill DP, Levine KK, Betensky RA, Codd PJ, Romany CA, Reavie LB, et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin Cancer Res 2007;13:2038–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunter C, Smith R, Cahill DP, Stephens P, Stevens C, Teague J, et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res 2006;66:3987–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jager PL, de Korte MA, Lub-de Hooge MN, van Waarde A, Koopmans KP, Perik PJ, et al. Molecular imaging: what can be used today. Cancer Imaging 2005;5 Spec No A:S27–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mamede M, Higashi T, Kitaichi M, Ishizu K, Ishimori T, Nakamoto Y, et al. [18F]FDG uptake and PCNA, Glut-1, and Hexokinase-II expressions in cancers and inflammatory lesions of the lung. Neoplasia 2005;7:369–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao S, Kuge Y, Mochizuki T, Takahashi T, Nakada K, Sato M, et al. Biologic correlates of intratumoral heterogeneity in 18F-FDG distribution with regional expression of glucose transporters and hexokinase-II in experimental tumor. J Nucl Med 2005;46:675–82 [PubMed] [Google Scholar]
- 24.Young H, Baum R, Cremerius U, Herholz K, Hoekstra O, Lammertsma AA, et al. Measurement of clinical and subclinical tumour response using [18F]-Fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. European Organization for Research and Treatment of Cancer (EORTC) PET Study Group. Eur J Cancer 1999;35:1773–82 [DOI] [PubMed] [Google Scholar]
- 25.Phelps ME, Mazziotta JC. Positron emission tomography: human brain function and biochemistry. Science 1985;228:799–809 [DOI] [PubMed] [Google Scholar]
- 26.Oriuchi N, Higuchi T, Ishikita T, Miyakubo M, Hanaoka H, Iida Y, et al. Present role and future prospects of positron emission tomography in clinical oncology. Cancer Sci 2006;97:1291–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhuang H, Yu JQ, Alavi A. Applications of fluorodeoxyglucose-PET imaging in the detection of infection and inflammation and other benign disorders. Radiol Clin North Am 2005;43:121–34 [DOI] [PubMed] [Google Scholar]
- 28.Heiss WD, Heindel W, Herholz K, Rudolf J, Bunke J, Jeske J, et al. Positron emission tomography of fluorine-18-deoxyglucose and image-guided phosphorus-31 magnetic resonance spectroscopy in brain tumors. J Nucl Med 1990;31:302–10 [PubMed] [Google Scholar]
- 29.Herholz K, Heindel W, Luyten PR, denHollander JA, Pietrzyk U, Voges J, et al. In vivo imaging of glucose consumption and lactate concentration in human gliomas. Ann Neurol 1992;31:319–27 [DOI] [PubMed] [Google Scholar]
- 30.Herholz K, Pietrzyk U, Voges J, Schroder R, Halber M, Treuer H, et al. Correlation of glucose consumption and tumor cell density in astrocytomas. A stereotactic PET study. J Neurosurg 1993;79:853–8 [DOI] [PubMed] [Google Scholar]
- 31.Delbeke D, Meyerowitz C, Lapidus RL, Maciunas RJ, Jennings MT, Moots PL, et al. Optimal cutoff levels of F-18 fluorodeoxyglucose uptake in the differentiation of low-grade from high-grade brain tumors with PET. Radiology 1995;195:47–52 [DOI] [PubMed] [Google Scholar]
- 32.Barker FG, 2nd, Chang SM, Valk PE, Pounds TR, Prados MD. 18-Fluorodeoxyglucose uptake and survival of patients with suspected recurrent malignant glioma. Cancer 1997;79:115–26 [PubMed] [Google Scholar]
- 33.Goldman S, Levivier M, Pirotte B, Brucher JM, Wikler D, Damhaut P, et al. Regional glucose metabolism and histopathology of gliomas. A study based on positron emission tomography-guided stereotactic biopsy. Cancer 1996;78:1098–106 [DOI] [PubMed] [Google Scholar]
- 34.Kim EE, Chung SK, Haynie TP, Kim CG, Cho BJ, Podoloff DA, et al. Differentiation of residual or recurrent tumors from post-treatment changes with F-18 FDG PET. Radiographics 1992;12:269–79 [DOI] [PubMed] [Google Scholar]
