

Abstract
Pulmonary lymphoproliferative disorders (LPD) are characterised by abnormal proliferation of indigenous cell lines or infiltration of lung parenchyma by lymphoid cells. They encompass a wide spectrum of focal or diffuse abnormalities, which may be classified as reactive or neoplastic on the basis of cellular morphology and clonality. The spectrum of reactive disorders results primarily from antigenic stimulation of bronchial mucosa-associated lymphoid tissue (MALT) and comprises three main entities: follicular bronchiolitis, lymphoid interstitial pneumonia and (more rarely) nodular lymphoid hyperplasia. Primary parenchymal neoplasms are most commonly extranodal marginal zone lymphomas of MALT origin (MALT lymphomas), followed by diffuse large B-cell lymphomas (DLBCLs) and lymphomatoid granulomatosis (LYG). Secondary lymphomatous parenchymal neoplasms (both Hodgkin and non-Hodgkin lymphomas) are far more prevalent than primary neoplasms. Acquired immune deficiency syndrome (AIDS)-related lymphoma (ARL) and post-transplantation lymphoproliferative disorder (PTLD) may also primarily affect the lung parenchyma. Modern advances in treatments for AIDS and transplant medicine are associated with an increase in the incidence of LPD and have heightened the need to understand the range of imaging appearance of these diseases. The multidetector CT (MDCT) findings of LPD are heterogeneous, thereby reflecting the wide spectrum of clinical manifestations of these entities. Understanding the spectrum of LPD and the various imaging manifestations is crucial because the radiologist is often the first one to suggest the diagnosis and has a pivotal role in differentiating these diseases. The current concepts of LPD are discussed together with a demonstration of the breadth of MDCT patterns within this disease spectrum.
The native lymphoid tissue in the lung was first described by Bienenstock et al [1] in 1973. It mainly comprises submucosal lymphoid aggregates distributed along the bifurcation of bronchioles and along lymphatic routes, termed bronchial mucosa-associated lymphoid tissue (MALT). Bronchial MALT is usually inconspicuous in healthy adult individuals, but may become hyperplastic secondary to antigenic stimulation. A wide array of inflammatory conditions might cause reactive changes of the MALT, such as infection, occupational exposure to organic/inorganic matter or cigarette smoking [2]. However, these represent normal and desired immunological reactions which are not considered in the category of lymphoproliferative disorders (LPDs).
The reactive LPDs are associated with immunological disturbances and are commonly seen in patients with immunodeficiency or autoimmune disorders. The neoplastic LPDs can be a manifestation of any of the lymphoid malignancies seen elsewhere in the body, but some specific entities are more commonly seen in the lungs. Not coincidentally, extranodal marginal zone lymphoma of bronchial MALT (MALT lymphoma) is the leading subtype and is also usually associated with immunological disturbances. The terminology used herein is based on the current 2008 World Health Organization (WHO) classification [3].
Multidetector CT (MDCT) has been established as the imaging modality of choice in the evaluation of diseases of the lung parenchyma. The MDCT findings of LPDs are heterogeneous, reflecting the wide spectrum of clinical manifestations of these entities. Imaging findings are frequently non-specific and require correlation with clinical findings. Histological confirmation is required for a definite diagnosis. Notably, histological diagnosis of lymphoma can be difficult on small biopsy specimens, and a multidisciplinary approach with imaging correlation may be necessary in these cases.
Classification of lymphoproliferative disorders
Figure 1 provides an overall classification of lymphoproliferative disorders, which are described further below.
Figure 1.
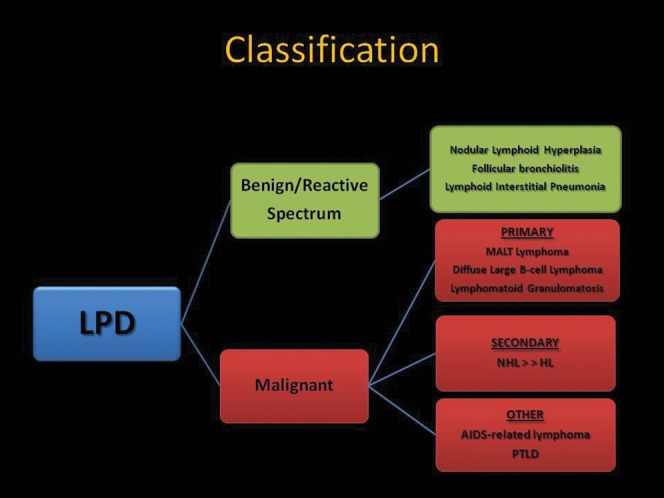
Schematic illustration of a classification system for the pulmonary lymphoproliferative disorders (LPDs). AIDS, acquired immune deficiency syndrome; HL Hodgkin lymphoma; MALT, mucosa–associated lymphoid tissue; NHL, non–Hodgkin lymphoma; PTLD, post-transplantation lymphoproliferative disorder.
Reactive/non-neoplastic lymphoid lesions
This group of benign rare diseases comprises a spectrum of three entities, each with differing patterns and distribution of a polyclonal lymphoid infiltrate:
nodular lymphoid hyperplasia (NLH): focal
follicular bronchiolitis (FB): peribronchial
lymphoid interstitial pneumonia (LIP): diffuse.
Malignant parenchymal lymphoproliferative lesions
Malignant LPDs can be primary (rare) or secondary (common). Although more common in the immunocompetent host, they are also frequently seen in immunocompromised patients, mainly post transplant and human immunodeficiency virus (HIV)-positive patients.
Primary malignant lymphoproliferative disorders
Primary pulmonary lymphomas represent only 0.5% of all primary lung neoplasms. Any given lymphoma can primarily involve the lung; however, most of them are exceedingly rare and beyond the scope of this review. The malignant LPDs that, albeit rare, occasionally present with primary lung involvement include:
extranodal marginal zone lymphoma of MALT origin (MALT lymphoma)
diffuse large B-cell lymphoma (DLBCL)
lymphomatoid granulomatosis (LYG).
Secondary malignant lymphoproliferative disorders
Secondary involvement of the lungs is much more common and originates from nodal or extrathoracic lymphoma:
Non-Hodgkin lymphoma (NHL) is the commonest type of lymphoma overall (80–90% of all cases), mainly represented by the multiple mature B-cell subtypes (follicular lymphoma, small lymphocytic lymphoma, etc.). Nearly half of these patients have intrathoracic disease at presentation. Pulmonary parenchymal disease occurs in 24% of NHL [4].
Hodgkin lymphoma (HL) represents 10–20% of all lymphoma cases; 85% have intrathoracic disease at presentation. Pulmonary parenchymal disease occurs in 38% [4]. In HL, lung involvement is almost always associated with intrathoracic lymph node enlargement. This is not the case in NHL, where pulmonary involvement frequently occurs in the absence of mediastinal disease.
Lymphoproliferative disorders in the immunocompromised patient
LPDs occurring in the immunocompromised patient warrant special consideration as they are generally more aggressive and present with more varied radiological appearance. These include:
acquired immune deficiency syndrome (AIDS)-related lymphoma (ARL):
usual lymphomas primarily involving the lung, DLBCL and Burkitt lymphoma being the commonest
others: primary effusion lymphoma (PEL), plasmablastic lymphoma
post-transplantation lymphoproliferative disorder (PTLD), which includes reactive processes, neoplasms and lymphoid proliferations of uncertain nature.
Overview
See Tables 1 and 2 for summaries of CT features in the reactive spectrum and the malignant spectrum of pulmonary lymphoproliferative disorders, respectively.