- 35.Wurker M, Herholz K, Voges J, Pietrzyk U, Treuer H, Bauer B, et al. Glucose consumption and methionine uptake in low-grade gliomas after iodine-125 brachytherapy. Eur J Nucl Med 1996;23:583–6 [DOI] [PubMed] [Google Scholar]
- 36.Chaiken L, Rege S, Hoh C, Choi Y, Jabour B, Juillard G, et al. Positron emission tomography with fluorodeoxyglucose to evaluate tumor response and control after radiation therapy. Int J Radiat Oncol Biol Phys 1993;27:455–64 [DOI] [PubMed] [Google Scholar]
- 37.Schelling M, Avril N, Nahrig J, Kuhn W, Romer W, Sattler D, et al. Positron emission tomography using [(18)F]Fluorodeoxyglucose for monitoring primary chemotherapy in breast cancer. J Clin Oncol 2000;18:1689–95 [DOI] [PubMed] [Google Scholar]
- 38.Kostakoglu L, Coleman M, Leonard JP, Kuji I, Zoe H, Goldsmith SJ. PET predicts prognosis after 1 cycle of chemotherapy in aggressive lymphoma and Hodgkin’s disease. J Nucl Med 2002;43:1018–27 [PubMed] [Google Scholar]
- 39.Weber WA, Petersen V, Schmidt B, Tyndale-Hines L, Link T, Peschel C, et al. Positron emission tomography in non-small-cell lung cancer: prediction of response to chemotherapy by quantitative assessment of glucose use. J Clin Oncol 2003;21:2651–7 [DOI] [PubMed] [Google Scholar]
- 40.Herholz K, Coope D, Jackson A. Metabolic and molecular imaging in neuro-oncology. Lancet Neurol 2007;6:711–24 [DOI] [PubMed] [Google Scholar]
- 41.Brock CS, Young H, O’Reilly SM, Matthews J, Osman S, Evans H, et al. Early evaluation of tumour metabolic response using [18F]fluorodeoxyglucose and positron emission tomography: a pilot study following the phase II chemotherapy schedule for temozolomide in recurrent high-grade gliomas. Br J Cancer 2000;82:608–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Charnley N, West CM, Barnett CM, Brock C, Bydder GM, Glaser M, et al. Early change in glucose metabolic rate measured using FDG-PET in patients with high-grade glioma predicts response to temozolomide but not temozolomide plus radiotherapy. Int J Radiat Oncol Biol Phys 2006;66:331–8 [DOI] [PubMed] [Google Scholar]
- 43.Reinhardt MJ, Kubota K, Yamada S, Iwata R, Yaegashi H. Assessment of cancer recurrence in residual tumors after fractionated radiotherapy: a comparison of fluorodeoxyglucose, L-methionine and thymidine. J Nucl Med 1997;38:280–7 [PubMed] [Google Scholar]
- 44.Vlassenko AG, Thiessen B, Beattie BJ, Malkin MG, Blasberg RG. Evaluation of early response to SU101 target-based therapy in patients with recurrent supratentorial malignant gliomas using FDG PET and Gd-DTPA MRI. J Neurooncol 2000;46:249–59 [DOI] [PubMed] [Google Scholar]
- 45.Rueger MA, Kracht LW, Hilker R, Thiel A, Sobesky J, Winkeler A, et al. Role of in vivo imaging of the central nervous system for developing novel drugs. Q J Nucl Med Mol Imaging 2007;51:164–81 [PubMed] [Google Scholar]
- 46.Sibtain NA, Howe FA, Saunders DE. The clinical value of proton magnetic resonance spectroscopy in adult brain tumours. Clin Radiol 2007;62:109–19 [DOI] [PubMed] [Google Scholar]
- 47.Howe FA, Opstad KS. 1H MR spectroscopy of brain tumours and masses. NMR Biomed 2003;16:123–31 [DOI] [PubMed] [Google Scholar]
- 48.Castillo M, Kwock L. Proton MR spectroscopy of common brain tumors. Neuroimaging Clin N Am 1998;8:733–52 [PubMed] [Google Scholar]
- 49.Maintz D, Heindel W, Kugel H, Jaeger R, Lackner KJ. Phosphorus-31 MR spectroscopy of normal adult human brain and brain tumours. NMR Biomed 2002;15:18–27 [DOI] [PubMed] [Google Scholar]
- 50.Jager PL, Vaalburg W, Pruim J, de Vries EG, Langen KJ, Piers DA. Radiolabeled amino acids: basic aspects and clinical applications in oncology. J Nucl Med 2001;42:432–45 [PubMed] [Google Scholar]
- 51.Kubota R, Kubota K, Yamada S, Tada M, Takahashi T, Iwata R, et al. Methionine uptake by tumor tissue: a microautoradiographic comparison with FDG. J Nucl Med 1995;36:484–92 [PubMed] [Google Scholar]
- 52.Miyagawa T, Oku T, Uehara H, Desai R, Beattie B, Tjuvajev J, et al. “Facilitated” amino acid transport is upregulated in brain tumors. J Cereb Blood Flow Metab 1998;18:500–9 [DOI] [PubMed] [Google Scholar]
- 53.Bergstrom M, Lundqvist H, Ericson K, Lilja A, Johnstrom P, Langstrom B, et al. Comparison of the accumulation kinetics of L-(methyl-11C)-methionine and D-(methyl-11C)-methionine in brain tumors studied with positron emission tomography. Acta Radiol 1987;28:225–9 [PubMed] [Google Scholar]
- 54.Kracht LW, Miletic H, Busch S, Jacobs AH, Voges J, Hoevels M, et al. Delineation of brain tumor extent with [11C]L-methionine positron emission tomography: local comparison with stereotactic histopathology. Clin Cancer Res 2004;10:7163–70 [DOI] [PubMed] [Google Scholar]
- 55.Kracht LW, Friese M, Herholz K, Schroeder R, Bauer B, Jacobs A, et al. Methyl-[11C]- l-methionine uptake as measured by positron emission tomography correlates to microvessel density in patients with glioma. Eur J Nucl Med Mol Imaging 2003;30:868–73 [DOI] [PubMed] [Google Scholar]
- 56.Sato N, Suzuki M, Kuwata N, Kuroda K, Wada T, Beppu T, et al. Evaluation of the malignancy of glioma using 11C-methionine positron emission tomography and proliferating cell nuclear antigen staining. Neurosurg Rev 1999;22:210–14 [DOI] [PubMed] [Google Scholar]
- 57.Sasaki M, Kuwabara Y, Yoshida T, Nakagawa M, Fukumura T, Mihara F, et al. A comparative study of thallium-201 SPET, carbon-11 methionine PET and fluorine-18 fluorodeoxyglucose PET for the differentiation of astrocytic tumours. Eur J Nucl Med 1998;25:1261–9 [DOI] [PubMed] [Google Scholar]
- 58.Herholz K, Holzer T, Bauer B, Schroder R, Voges J, Ernestus RI, et al. 11C-methionine PET for differential diagnosis of low-grade gliomas. Neurology 1998;50:1316–22 [DOI] [PubMed] [Google Scholar]
- 59.Nuutinen J, Sonninen P, Lehikoinen P, Sutinen E, Valavaara R, Eronen E, et al. Radiotherapy treatment planning and long-term follow-up with [(11)C]methionine PET in patients with low-grade astrocytoma. Int J Radiat Oncol Biol Phys 2000;48:43–52 [DOI] [PubMed] [Google Scholar]
- 60.Thiel A, Pietrzyk U, Sturm V, Herholz K, Hovels M, Schroder R. Enhanced accuracy in differential diagnosis of radiation necrosis by positron emission tomography-magnetic resonance imaging coregistration: technical case report. Neurosurgery 2000;46:232–4 [PubMed] [Google Scholar]
- 61.Tang BN, Sadeghi N, Branle F, De Witte O, Wikler D, Goldman S. Semi-quantification of methionine uptake and flair signal for the evaluation of chemotherapy in low-grade oligodendroglioma. J Neurooncol 2005;71:161–8 [DOI] [PubMed] [Google Scholar]
- 62.Herholz K, Kracht LW, Heiss WD. Monitoring the effect of chemotherapy in a mixed glioma by C-11-methionine PET. J Neuroimaging 2003;13:269–71 [PubMed] [Google Scholar]
- 63.Galldiks N, Kracht LW, Burghaus L, Thomas A, Jacobs AH, Heiss WD, et al. Use of 11C-methionine PET to monitor the effects of temozolomide chemotherapy in malignant gliomas. Eur J Nucl Med Mol Imaging 2006;33:516–24 [DOI] [PubMed] [Google Scholar]
- 64.Pöpperl G, Goldbrunner R, Gildehaus FJ, Kreth FW, Tanner P, Holtmannspötter M, et al. O-(2-[18F]fluoroethyl)-L-tyrosine PET for monitoring the effects of convection-enhanced delivery of paclitaxel in patients with recurrent glioblastoma. Eur J Nucl Med Mol Imaging 2005;32:1018–25 [DOI] [PubMed] [Google Scholar]
- 65.Singhal T, Narayanan TK, Jain V, Mukherjee J, Mantil J. 11C-L-methionine positron emission tomography in the clinical management of cerebral gliomas. Mol Imaging Biol 2008;10:1–18 [DOI] [PubMed] [Google Scholar]