Table 1. Summary of CT features in the reactive spectrum of pulmonary lymphoproliferative disorders.
| Benign spectrum | CT features |
| Nodular lymphoid hyperplasia | Discrete nodular mass (usually 2–3 cm in diameter)Mild focal lymphangitic extension |
| Follicular bronchiolitis | Diffuse small airways involvementNodules 1–3 mm in diameterNodules which can mimic “tree-in-bud”Bronchial dilatation |
| Lymphoid interstitial pneumonia | Diffuse interstitial involvementGround-glass opacityNodulesCystsPeribronchovascular thickening |
Table 2. Summary of CT features in the malignant spectrum of pulmonary lymphoproliferative disorders.
| Malignant spectrum | CT features |
| Primary malignant | |
| 1. MALT lymphoma | Nodules or consolidation |
| Multiple/bilateral (>70%) | |
| Peribronchovascular | |
| Hilar/mediastinal nodes (30%) | |
| 2. Diffuse large B-cell lymphoma | Nodule or mass |
| Cavitation in 50% | |
| 3. Lymphomatoid granulomatosis | Bilateral nodules/masses |
| Basal predominance | |
| Peribronchovascular | |
| Coalesce and/or cavitate | |
| Migratory | |
| Secondary malignant | |
| 1. Hodgkin lymphoma | Similar findings in both HL and NHL: |
| 2. Non-Hodgkin lymphoma | Varied and non-specific CT presentation |
| Nodules/masses with or without air bronchograms | |
| Lymphangitic spread | |
| Pleural effusion | |
| Lymphadenopathy HL>NHL | |
| LPD in immunocompromised | |
| 1. AIDS-related lymphoma | Well-defined nodules |
| Cavitating pulmonary mass | |
| Pleural effusion | |
| Lymphadenopathy | |
| 2. Post-transplantation lymphoproliferative disorder | Nodules: single>multiple |
| Usually well defined | |
| “Halo sign” | |
| Peribronchovascular/subpleural | |
| Lymphadenopathy (30–60%) |
HL, Hodgkin lymphoma; LPD, lymphoproliferative disorder; MALT, mucosa-associated lymphoid tissue; NHL, non-Hodgkin lymphoma.
Nodular lymphoid hyperplasia
The concept of reactive localised masses of lymphoid tissue in the lung was proposed in 1963 by Saltzstein [5] and initially called pseudolymphoma. However, this concept fell into disrepute with the discovery of marginal zone lymphoma of MALT origin. Abbondanzo et al [6] later validated the existence of nodular lymphoid hyperplasia (NLH; Figure 2) using immunohistochemistry and molecular genetic analysis in a small series of 14 cases. The term nodular lymphoid hyperplasia was suggested by Kradin and Mark in 1983 [7].
Figure 2.
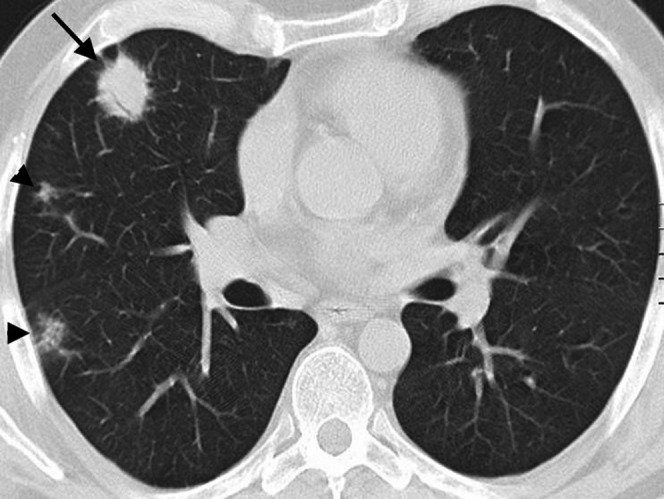
Multifocal nodular lymphoid hyperplasia in a 30-year-old female. Multidetector CT scan with lung window settings demonstrates a well-circumscibed pulmonary nodule (arrow) with an air bronchogram in the periphery of the right upper lobe. Note the mild surrounding linear opacities indicating focal lymphangitic extension. In addition two smaller nodules (arrowheads) are seen in the right lung. Biopsy of the larger nodule was consistent with nodular lymphoid hyperplasia.
NLH is a benign, localised, reactive polyclonal lymphoproliferative lesion, the aetiopathogenesis of which is unclear. Little is known about its incidence or natural history as it is a relatively rare entity [8]. The existence of a primary reactive focal lymphoid hyperplasia has traditionally been a controversial topic, most of the cases representing secondary reactions to focal inflammatory conditions. The median age at presentation is 65 years (age range 19–80 years), with an even sex distribution.
NLH is usually asymptomatic and found incidentally on imaging studies (70%) [6]. Occasionally, it can present with shortness of breath, cough and/or pleuritic chest pain. Surgical resection has proved curative in the few cases described.
Histopathology
NLH is characterised by a dense nodular infiltration of mature, polyclonal lymphocytes and plasma cells with multiple reactive germinal centres, sharply demarcated from surrounding parenchyma and with central areas of scarring. Foci of organising pneumonia are frequently seen in the periphery. Mild, local lymphangitic spread of lymphocytes may be present, permeating into the perivascular interstitium.
Imaging findings
Because of its rarity, no CT studies of a series of patients are available. Most of the imaging descriptions refer to the appearances on chest radiographs or single case reports. NLH has been described most commonly as a solitary lesion, although multiple pulmonary nodules may be seen. It can also manifest with focal consolidation and may regress or remain stable for years.
Although it may be difficult to distinguish NLH and MALT lymphoma radiologically and pathologically, there are some features that can help in the radiological differential diagnosis;
NLH is more often seen as a solitary nodule unlike MALT lymphoma, which usually presents with multiple, bilateral nodules.
Pleural invasion favours MALT lymphoma.
Prominent lymphangitic extension favours MALT lymphoma (however, occasional, limited focal lymphangitic extension can be seen in NLH).
Moreover, NLH is rare whereas MALT lymphoma is relatively common. Importantly, as the natural history of NLH has not been well described, biopsy-proven patients with multiple persistent lesions usually undergo continued surveillance scans for a considerable time period after the initial diagnosis.
Multidetector CT: key features
Well-circumscribed nodular lesion: solitary pulmonary nodule or focal consolidation—average diameter 2 cm (range 0.6–6 cm).
Occasionally 2–3 nodules coalesce to form a discrete mass.
Very mild, focal lymphangitic spread may be seen.
Differs from LIP/FB as it forms a discrete nodular mass rather than affecting the lung diffusely.
Follicular bronchiolitis
FB (Figure 3) comprises a benign, polyclonal bronchial MALT hyperplasia in and around the bronchioles. Primary FB is usually associated with collagen vascular diseases, especially rheumatoid arthritis and Sjögren syndrome, or immunodeficiency syndromes. Secondary FB is a frequent incidental finding on lung biopsy and is usually related to chronic bronchial inflammation (e.g. bronchiectasis). The predominantly peribronchiolar pattern of FB and the more diffuse pattern of LIP are considered to be a continuum of the same entity.
Figure 3.

Follicular bronchiolitis. (a) A high-resolution CT demonstrates tiny parenchymal nodules (arrows) and subtle mosaic attenuation in a 56-year-old female with rheumatoid arthritis. There is a mosaic pattern due to small airways disease. The patient presented with progressively worsening shortness of breath. (b) Follicular bronchiolitis in a patient with thymoma. The image shows chronic inflammation of membranous bronchioles (arrows) with reactive lymphoid follicles (arrowheads). Note the normal appearing pulmonary artery (curved arrow) adjacent to an abnormal bronchiole. The intervening lung parenchyma is preserved. Haematoxylin–eosin stain; original magnification ×40.
In the setting of collagen vascular disease, FB occurs mostly in adults, with a mean age of 44 years, while in patients who are immunocompromised FB tends to occur at a younger age (mean age, 16 years) [9].
Typically, progressive dyspnoea and cough are the manifestations of primary FB. Fever and recurrent bronchopneumonia are recognised. Patients under 30 years of age have a tendency to have progressive disease [9]. Treatment regimes are usually steroid based.
Histopathology
Lymphoid follicular hyperplasia in the walls of bronchioles with narrowing of the lumen is the defining feature of FB. The lymphocytie infiltrate may also extend into the peribronchiolar interstitium, but no significant extension into the alveolar septa should be noted (the hallmark of LIP). Reactive follicles may also be seen along interlobular septae and subpleural regions.
Imaging findings
Howling et al [10], reviewed the high-resolution CT (HRCT) manifestations of 12 patients with biopsy-proven FB, and the commonest findings were small nodules (usually 1–3 mm in diameter, range 1–12 mm) and patchy bilateral areas of ground-glass opacity (75%). The nodules had a centrilobular (100%) or peribronchial (42%) distribution. Less common findings include bronchial dilatation and mild interlobular septal thickening [11].
Multidetector CT: key features
Bilateral 1–3 mm nodules—centrilobular/peribronchial distribution.
Bilateral patchy ground-glass opacities.
Nodules which mimic a tree-in-bud pattern (i.e. peribronchiolar distribution).
Bronchial dilatation.
Disease is limited to the airways (i.e. no diffuse interstitial involvement as in LIP).
Lymphoid interstitial pneumonia
LIP (Figures 4–7) represents a diffuse lung disorder characterised by lymphoid hyperplasia and an interstitial polyclonal inflammatory infiltrate of unknown pathogenesis. It is almost invariably associated with systemic immunological disturbances, with Epstein–Barr virus DNA and HIV RNA being occasionally demonstrated.