- 66.Sherley JL, Kelly TJ. Regulation of human thymidine kinase during the cell cycle. J Biol Chem 1988;263:8350–8 [PubMed] [Google Scholar]
- 67.Shields AF, Mankoff D, Graham MM, Zheng M, Kozawa SM, Link JM, et al. Analysis of 2-carbon-11-thymidine blood metabolites in PET imaging. J Nucl Med 1996;37:290–6 [PubMed] [Google Scholar]
- 68.Wells P, Aboagye E, Gunn RN, Osman S, Boddy AV, Taylor GA, et al. 2-[11C]thymidine positron emission tomography as an indicator of thymidylate synthase inhibition in patients treated with AG337. J Natl Cancer Inst 2003;95:675–82 [DOI] [PubMed] [Google Scholar]
- 69.Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Lawhorn-Crews JM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med 1998;4:1334–6 [DOI] [PubMed] [Google Scholar]
- 70.Been LB, Suurmeijer AJ, Cobben DC, Jager PL, Hoekstra HJ, Elsinga PH. [18F]FLT-PET in oncology: current status and opportunities. Eur J Nucl Med Mol Imaging 2004;31:1659–72 [DOI] [PubMed] [Google Scholar]
- 71.Vesselle H, Grierson J, Muzi M, Pugsley JM, Schmidt RA, Rabinowitz P, et al. In vivo validation of 3′deoxy-3′-[(18)F]fluorothymidine ([(18)F]FLT) as a proliferation imaging tracer in humans: correlation of [(18)F]FLT uptake by positron emission tomography with Ki-67 immunohistochemistry and flow cytometry in human lung tumors. Clin Cancer Res 2002;8:3315–23 [PubMed] [Google Scholar]
- 72.Wells P, Gunn RN, Alison M, Steel C, Golding M, Ranicar AS, et al. Assessment of proliferation in vivo using 2-[(11)C]thymidine positron emission tomography in advanced intra-abdominal malignancies. Cancer Res 2002;62:5698–702 [PubMed] [Google Scholar]
- 73.Buck AK, Bommer M, Stilgenbauer S, Juweid M, Glatting G, Schirrmeister H, et al. Molecular imaging of proliferation in malignant lymphoma. Cancer Res 2006;66:11055–61 [DOI] [PubMed] [Google Scholar]
- 74.Buck AK, Schirrmeister H, Hetzel M, Von DerHeide M, Halter G, Glatting G, et al. 3-deoxy-3-[(18)F]fluorothymidine-positron emission tomography for noninvasive assessment of proliferation in pulmonary nodules. Cancer Res 2002;62:3331–4 [PubMed] [Google Scholar]
- 75.Wagner M, Seitz U, Buck A, Neumaier B, Schultheiss S, Bangerter M, et al. 3′-[18F]fluoro-3′-deoxythymidine ([18F-FLT) as positron emission tomography tracer for imaging proliferation in a murine B-Cell lymphoma model and in the human disease. Cancer Res 2003;63:2681–7 [PubMed] [Google Scholar]
- 76.Kenny LM, Vigushin DM, Al-Nahhas A, Osman S, Luthra SK, Shousha S, et al. Quantification of cellular proliferation in tumor and normal tissues of patients with breast cancer by [18F]fluorothymidine-positron emission tomography imaging: evaluation of analytical methods. Cancer Res 2005;65:10104–12 [DOI] [PubMed] [Google Scholar]
- 77.Muzi M, Vesselle H, Grierson JR, Mankoff DA, Schmidt RA, Peterson L, et al. Kinetic analysis of 3′-deoxy-3′-fluorothymidine PET studies: validation studies in patients with lung cancer. J Nucl Med 2005;46:274–82 [PubMed] [Google Scholar]
- 78.Jacobs AH, Thomas A, Kracht LW, Li H, Dittmar C, Garlip G, et al. 18F-fluoro-L-thymidine and 11C-methylmethionine as markers of increased transport and proliferation in brain tumors. J Nucl Med 2005;46:1948–58 [PubMed] [Google Scholar]
- 79.Ullrich R, Backes H, Li H, Kracht L, Miletic H, Kesper K, et al. Glioma proliferation as assessed by 3′-fluoro-3′-deoxy-L-thymidine positron emission tomography in patients with newly diagnosed high-grade glioma. Clin Cancer Res 2008;14:2049–55 [DOI] [PubMed] [Google Scholar]
- 80.Chen W, Delaloye S, Silverman DH, Geist C, Czernin J, Sayre J, et al. Predicting treatment response of malignant gliomas to bevacizumab and irinotecan by imaging proliferation with [18F] fluorothymidine positron emission tomography: a pilot study. J Clin Oncol 2007;25:4714–21 [DOI] [PubMed] [Google Scholar]
- 81.Herrmann K, Wieder HA, Buck AK, Schoffel M, Krause BJ, Fend F, et al. Early response assessment using 3′-deoxy-3′-[18F]fluorothymidine-positron emission tomography in high-grade non-Hodgkin’s lymphoma. Clin Cancer Res 2007;13:3552–8 [DOI] [PubMed] [Google Scholar]
- 82.Ullrich RT, Zander T, Neumaier B, Koker M, Shimamura T, Waerzeggers Y, et al. Early detection of erlotinib treatment response in NSCLC by 3′-Deoxy-3′- [18F-Fluoro-L-thymidine ([18F]FLT) positron emission tomography (PET). PlosOne 2008;3:e3908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Scott JN, Brasher PM, Sevick RJ, Rewcastle NB, Forsyth PA. How often are nonenhancing supratentorial gliomas malignant? A population study. Neurology 2002;59:947–9 [DOI] [PubMed] [Google Scholar]
- 84.Kumar AJ, Leeds NE, Fuller GN, Van Tassel P, Maor MH, Sawaya RE, et al. Malignant gliomas: MR imaging spectrum of radiation therapy- and chemotherapy-induced necrosis of the brain after treatment. Radiology 2000;217:377–84 [DOI] [PubMed] [Google Scholar]
- 85.Vos MJ, Uitdehaag BM, Barkhof F, Heimans JJ, Baayen HC, Boogerd W, et al. Interobserver variability in the radiological assessment of response to chemotherapy in glioma. Neurology 2003;60:826–30 [DOI] [PubMed] [Google Scholar]
- 86.Jacobs AH, Voges J, Kracht LW, Dittmar C, Winkeler A, Thomas A, et al. Imaging in gene therapy of patients with glioma. J Neurooncol 2003;65:291–305 [DOI] [PubMed] [Google Scholar]
- 87.Sugahara T, Korogi Y, Kochi M, Ikushima I, Shigematu Y, Hirai T, et al. Usefulness of diffusion-weighted MRI with echo-planar technique in the evaluation of cellularity in gliomas. J Magn Reson Imaging 1999;9:53–60 [DOI] [PubMed] [Google Scholar]
- 88.Pauleit D, Langen KJ, Floeth F, Hautzel H, Riemenschneider MJ, Reifenberger G, et al. Can the apparent diffusion coefficient be used as a noninvasive parameter to distinguish tumor tissue from peritumoral tissue in cerebral gliomas? J Magn Reson Imaging 2004;20:758–64 [DOI] [PubMed] [Google Scholar]
- 89.Price DT, Coleman RE, Liao RP, Robertson CN, Polascik TJ, DeGrado TR. Comparison of [18 F]fluorocholine and [18 F]fluorodeoxyglucose for positron emission tomography of androgen dependent and androgen independent prostate cancer. J Urol 2002;168:273–80 [PubMed] [Google Scholar]
- 90.Kwee SA, Coel MN, Lim J, Ko JP. Combined use of F-18 fluorocholine positron emission tomography and magnetic resonance spectroscopy for brain tumor evaluation. J Neuroimaging 2004;14:285–9 [DOI] [PubMed] [Google Scholar]
- 91.Glunde K, Jie C, Bhujwalla ZM. Molecular causes of the aberrant choline phospholipid metabolism in breast cancer. Cancer Res 2004;64:4270–6 [DOI] [PubMed] [Google Scholar]
- 92.Moller-Hartmann W, Herminghaus S, Krings T, Marquardt G, Lanfermann H, Pilatus U, et al. Clinical application of proton magnetic resonance spectroscopy in the diagnosis of intracranial mass lesions. Neuroradiology 2002;44:371–81 [DOI] [PubMed] [Google Scholar]
- 93.Kuesel AC, Sutherland GR, Halliday W, Smith IC. 1H MRS of high grade astrocytomas: mobile lipid accumulation in necrotic tissue. NMR Biomed 1994;7:149–55 [DOI] [PubMed] [Google Scholar]
- 94.Rock JP, Hearshen D, Scarpace L, Croteau D, Gutierrez J, Fisher JL, et al. Correlations between magnetic resonance spectroscopy and image-guided histopathology, with special attention to radiation necrosis. Neurosurgery 2002;51:912–9; discussion 919–20 [DOI] [PubMed] [Google Scholar]
- 95.Graves EE, Nelson SJ, Vigneron DB, Verhey L, McDermott M, Larson D, et al. Serial proton MR spectroscopic imaging of recurrent malignant gliomas after gamma knife radiosurgery. AJNR Am J Neuroradiol 2001;22:613–24 [PMC free article] [PubMed] [Google Scholar]