Figure 4.
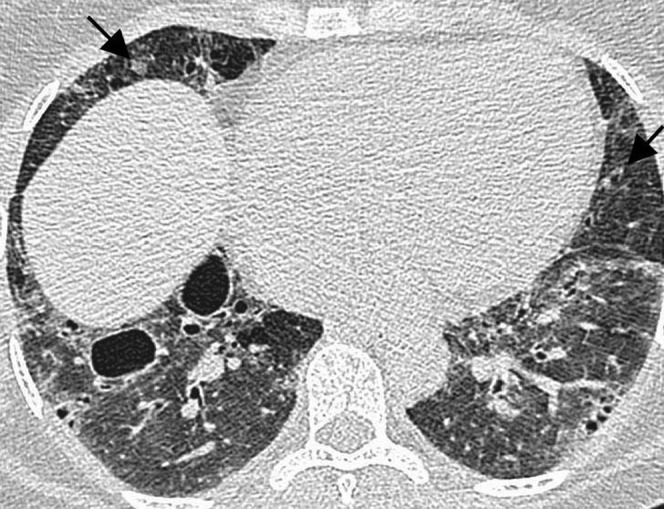
Lymphoid interstitial pneumonia in a 53-year-old female with Sjogren syndrome. High-resolution CT demonstrates bilateral diffuse ground-glass opacity, cystic airspaces and small nodules (arrows). This was diagnosed with an open lung biopsy.
Figure 7.

Lymphoid interstital pneumonitis (LIP) with amyloid. (a) A high-resolution CT scan in a 40-year-old female demonstrates cysts and soft tissue nodules (arrows) in the left lung. The nodule in the lingula contains calcium and was proven on percutaneous core biopsy to represent amyloid. The presence of LIP was confirmed with open lung biopsy. (b) A multidetector CT scan with mediastinal window setting in the same patient demonstrates calcification in two further soft tissue lung nodules (arrows). Such nodules in this context should raise suspicion for coexistent pulmonary amyloidosis.
LIP is most commonly seen in the context of HIV infection and is considered an AIDS-defining illness. In this setting, it primarily affects children, being relatively rare in HIV-infected adults. In non-AIDS patients, LIP affects most frequently females between 40 and 60 years of age with systemic conditions, typically connective tissue disorders.
The underlying disease usually dominates the clinical picture. The most common respiratory symptoms include cough and gradually progressive dyspnoea.
Histopathology
LIP is characterised by a diffuse inflammatory infiltrate within the alveolar interstitium that comprises mainly T-lymphocytes, plasma cells and histiocytes. The B-cells are limited to the frequent reactive lymphoid follicles. Loosely formed epithelioid granulomas are commonly seen, but necrosis is not a feature. LIP can be regarded as FB with diffuse extension of the lymphocytic infiltrate to the distal parenchyma, and indeed they seem to represent opposite ends of the same spectrum.
Imaging findings
In a series of 22 patients with biopsy-proven LIP the primary HRCT features were [12]:
uniform or patchy areas of bilateral ground-glass opacity (n=22/22)
poorly defined centrilobular nodules (n=22/22) with or without subpleural nodules (n=19/22)
thin-walled cystic air spaces: 1–30 mm diameter (n=15/22)
thickened peribronchovascular interstitium (n=19/22)
mild interlobular septal thickening (n=18/22)
mediastinal lymph node enlargement (n=15/22).
Ichikawa et al [13] described cystic airspaces, and the associated lung biopsy specimens showed bronchiolar stenosis and obstruction caused by peribronchiolar lymphocytic infiltration, suggesting that the cyst formation in LIP could well be due to a partial “check-valve” bronchiolar obstruction.
The MDCT findings of LIP vary considerably depending on the underlying associated disease. Cysts are typically seen in association with underlying Sjögren syndrome and are often the predominant finding (Figures 4 and 5). In congenital immunodeficiencies, LIP most often appears as patchy ground-glass opacity, whereas in patients with AIDS it typically manifests with multiple nodules (Figure 6).
Figure 5.

Lymphoid interstitial pneumonitis. (a) A high-resolution CT in a 52-year-old female with mixed connective tissue disease demonstrates extensive ground-glass opacity with small centrilobular nodules (black arrows) and lung cysts. A bizarrely shaped nodule in the left upper lobe (white arrow) was found to represent an amyloid deposit on percutaneous core biopsy. (b) Histopathological specimen: there is a prominent and diffuse lymphocytic interstitial infiltrate, involving both the airways (thick arrow) and the alveolar septa (thin arrows), which are remarkably thickened. A reactive lymphoid follicle with germinal centre is also noted (asterisk). Haematoxylin–eosin stain; original magnification ×40.
Figure 6.
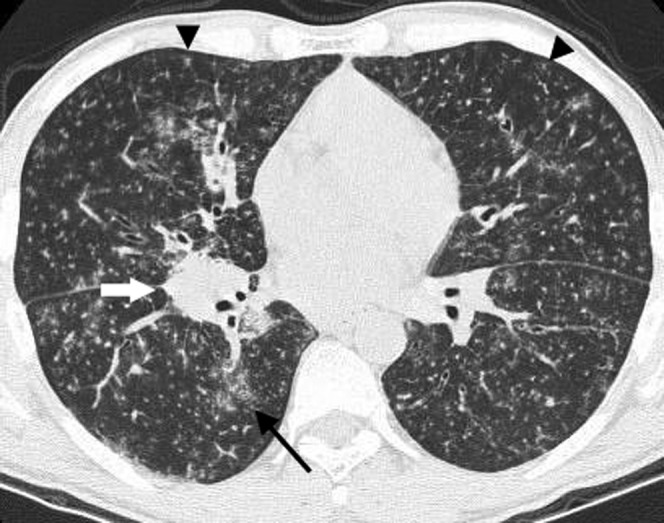
Lymphoid interstitial pneumonitis. A high-resolution CT in a 38-year-old male with human immunodeficiency virus infection demonstrates innumerable small bilateral centrilobular nodules (arrowheads) and patchy ground-glass opacities (arrow) with no lung cysts seen. The diagnosis was confirmed with a transbronchial lung biopsy. There is also a right hilar nodal mass (white arrow).
LIP can occasionally be seen in association with pulmonary amyloidosis, particularly in the context of Sjögren syndrome [14]. The presence of large soft tissue or calcified pulmonary nodules in conjunction with LIP should raise this suspicion (Figures 5 and 7).
Multidetector CT: key features
Ground-glass opacity.
Nodules—poorly defined, centrilobular and subpleural.
Cystic airspaces.
Peribronchovascular interstitial thickening.
Lymphoid interstitial pneumonia vs marginal zone lymphoma of mucosa-associated lymphoid tissue origin
MALT lymphoma is the most commonly considered differential diagnosis for LIP. It can be very difficult to distinguish between the two entities, which often are very similar clinically, histologically and radiologically.
There are, however, a number of pathological features that favour LIP over marginal zone lymphoma. Immunohistochemical analysis is particularly important to demonstrate the primarily follicular distribution of B-cells and the polyclonality of the lymphocytic proliferation in LIP. Pathological features favouring MALT lymphoma may include distortion of lung architecture, a more dense infiltrate, frequent lymphoepithelial lesions and the presence of pleural infiltration. Intranuclear B-lymphocyte inclusions, known as Dutcher bodies, are not usually found in benign processes and suggest MALT lymphoma but not in LIP.
LIP and MALT lymphoma may coexist (Figure 8); however, LIP is not thought to directly progress to lymphoma. It is believed that in cases where transformation from LIP to lymphoma has been suggested, lymphoma was initially present but wrongly diagnosed as LIP [15].
Figure 8.
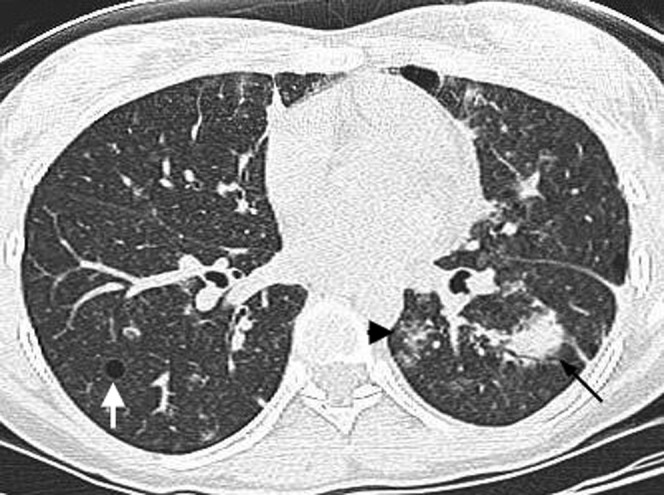
A thin-section CT scan in a 28-year-old female demonstrating a peribronchial left lower lobe soft tissue mass (black arrow) proven to represent lymphoma. There is a background of ground-glass opacities (black arrowhead) and cysts (white arrow), consistent with coexistent lymphoid interstitial pneumonitis.
CT can help differentiate LIP from lymphoma. Cysts are characteristic of LIP, whereas large nodules, consolidation and effusions favour lymphoma. A comparative study between the HRCT findings in LIP (n=17) and malignant lymphoma (n=44) [16] found that certain morphological features allowed a presumptive diagnosis based on the presence of specific features:
cysts: LIP (82%) > lymphoma (2%)
consolidation: lymphoma (66%) > LIP (18%)
large nodules (11–30 mm): lymphoma (41%) > LIP (6%)
pleural effusions: lymphoma (25%) > LIP (0%)
distribution of parenchymal lesions: no difference found.
Primary pulmonary lymphoma
Primary pulmonary lymphoma (PPL; Figures 9–12) represents a monoclonal lymphoid proliferation affecting the lungs in a patient with no detectable extrathoracic lymphoma for at least 3 months after the initial diagnosis. PPL ranges from a relatively indolent MALT lymphoma to more aggressive forms of DLBCL. These two entities make up the majority of cases of PPL, which is overall a rare disorder. LYG is also within the spectrum of PPL, but as it is a rare entity with distinctive radiological features and unusual pathological characteristics it is worthy of consideration as a separate lymphoproliferative disorder.
Figure 9.
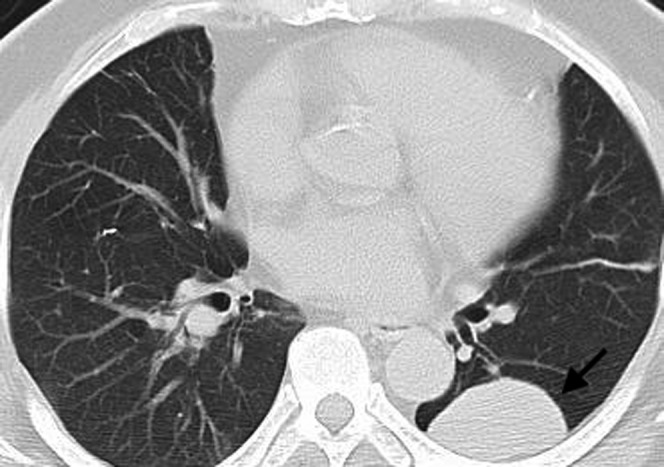
Mucosa-associated lymphoid tissue (MALT) lymphoma in a 31-year-old male. The multidetector CT scan demonstrates a large, well-circumscribed soft tissue peripheral mass in the left lung posteriorly (arrow). Percutaneous core needle biopsy demonstrated MALT lymphoma.
Figure 12.

(a) Diffuse large B-cell lymphoma in a 32-year-old male. A chest radiograph demonstrates a large opacity in the right middle lobe obscuring the right heart border. (b) A multidetector CT scan in the same patient demonstrates a well-circumscribed right middle lobe mass with central low attenuation, consistent with necrosis (arrow). This was proven on biopsy to represent a primary form of diffuse large B-cell lymphoma.
Figure 10.
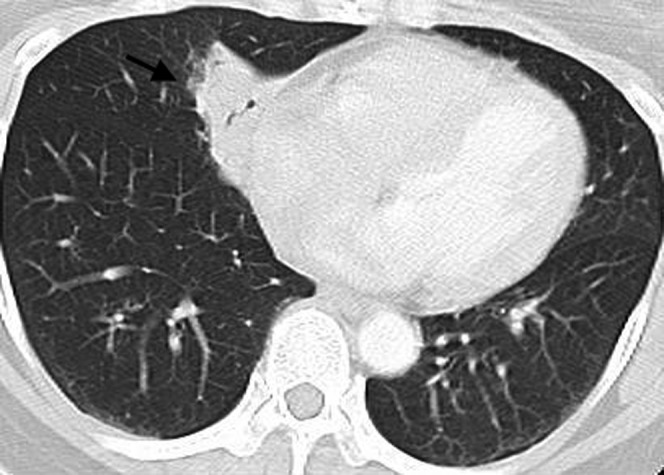
Mucosa-associated lymphoid tissue (MALT) lymphoma. A multidetector CT scan in a 43-year-old female demonstrates a wedge-shaped soft tissue opacity with an air bronchogram (arrow) adjacent to the right cardiac border. This was proven to be a MALT lymphoma on percutaneous core biopsy.
Non-Hodgkin B-cell lymphoma (NHL-B) is the most frequent type and accounts for roughly 80% of all PPL. The majority of these are MALT lymphomas, which are frequently associated with autoimmune diseases. DLBCL accounts for the majority of the other patients with PPL [17] and it is characteristically seen in patients with underlying immunodeficiency, such as transplant patients on ciclosporin. The incidence of DLBCL may well be underestimated, as it could potentially spread rapidly from the lung into mediastinal and extrathoracic locations.
Clinical findings
MALT lymphomas are usually asymptomatic and discovered as an incidental finding. They are typically indolent lesions and have a good prognosis. Immunocompromised patients with DLBCL are usually symptomatic with dyspnoea, fever and weight loss. Overall survival is poor and the prognosis is worse if there is an underlying disorder. Progression and relapse also occur much earlier in this group. DLBCL tends to be more diffuse and destructive, with 5-year survival rates ranging from 0–60% compared with 84–94% for MALT lymphomas.
Histopathology
Mucosa-associated lymphoid tissue lymphoma
This is a form of extranodal marginal zone B-cell lymphoma of MALT origin that is characterised by a massive interstitial infiltrate of small lymphocytes forming a mass-like lesion. A variable number of admixed plasma cells and histiocytes, and occasional small germinal centres, are also seen. The cellular infiltrate extends into the alveolar septa in the periphery and also along the adjacent bronchovascular bundles, interlobular septa and visceral pleura (lymphatic distribution). The characteristic lymphoepithelial lesions can be seen in the bronchiolar mucosa, where the neoplastic cells infiltrate the respiratory epithelium. The diagnosis of MALT lymphoma is morphological; however, the frequent aberrant immunohistochemical coexpression of CD20 (cluster of differentation 20) and CD43 by the intraepithelial lymphocytes, a B-cell and a T-cell marker respectively, rules out a reactive process. This is especially helpful in small biopsies, where the histological differentiation between reactive and neoplastic conditions can be difficult. The tissue can also be tested for immunoglobulin gene rearrangements which are seen in MALT lymphomas but not in benign lymphocytic proliferations.
Diffuse large B-cell lymphoma
DLBCL presents as sheets of medium to large atypical lymphocytes forming a solid lesion that replaces the underlying lung architecture. The neoplastic cells show frequent mitotic figures, and necrosis is a common feature, occasionally with central cavitation. There is usually a sharp demarcation with the normal adjacent lung parenchyma and the neoplastic nature of the lesion is readily recognised.
Imaging findings
Mucosa-associated lymphoid tissue lymphoma
McCulloch et al [18] assessed the CT appearances of five cases of MALT lymphoma (Figures 9–11), and the lesions were usually multifocal and comprised ill-defined nodules containing air bronchograms (n=4/5). Focal lobar consolidation was also seen (n=1/5), in addition to interlobular septal thickening, centrilobular micronodules and bronchial wall thickening (n=2/5). Mediastinal lymphadenopathy and pleural reaction are uncommon.
Figure 11.
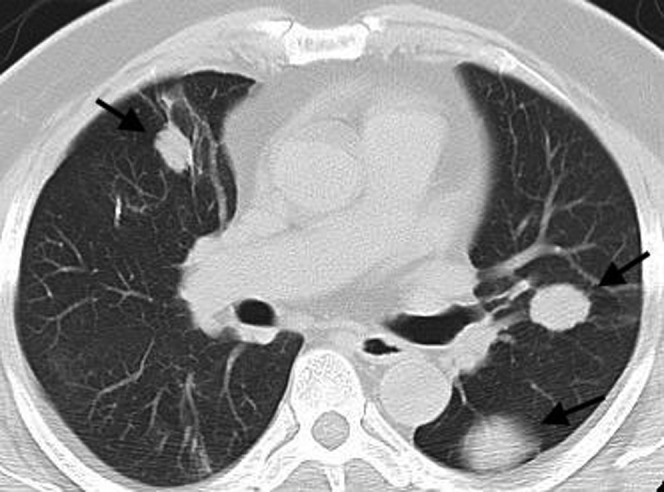
Mucosa-associated lymphoid tissue (MALT) lymphoma. A multidetector CT scan in a 40-year-old female demonstrates multiple large bilateral lung nodules (arrows). These were slowly growing on 6-monthly follow-up scans. Biopsy revealed a diagnosis of MALT lymphoma.