- 96.Hakumaki JM, Poptani H, Sandmair AM, Yla-Herttuala S, Kauppinen RA. 1H MRS detects polyunsaturated fatty acid accumulation during gene therapy of glioma: implications for the in vivo detection of apoptosis. Nat Med 1999;5:1323–7 [DOI] [PubMed] [Google Scholar]
- 97.Chung YL, Troy H, Banerji U, Jackson LE, Walton MI, Stubbs M, et al. Magnetic resonance spectroscopic pharmacodynamic markers of the heat shock protein 90 inhibitor 17-allylamino,17-demethoxygeldanamycin (17AAG) in human colon cancer models. J Natl Cancer Inst 2003;95:1624–33 [DOI] [PubMed] [Google Scholar]
- 98.Al-Saffar NM, Troy H, Ramirez deMolina A, Jackson LE, Madhu B, Griffiths JR, et al. Noninvasive magnetic resonance spectroscopic pharmacodynamic markers of the choline kinase inhibitor MN58b in human carcinoma models. Cancer Res 2006;66:427–34 [DOI] [PubMed] [Google Scholar]
- 99.Beloueche-Babari M, Jackson LE, Al-Saffar NM, Workman P, Leach MO, Ronen SM. Magnetic resonance spectroscopy monitoring of mitogen-activated protein kinase signaling inhibition. Cancer Res 2005;65:3356–63 [DOI] [PubMed] [Google Scholar]
- 100.Jouanneau E. Angiogenesis and gliomas: current issues and development of surrogate markers. Neurosurgery 2008;62:31–50; discussion 50–2 [DOI] [PubMed] [Google Scholar]
- 101.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996;86:353–64 [DOI] [PubMed] [Google Scholar]
- 102.Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat Med 2001;7:1194–201 [DOI] [PubMed] [Google Scholar]
- 103.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature 2000;407:249–57 [DOI] [PubMed] [Google Scholar]
- 104.Michor F, Nowak MA, Iwasa Y. Evolution of resistance to cancer therapy. Curr Pharm Des 2006;12:261–71 [DOI] [PubMed] [Google Scholar]
- 105.Gaya AM, Rustin GJ. Vascular disrupting agents: a new class of drug in cancer therapy. Clin Oncol (R Coll Radiol) 2005;17:277–90 [DOI] [PubMed] [Google Scholar]
- 106.O’Hanlon LH. Taking down tumors: vascular disrupting agents entering clinical trials. J Natl Cancer Inst 2005;97:1244–5 [DOI] [PubMed] [Google Scholar]
- 107.Kabbinavar FF, Hambleton J, Mass RD, Hurwitz HI, Bergsland E, Sarkar S. Combined analysis of efficacy: the addition of bevacizumab to fluorouracil/leucovorin improves survival for patients with metastatic colorectal cancer. J Clin Oncol 2005;23:3706–12 [DOI] [PubMed] [Google Scholar]
- 108.Dijkhuizen RM, Nicolay K. Magnetic resonance imaging in experimental models of brain disorders. J Cereb Blood Flow Metab 2003;23:1383–402 [DOI] [PubMed] [Google Scholar]
- 109.Tofts PS. Modeling tracer kinetics in dynamic Gd-DTPA MR imaging. J Magn Reson Imaging 1997;7:91–101 [DOI] [PubMed] [Google Scholar]
- 110.Cao Y, Shen Z, Chenevert TL, Ewing JR. Estimate of vascular permeability and cerebral blood volume using Gd-DTPA contrast enhancement and dynamic T2*-weighted MRI. J Magn Reson Imaging 2006;24:288–96 [DOI] [PubMed] [Google Scholar]
- 111.Li KL, Zhu XP, Waterton J, Jackson A. Improved 3D quantitative mapping of blood volume and endothelial permeability in brain tumors. J Magn Reson Imaging 2000;12:347–57 [DOI] [PubMed] [Google Scholar]
- 112.Vonken EP, van Osch MJ, Bakker CJ, Viergever MA. Simultaneous quantitative cerebral perfusion and Gd-DTPA extravasation measurement with dual-echo dynamic susceptibility contrast MRI. Magn Reson Med 2000;43:820–7 [DOI] [PubMed] [Google Scholar]
- 113.Brasch R, Turetschek K. MRI characterization of tumors and grading angiogenesis using macromolecular contrast media: status report. Eur J Radiol 2000;34:148–55 [DOI] [PubMed] [Google Scholar]
- 114.Pathak AP, Schmainda KM, Ward BD, Linderman JR, Rebro KJ, Greene AS. MR-derived cerebral blood volume maps: issues regarding histological validation and assessment of tumor angiogenesis. Magn Reson Med 2001;46:735–47 [DOI] [PubMed] [Google Scholar]
- 115.Cha S, Johnson G, Wadghiri YZ, Jin O, Babb J, Zagzag D, et al. Dynamic, contrast-enhanced perfusion MRI in mouse gliomas: correlation with histopathology. Magn Reson Med 2003;49:848–55 [DOI] [PubMed] [Google Scholar]
- 116.Law M, Yang S, Babb JS, Knopp EA, Golfinos JG, Zagzag D, et al. Comparison of cerebral blood volume and vascular permeability from dynamic susceptibility contrast-enhanced perfusion MR imaging with glioma grade. AJNR Am J Neuroradiol 2004;25:746–55 [PMC free article] [PubMed] [Google Scholar]
- 117.Law M, Yang S, Wang H, Babb JS, Johnson G, Cha S, et al. Glioma grading: sensitivity, specificity, and predictive values of perfusion MR imaging and proton MR spectroscopic imaging compared with conventional MR imaging. AJNR Am J Neuroradiol 2003;24:1989–98 [PMC free article] [PubMed] [Google Scholar]
- 118.Law M, Oh S, Johnson G, Babb JS, Zagzag D, Golfinos J, et al. Perfusion magnetic resonance imaging predicts patient outcome as an adjunct to histopathology: a second reference standard in the surgical and nonsurgical treatment of low-grade gliomas. Neurosurgery 2006;58:1099–1107; Discussion 1099–107 [DOI] [PubMed] [Google Scholar]
- 119.Morgan B, Thomas AL, Drevs J, Hennig J, Buchert M, Jivan A, et al. Dynamic contrast-enhanced magnetic resonance imaging as a biomarker for the pharmacological response of PTK787/ZK 222584, an inhibitor of the vascular endothelial growth factor receptor tyrosine kinases, in patients with advanced colorectal cancer and liver metastases: results from two phase I studies. J Clin Oncol 2003;21:3955–64 [DOI] [PubMed] [Google Scholar]
- 120.Wedam SB, Low JA, Yang SX, Chow CK, Choyke P, Danforth D, et al. Antiangiogenic and antitumor effects of bevacizumab in patients with inflammatory and locally advanced breast cancer. J Clin Oncol 2006;24:769–77 [DOI] [PubMed] [Google Scholar]
- 121.Akella NS, Twieg DB, Mikkelsen T, Hochberg FH, Grossman S, Cloud GA, et al. Assessment of brain tumor angiogenesis inhibitors using perfusion magnetic resonance imaging: quality and analysis results of a phase I trial. J Magn Reson Imaging 2004;20:913–22 [DOI] [PubMed] [Google Scholar]
- 122.Cha S, Knopp EA, Johnson G, Litt A, Glass J, Gruber ML, et al. Dynamic contrast-enhanced T2-weighted MR imaging of recurrent malignant gliomas treated with thalidomide and carboplatin. AJNR Am J Neuroradiol 2000;21:881–90 [PMC free article] [PubMed] [Google Scholar]
- 123.Laking GR, Price PM. Positron emission tomographic imaging of angiogenesis and vascular function. Br J Radiol 2003;76(Spec. No. 1):S50–9 [DOI] [PubMed] [Google Scholar]
- 124.McDonald DM, Choyke PL. Imaging of angiogenesis: from microscope to clinic. Nat Med 2003;9:713–25 [DOI] [PubMed] [Google Scholar]
- 125.Neeman M, Dafni H, Bukhari O, Braun RD, Dewhirst MW. In vivo BOLD contrast MRI mapping of subcutaneous vascular function and maturation: validation by intravital microscopy. Magn Reson Med 2001;45:887–98 [DOI] [PubMed] [Google Scholar]
- 126.Giaccia AJ, Simon MC, Johnson R. The biology of hypoxia: the role of oxygen sensing in development, normal function, and disease. Genes Dev 2004;18:2183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hopfl G, Ogunshola O, Gassmann M. HIFs and tumors–causes and consequences. Am J Physiol Regul Integr Comp Physiol 2004;286:R608–23 [DOI] [PubMed] [Google Scholar]
- 128.Coleman CN, Mitchell JB, Camphausen K. Tumor hypoxia: chicken, egg, or a piece of the farm? J Clin Oncol 2002;20:610–15 [DOI] [PubMed] [Google Scholar]