Bubble-like lucencies, ground-glass opacities and bronchial dilatation have also been described on CT [19].
A positive “angiogram” sign, consisting of an enhancing pulmonary vessel within a homogeneous area of consolidation, and a halo of ground-glass shadowing at the lesion margin have been reported in MALT lymphoma [20], but these are non-specific findings seen in a number of other conditions, including bronchoalveolar cell carcinoma. Parenchymal lesions are usually bilateral (60–70%) and multiple (70–77%), and nearly all the lesions contain air bronchograms [21]. The presence of distended bronchi within the lesions is a good diagnostic sign, although the underlying mechanism for this is unexplained.
Another series reviewed the CT imaging findings in 21 patients with MALT lymphoma [22], and this group identified the following four radiological patterns of disease:
single nodule or single consolidative opacity (33%)
multiple nodules or multiple areas of consolidation (43%)
bronchiectasis and bronchiolitis (14%)
diffuse interstitial lung disease (10%).
Diffuse large B-cell lymphoma
The commonest radiological presentation of DLBCL (Figure 12) is solitary or multiple pulmonary nodules. Cavitation is a common feature.
Multidetector CT: key features
Mucosa-associated lymphoid tissue lymphoma:
exhibit diverse patterns of lung abnormality on CT scan
single or multiple nodules or areas of consolidation are the commonest patterns (>70%)
multiple, bilateral lesions (>70%)
lesions tend to be peribronchovascular
bronchial dilatation is commonly seen and due to peribronchial location
hilar/mediasitinal lymphadenopathy is present in approximately 30% of cases.
Diffuse large B-cell lymphoma:
Single or multiple solid pulmonary nodules or masses.
Cavitation—common, regardless of immune status.
Mediastinal lymph node enlargement may be present.
Lymphomatoid granulomatosis
Lymphomatoid granulomatosis (LYG; Figures 13 and 14) is a rare Epstein–Barr virus (EBV)-associated lymphoproliferative disorder with a propensity for blood vessel destruction. Although part of the PPL spectrum, LYG merits special consideration as (1) the lung is the most common primary site of involvement, (2) it is often difficult to prove clonality, and (3) it has an angiocentric distribution.
Figure 13.

Lymphomatoid granulomatosis. (a) A chest radiograph in a 37-year-old male demonstrates multiple large bilateral pulmonary nodules with a lower zone predominance. (b) A high-resolution CT in the same patient demonstrates large, irregularly marginated pulmonary nodules. Some of the nodules are coalescent (arrow) and others are of ground-glass opacity, consistent with the “migratory” phenomenon associated with nodules in this disease.
Figure 14.

(a) Lymphomatoid granulomatosis. A multidetector CT scan in a 46-year-old male demonstrates multiple pulmonary nodules coalescing in the lower lobes to form larger masses (arrows). Percutaneous core biopsy of the mass-like consolidation in the left lower lobe revealed lymphomatoid granulomatosis. (b) Lymphomatoid granulomatosis histopathological specimen in the same patient: well-demarcated cellular nodule with a central area of necrosis (asterisk). The preserved alveolar parenchyma is shown in the upper part of the image, above the dashed line. Inset shows the angiocentric nature of the lesion. Inset A: infiltration of the intima by lymphoid cells. The muscular wall of the artery is indicated by the arrow. Most of the lymphocytes are small reactive T-cells and atypical B-cells are rare (thin arrows). Haematoxylin–eosin stain; original magnification ×200. Inset B: elastin stain shows the elastic lamina of the artery (arrows). Elastin stain; original magnification ×200.
LYG is composed of EBV-positive B-cells and reactive T-cells, and is considered an EBV-driven T-cell-rich B-cell lymphoma. Although the commonest site of involvement is the lung parenchyma (>90%), synchronous extrapulmonary involvement is common, involving mainly the central nervous system (CNS) and skin.
This is a rare condition, usually affecting males aged 30–50 years. There is a variable natural history: generally it is associated with a poor prognosis and a median survival of less than 2 years, but spontaneous remission has been reported [23].
Clinical findings
LYG is a systemic multiorgan disease. Lung involvement is usually present, with skin (50%), CNS (25%) and kidneys less commonly affected. Lung involvement can cause cough and dyspnoea with haemoptysis, usually indicating that the parenchymal lung disease has cavitated.
Histopathology
Microscopically, LYG is an angiocentric polymorphous mononuclear infiltrate composed of numerous small lymphocytes, plasma cells, histiocytes and large atypical EBV-positive B-cells, which resemble immunoblasts (Figure 14b). The small lymphocytes are mainly T-cells, which are negative for EBV. The atypical lymphoid cells tend to cluster around and within vessels, frequently obstructing the lumen, which may result in areas of coagulative necrosis. Fibrinoid necrosis of the vessel wall may also occur. Despite the name, giant cells or true granulomas are not a histological feature.
In the current WHO classification [3], LYG is grouped along with NHL as a neoplasm of mature B-cells. LYG is graded from 1 to 3 based on the proportion of large atypical EBV-positive B-cells present: Grade 1 containing rare EBV-positive B-cells and Grade 3 consisting almost entirely of large atypical cells and undisputedly regarded as an aggressive lymphoma. Clinical behaviour correlates with the proportion of neoplastic B-cells.
Imaging findings
The most common radiographic feature is multiple lung nodules, occurring in approximately 80% of all cases [24], predominantly involving the lung bases. The lesions can progress rapidly, coalesce and commonly cavitate, therefore mimicking Wegener's granulomatosis or metastases.
Dee et al [25] described two distinct radiographic manifestations of LYG. In their series of five patients, diffuse reticulonodular opacities correlated microscopically with angiocentric granulomatous infiltration without pulmonary infarction, whereas larger mass-like opacities corresponded to biopsy-proven pulmonary infarcts. There is a wide range in the number (5–60) and the diameter of nodules (up to 6.5 cm) found in patients with LYG [26], but generally the nodules themselves measure <1 cm and tend to be located along the bronchovascular bundles and interlobular septa. Less common radiological appearances include coarse linear opacities along the bronchovascular bundles and thin-walled cysts.
Nodules can disappear or migrate spontaneously [27], and may display central ground-glass opacity surrounded by denser consolidation at least 2 mm thick—the so called “reversed halo sign”. However, this is a non-specific sign, most commonly seen in organising pneumonia [28].
Multidetector CT: key features
Bilateral, round, poorly marginated nodules 0.5–8 cm in diameter.
Basal predominance.
Peribronchovascular distribution.
Can coalesce and cavitate.
“Reversed halo sign”.
“Migratory” nodules due to “waxing and waning”.
Secondary pulmonary lymphoma
The pathogenesis of secondary pulmonary lymphoma (SPL; Figures 15–19) depends on the type of primary lymphoma, and is therefore quite variable.
Figure 15.
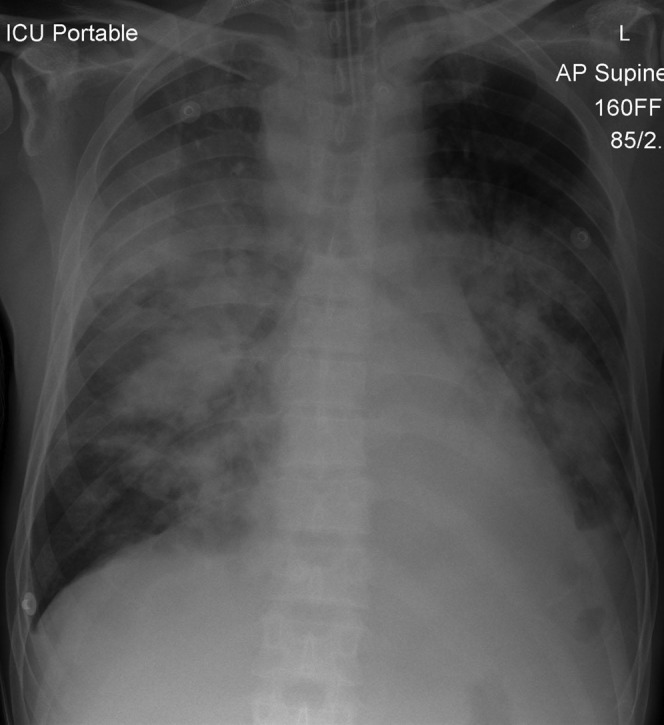
Secondary pulmonary lymphoma in a 40-year-old male. A chest radiograph demonstrates bilateral parenchymal consolidation with air bronchograms. Note the presence of abnormal paratracheal soft tissue opacity, in keeping with lymphadenopathy. This combination of findings should raise suspicion of lymphoma.
Figure 19.

(a) Secondary pulmonary lymphoma in a 42-year-old male with a history of non-Hodgkin lymphoma. Multidetector CT scan demonstrates two peripheral pulmonary masses (arrows). This was confirmed to represent recurrence of non-Hodgkin lymphoma on percutaneous core biopsy. (b) A multidetector CT scan at a different level in the same patient shows cavitation of a recurrent non-Hodgkin lymphoma nodule in the medial basal segment of the right lower lobe (arrow).
Figure 16.
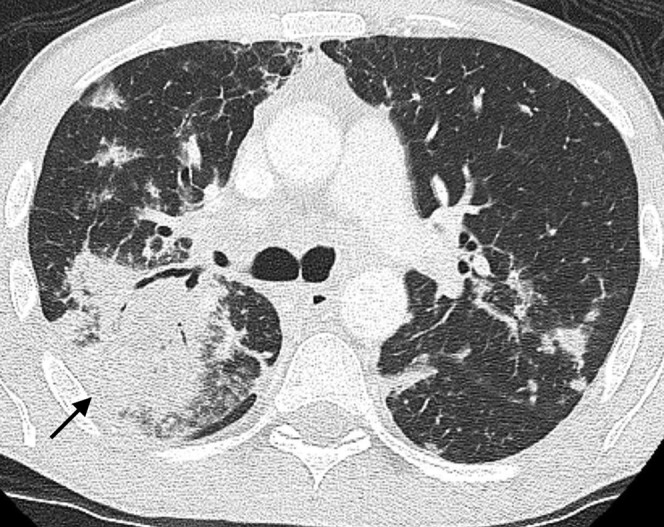
Secondary pulmonary lymphoma. An axial multidetector CT scan in the same patient in Figure 15 demonstrates the mass-like right upper lobe consolidation with air bronchograms (arrow) and this is associated with further bilateral areas of parenchymal involvement. The alveolar pattern of secondary lymphoma is associated with a poor prognosis.
Figure 17.

(a) Secondary pulmonary lymphoma. A multidetector CT scan in a 26-year-old male demonstrates a large anterior mediastinal soft tissue mass (arrow) displacing the mediastinal structures posteriorly and abutting the pleura on the left. There is a small left pleural effusion (arrowhead). (b) Integrated fluorodeoxyglucose (FDG) positron emission tomography CT scan in the same patient demonstrates parenchymal extension (arrow) from the large anterior mediastinal mass which demonstrates markedly increased FDG uptake.
Figure 18.
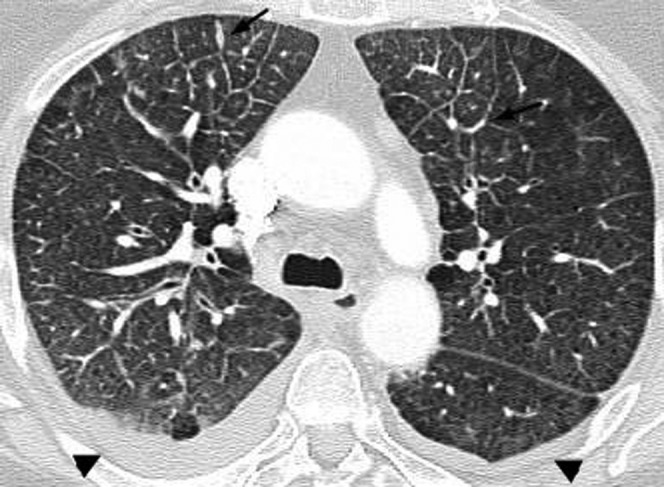
Secondary pulmonary lymphoma. A thin-section CT scan in a 63-year-old female demonstrates nodular thickening of interlobular septa in both anterior upper lobes (arrows), consistent with lymphangitic spread of lymphoma. Small bilateral pleural effusions are also seen (arrowheads).
All the different forms of lymphoma may secondarily involve the lungs, but the mature B-cell neoplasms are the most frequent. The occurrence of parenchymal involvement is difficult to quantify due to a lack of recent CT series. Although a greater proportion of patients with HL rather than NHL have lung involvement, more NHL SPL is seen in clinical practice because NHL is more prevalent. A CT series from 1988 indicates a 12% prevalence of lung parenchymal disease in patients with suspected relapse of HL [29]. Early autopsy series, however, indicate that parenchymal involvement may be as high as 62% [30].
Clinical findings
Lymphomatous involvement of the lung itself is often asymptomatic. If symptoms are present, they tend to be non-specific. Symptoms more often relate to the presence of mediastinal lymphadenopathy, particularly in the case of HL, in which case B-symptoms are often present as well.
Histopathology
Secondary pulmonary lymphoma cannot be reliably distinguished from primary pulmonary lymphoma solely on the basis of pathological tissue analysis as they show identical morphological features.
Imaging findings
Intrathoracic involvement is common in both HL and NHL. Since any form of lymphoma can secondarily involve the lungs, the imaging features are varied and non-specific, ranging from a solitary nodule to lymphangitic parenchymal involvement. Although non-specific, the radiological features described below should raise the possibility of secondary lung involvement in any patient with a known history of lymphoma. The commonest intrathoracic manifestation is mediastinal lymph node enlargement.
In HL, parenchymal involvement is almost always associated with mediastinal and/or hilar lymphadenopathy, and usually there is contiguous spread of the disease from the adenopathy. Isolated parenchymal HL is exceedingly rare. The findings in NHL are more variable and isolated lung disease without mediastinal involvement can occur. Mentzer et al [31] reviewed the pattern of lung parenchymal involvement in 651 patients with malignant lymphoma and identified 54 patients (8%) with biopsy-proven pulmonary involvement. Based on a radiological–surgical correlation, the patterns of lung involvement were characterised as (1) lymphangitic, (2) nodular, or (3) alveolar. Furthermore, distinct clinical courses were associated with each of the three patterns of disease with the alveolar pattern associated with particularly poor prognosis.
Lewis et al [32] reviewed 31 patients with either recurrent or secondary NHL/HL who had parenchymal abnormalities on CT of the chest: the commonest abnormality in HL was a mass or mass-like consolidation, seen in 12 of 15 (80%) patients. In contrast, the commonest CT abnormality in NHL was peribronchial and/or perivascular interstitial thickening, mimicking lymphangitis, which was seen in 11 of 16 (69%) patients. Nodules of <1 cm in diameter, alveolar or interstitial infiltrates, pleural-based masses and pleural effusions were seen with fairly equal frequency in both HL and NHL. Lymph node enlargement within the chest was seen in 8 of 15 (53%) HL cases and in 3 of 16 (19%) NHL cases.
Multidetector CT: key feature
Nodules <1 cm in diameter, solitary or multiple.
Mass/mass-like consolidation.
Bronchovascular thickening mimicking lymphangitis carcinomatosa.
Cavitation may occur.
Air bronchograms (61% NHL and 47% HL).
Pleural effusion.
Nodal enlargement: HL>NHL.
HRCT appearances of HL and NHL are similar.
Nodules/masses with or without air bronchograms are the commonest HRCT features.
Lymphangitic spread is frequently seen.
Acquired immune deficiency syndrome-related lymphoma
Lymphoma is the second most common tumour occurring in patients with AIDS , after Kaposi's sarcoma. Lymphoma is thought to occur as a consequence of B-lymphocyte proliferation due to long-term stimulation by the HIV and Epstein–Barr virus infection. ARL (Figures 20 and 21) is typically and almost exclusively a NHL, usually of an aggressive B-cell type [33].
Figure 20.