- 129.Rajendran JG, Mankoff DA, O’Sullivan F, Peterson LM, Schwartz DL, Conrad EU, et al. Hypoxia and glucose metabolism in malignant tumors: evaluation by [18F] fluoromisonidazole and [18F]fluorodeoxyglucose positron emission tomography imaging. Clin Cancer Res 2004;10:2245–52 [DOI] [PubMed] [Google Scholar]
- 130.Amellem O, Pettersen EO. Cell inactivation and cell cycle inhibition as induced by extreme hypoxia: the possible role of cell cycle arrest as a protection against hypoxia-induced lethal damage. Cell Prolif 1991;24:127–41 [DOI] [PubMed] [Google Scholar]
- 131.Bruehlmeier M, Roelcke U, Schubiger PA, Ametamey SM. Assessment of hypoxia and perfusion in human brain tumors using PET with 18F-fluoromisonidazole and 15O-H2O. J Nucl Med 2004;45:1851–9 [PubMed] [Google Scholar]
- 132.Cher LM, Murone C, Lawrentschuk N, Ramdave S, Papenfuss A, Hannah A, et al. Correlation of hypoxic cell fraction and angiogenesis with glucose metabolic rate in gliomas using 18F-fluoromisonidazole, 18F-FDG PET, and immunohistochemical studies. J Nucl Med 2006;47:410–18 [PubMed] [Google Scholar]
- 133.Valk PE, Mathis CA, Prados MD, Gilbert JC, Budinger TF. Hypoxia in human gliomas: demonstration by PET with fluorine-18-fluoromisonidazole. J Nucl Med 1992;33:2133–7 [PubMed] [Google Scholar]
- 134.Robinson SP, Howe FA, Rodrigues LM, Stubbs M, Griffiths JR. Magnetic resonance imaging techniques for monitoring changes in tumor oxygenation and blood flow. Semin Radiat Oncol 1998;8:197–207 [DOI] [PubMed] [Google Scholar]
- 135.Buchholz TA, Davis DW, McConkey DJ, Symmans WF, Valero V, Jhingran A, et al. Chemotherapy-induced apoptosis and Bcl-2 levels correlate with breast cancer response to chemotherapy. Cancer J 2003;9:33–41 [DOI] [PubMed] [Google Scholar]
- 136.Dubray B, Breton C, Delic J, Klijanienko J, Maciorowski Z, Vielh P, et al. In vitro radiation-induced apoptosis and early response to low-dose radiotherapy in non-Hodgkin’s lymphomas. Radiother Oncol 1998;46:185–91 [DOI] [PubMed] [Google Scholar]
- 137.Williams SN, Anthony ML, Brindle KM. Induction of apoptosis in two mammalian cell lines results in increased levels of fructose-1,6-bisphosphate and CDP-choline as determined by 31P MRS. Magn Reson Med 1998;40:411–20 [DOI] [PubMed] [Google Scholar]
- 138.Blankenberg FG, Storrs RW, Naumovski L, Goralski T, Spielman D. Detection of apoptotic cell death by proton nuclear magnetic resonance spectroscopy. Blood 1996;87:1951–6 [PubMed] [Google Scholar]
- 139.Blankenberg FG. In vivo detection of apoptosis. J Nucl Med 2008;49(Suppl. 2):81S–95S [DOI] [PubMed] [Google Scholar]
- 140.Kettunen MI, Grohn OH. Tumour gene therapy monitoring using magnetic resonance imaging and spectroscopy. Curr Gene Ther 2005;5:685–96 [DOI] [PubMed] [Google Scholar]
- 141.Chenevert TL, Stegman LD, Taylor JM, Robertson PL, Greenberg HS, Rehemtulla A, et al. Diffusion magnetic resonance imaging: an early surrogate marker of therapeutic efficacy in brain tumors. J Natl Cancer Inst 2000;92:2029–36 [DOI] [PubMed] [Google Scholar]
- 142.Lemort M, Canizares-Perez AC, Van derStappen A, Kampouridis S. Progress in magnetic resonance imaging of brain tumours. Curr Opin Oncol 2007;19:616–22 [DOI] [PubMed] [Google Scholar]
- 143.Moffat BA, Chenevert TL, Lawrence TS, Meyer CR, Johnson TD, Dong Q, et al. Functional diffusion map: a noninvasive MRI biomarker for early stratification of clinical brain tumor response. Proc Natl Acad Sci U S A 2005;102:5524–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hamstra DA, Chenevert TL, Moffat BA, Johnson TD, Meyer CR, Mukherji SK, et al. Evaluation of the functional diffusion map as an early biomarker of time-to-progression and overall survival in high-grade glioma. Proc Natl Acad Sci U S A 2005;102:16759–64 [DOI] [PMC free article] [PubMed] [Google Scholar]