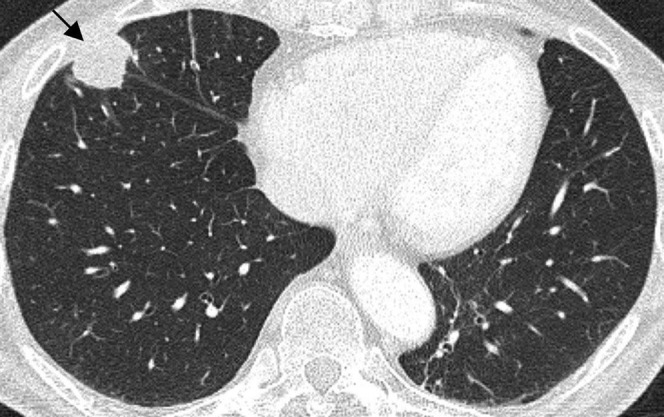
Acquired immune deficiency syndrome-related lymphoma: thin-section CT scan in a 49-year-old who is human immunodeficiency virus-positive with a CD4 (cluster of differentiation 4) count of 68 dl–1 demonstrates a single, well-defined, peripheral large pulmonary nodule (arrow), proven to represent non-Hodgkin lymphoma on percutaneous core biopsy.
Figure 21.
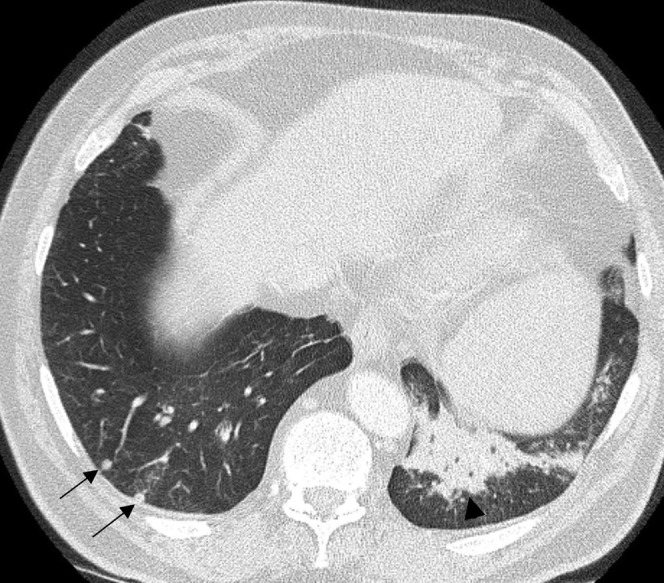
Acquired immune deficiency syndrome (AIDS)-related lymphoma. A CT scan in a 36-year-old male with AIDS reveals scattered small pulmonary nodules (arrows) with a focus of chronic left lower lobe consolidation (arrowhead). Biopsy of the consolidation revealed an aggressive non-Hodgkin lymphoma.
The prevalence of lymphoma in AIDS is 40–100 times that of the general population. It tends to be associated with advanced AIDS and very low CD4 cell counts, with a median CD4 count of <55 dl–1 [34]. Lymphoma is the cause of death in up to 20% of HIV-infected patients and primary pulmonary involvement accounts for approximately 10% of these.
The reported incidence of ARL overall varies between 5% and 20%, but evidence suggests that it may be underestimated as parenchymal lung disease is frequently undetected: in an autopsy series of patients with AIDS with systemic NHL, 71% (n=20/28) had pulmonary involvement in the form of nodules, infiltrates or masses. This was compared with a recorded incidence of only 5.8% in the corresponding clinical database [35].
Clinical findings
Most patients are symptomatic, presenting with cough and shortness of breath. In contrast to lymphoma in the non-AIDS population, ARL tend to affect younger patients with a greater spectrum of clinical manifestations, more aggressive course and poorer prognosis.
Ioachim et al [33] reviewed 111 cases of ARL, and found that both NHL (n=100/111) and HL (n=11/111) patients presented at a more advanced clinical stage and frequently had bone marrow involvement.
Thoracic manifestations of ARL tend to be diffuse in distribution, by virtue of the fact that the disease is frequently extranodal, in contrast to the predominant nodal disease seen in non-AIDS patients.
Histopathology
This subset of patients usually develops aggressive B-cell lymphomas. The wide array includes lymphomas usually diagnosed in immunocompetent patients and lymphomas that are predominantly HIV associated. The first group is commonest and comprises mainly Burkitt lymphoma, DLBCL and HL. The second group is mainly represented by PEL and plasmablastic lymphoma. Of particular interest is the frequent presence of concomitant viral infection in the neoplastic cells, with EBV-DNA and human herpesvirus 8 (HHV8) demonstrated in a fair number of cases [36].
Imaging findings
Thoracic involvement is common in ARL, and in fact the presence of pleural effusion, pulmonary nodules and lymphadenopathy in HIV-positive patients should suggest the diagnosis of lymphoma. Again, since different types of lymphomas may involve the lung, the radiological features vary accordingly.
Pulmonary nodules are the commonest finding on CT. These nodules are usually well circumscribed, >1 cm in diameter (range 0.5–5 cm), multiple, frequently peripheral and may demonstrate central cavitation [37].
In two CT series of patients with ARL, the commonest finding was pleural effusion, and most patients also had multifocal parenchymal disease with nodules, interstitial infiltrates or air-space opacities with no particular predominant pattern [35,38]. The proportion of patients with ARL with thoracic lymphadenopathy varies in different CT series, ranging from 3% to 54% [33,35]. The extent of mediastinal lymphadenopathy, however, is typically less than in lymphoma in patients who don't have AIDS.
Multidetector CT: key features
Well-defined pulmonary nodules (0.5–5 cm) with or without cavitation or a large solitary pulmonary nodule/mass (2–5 cm).
Pleural effusions—commonest thoracic manifestation.
Pericardial effusions can be present.
Focus of consolidation and/or ground-glass opacity.
Nodules + effusions + adenopathy + HIV = highly suggestive of ARL.
An unusual, rare form of ARL that deserves mention is PEL, also known as body-cavity based lymphoma. First described in 1989 [39], this is an aggressive form of NHL, which is universally associated with HHV8. PEL is typically seen in an immunocompromised host, often in young to middle-aged males with HIV infection, and there is frequently coinfection with monoclonal EBV. In one study looking at 277 patients with systemic NHL in the setting of HIV infection, 4% were diagnosed with PEL [40]. However, PEL is not exclusively seen in HIV patients and has also been reported in recipients of solid organ transplants [3,41]. In regions with a high prevalence of HHV8, such as the Mediterranean, PEL can also be seen in the absence of immunodeficiency [42]. PEL is essentially a lymphomatous effusion in the absence of a solid tumour mass and typically only one body cavity is involved. The commonest sites of involvement are the pleural, pericardial and peritoneal cavities. Some patients can secondarily develop solid masses in adjacent structures underlying the serous cavities and extracavitary involvement has been reported in the lung parenchyma [3]. The prognosis is extremely poor, with median survival less than 6 months.
Post-transplantation lymphoproliferative disorder
PTLD (Figure 22) is a lymphoid/plasmacytic proliferation that occurs following solid organ or haematopoietic stem cell transplantation, manifesting as a spectrum of disorders ranging from polyclonal benign proliferations to monoclonal and monomorphic aggressive lymphomas.
Figure 22.
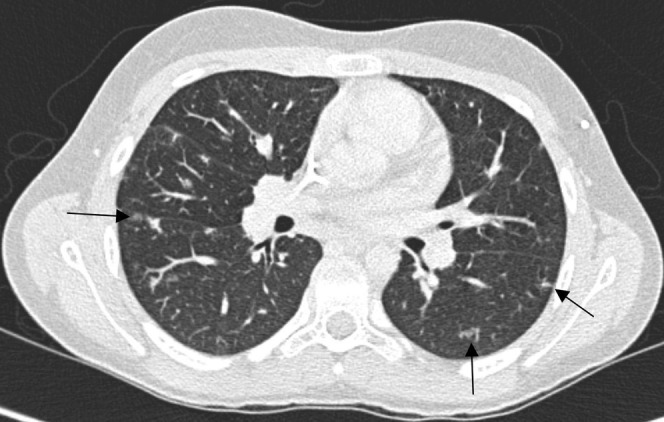
Post-transplantation lymphoproliferative disorder. A CT scan in a 14-year-old stem cell transplant recipient demonstrates multiple small lung nodules, many of which are subpleural (arrows). There was also hilar adenopathy, which is not well seen on the lung window setting. This patient presented 7 months post transplant with fever and an intractable infectious mononucleosis-type syndrome.
The aetiology of PTLD is thought to be closely associated with EBV infection. EBV-induced B-cell proliferation continues unopposed due to the host's pharmacologically suppressed T-cells, and leads to a spectrum of predominantly B-lymphocyte proliferations [43].
A much smaller category of typically EBV-negative PTLD (14%) originates from T-cells and plasma cell proliferations. These subtypes usually present later (50–60 months post transplant) and are generally more aggressive [43,44].
PTLD is seen in less than 2% of all transplant cases and there is a clear association with the specific organ transplanted, with the highest incidence in lung transplantation (6–9%), followed by cardiac (2–5%), liver (2–5%), pancreas (2%), renal (1%) and stem cell transplant (0.5–1%) [45]. The risk of children developing PTLD is two to three times greater than that of adults. Besides the organ type, risk factors include immunosuppressive drug regime, age and EBV-status pre-transplantation.
Clinical findings
The majority of cases of PTLD occur within 2 years of transplantation, but it may be seen as early as 60 days post transplant. Late-onset PTLD can be seen as late as 20 years after transplantation, is usually monoclonal and heralds a worse prognosis [46].
PTLD, irrespective of histology, is a significant cause of morbidity and mortality in transplant patients and can be fatal if untreated. PTLD involves multiple organ systems at presentation in 50% of cases and extranodal disease is more frequent than in lymphoma.
The clinical presentation is variable and includes fever, lymphadenopathy, abdominal pain with diarrhoea, a mononucleosis-like syndrome, upper respiratory tract infection, central nervous system symptoms and weight loss. Unexplained infectious syndrome in a transplant recipient should always raise the suspicion of a PTLD. Up to 25% of patients will present with allograft failure, and this can mimic primary allograft rejection.
The majority of polyclonal PTLD cases are indolent and either resolve completely or improve significantly with reduction in immunosuppression alone (60–70%). However, mortality rates as high as 60–100% have been described in more aggressive forms of PTLD [43]. Early diagnosis is crucial, and the radiologist may be the first one to consider this diagnosis in post-transplant patients.
Histopathology
B-cell NHL is the most common type of lymphoma associated with PTLD. However, 14% of PTLD are of T-cell rather than B-cell origin [47]. Four main types of lymphoproliferation are recognised in PTLD [48]:
Early lesions: “hyperplastic” proliferations, usually corresponding to an infectious mononucleosis-like pattern or a plasmacytic proliferation.
Polymorphic PTLD: polymorphic infiltrates forming destructive lesions that do not fulfil the criteria for any type of lymphoma.
Monomorphic PTLD: morphologically similar to lymphomas seen in immunocompetent patients and classified accordingly. The most common subtypes are DLBCL, Burkitt lymphoma and plasma cell neoplasms.
Classic HL-type PTLD: morphologically similar to HL seen in immunocompetent patients and classified accordingly.
Imaging findings
The commonest intrathoracic manifestations of PTLD are randomly distributed, well-circumscribed pulmonary nodules (0.3–5 cm in diameter), mediastinal and hilar adenopathy, and patchy air-space consolidation [49,50]. The well-defined nodules have a peripheral, basal predominance, and occasionally have a ground-glass halo. Less commonly, PTLD may manifest as a solitary parenchymal mass [51]. In a study of 17 organ transplant patients with PTLD, of whom 9 had monoclonal malignant lymphoma and 8 had polyclonal PTLD [52], the commonest overall CT finding was multiple pulmonary nodules (88%) of varying sizes with a predominantly peribronchovascular and subpleural distribution. None of these nodules was seen to cavitate. Other findings included septal thickening (35%), lymphadenopathy (29%) and patchy ground-glass opacity (29%). There was no significant difference in the findings between the monoclonoal and polyclonal groups.
Multidetector CT: key features
Nodules: more single than multiple, diameter range 0.3–5 cm.
More well-defined than ill-defined nodules.
“Halo sign” around nodules.
Patchy/focal consolidation or ground-glass opacity.
More peribronchial/subpleural than diffuse distribution of nodules.
Lymphadenopathy (30–60%).
Conclusions
Benign LPDs and primary pulmonary lymphoma of the lung are relatively rare, whereas secondary lymphomatous involvement is far more common. The radiological appearance of the LPDs is not pathognomonic and a multidisciplinary approach to diagnosis is vital.
The commonest parenchymal findings are multiple nodules and masses, often with air bronchograms, and ancillary features such as pleural effusions, cysts and mediastinal disease may help to narrow the differential diagnosis. Lesions can be solitary, solid and liable to cavitate, and it is important to remember that the presence of parenchymal lung cysts does not exclude malignant disease.
In immunosuppressed patients, recognising lymphoproliferative disease as a potential cause of pulmonary parenchymal pathology may expedite treatment and potentially improve prognosis, as this group frequently has more aggressive disease.
Appreciating the radiological spectrum of morphological guises is helpful for disease detection, but the majority of those with clinically and radiologically suspected lymphoproliferative disease will then require histological correlation to confirm the diagnosis and refine treatment options.
References
- 1.Bienenstock J, Johnston N, Perey DY. Bronchial lymphoid tissue. I. Morphologic characteristics. Lab Invest 1973;28:686–92 [PubMed] [Google Scholar]
- 2.Rangel-Moreno J, Hartson L, Navarro C, Gaxiola M, Selman M, Randall TD. Inducible bronchus-associated lymphoid tissue (iBALT) in patients with pulmonary complications of rheumatoid arthritis. J Clin Invest 2006;116:3183–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. , eds WHO classification of tumours of haematopoietic and lymphoid tissues. 4th edn. Lyon, France: IARC; 2008 [Google Scholar]
- 4.Berkman N, Breuer R, Kramer MR, Polliack A. Pulmonary involvement in lymphoma. Leuk Lymphoma 1996;20:229–37 [DOI] [PubMed] [Google Scholar]
- 5.Saltzstein SL. Pulmonary malignant lymphomas and pseudolymphomas: classification, therapy, and prognosis. Cancer 1963;16:928–55 [DOI] [PubMed] [Google Scholar]
- 6.Abbondanzo SL, Rush W, Bijwaard KE, Koss MN. Nodular lymphoid hyperplasia of the lung: a clinicopathologic study of 14 cases. Am J Surg Pathol 2000;24:587–97 [DOI] [PubMed] [Google Scholar]
- 7.Kradin RL, Mark EJ. Benign lymphoid disorders of the lung, with a theory regarding their development. Hum Pathol 1983;14:857–67 [DOI] [PubMed] [Google Scholar]
- 8.Sakurai H, Hada M, Oyama T. Nodular lymphoid hyperplasia of the lung: a very rare disease entity. Ann Thorac Surg 2007;83:2197–9 [DOI] [PubMed] [Google Scholar]
- 9.Yousem SA, Colby TV, Carrington CB. Follicular bronchitis/bronchiolitis. Hum Pathol 1985;16:700–6 [DOI] [PubMed] [Google Scholar]
- 10.Howling SJ, Hansell DM, Wells AU, Nicholson AG, Flint JD, Muller NL. Follicular bronchiolitis: thin-section CT and histologic findings. Radiology 1999;212:637–42 [DOI] [PubMed] [Google Scholar]
- 11.Pipavath SJ, Lynch DA, Cool C, Brown KK, Newell JD. Radiologic and pathologic features of bronchiolitis. AJR Am J Roentgenol 2005;185:354–63 [DOI] [PubMed] [Google Scholar]
- 12.Johkoh T, Muller NL, Pickford HA, Hartman TE, Ichikado K, Akira M, et al. Lymphocytic interstitial pneumonia: thin-section CT findings in 22 patients. Radiology 1999;212:567–72 [DOI] [PubMed] [Google Scholar]
- 13.Ichikawa Y, Kinoshita M, Koga T, Oizumi K, Fujimoto K, Hayabuchi N. Lung cyst formation in lymphocytic interstitial pneumonia: CT features. J Comput Assist Tomogr 1994;18:745–8 [DOI] [PubMed] [Google Scholar]
- 14.Jeong YJ, Lee KS, Chung MP, Han J, Chung MJ, Kim KI, et al. Amyloidosis and lymphoproliferative disease in Sjogren syndrome: thin-section computed tomography findings and histopathologic comparisons. J Comput Assist Tomogr 2004;28:776–81 [DOI] [PubMed] [Google Scholar]
- 15.Teruya-Feldstein J, Temeck BK, Sloas MM, Kingma DW, Raffeld M, Pass HI, et al. Pulmonary malignant lymphoma of mucosa-associated lymphoid tissue (MALT) arising in a pediatric HIV-positive patient. Am J Surg Pathol 1995;19:357–63 [DOI] [PubMed] [Google Scholar]
- 16.Honda O, Johkoh T, Ichikado K, Tomiyama N, Maeda M, Mihara N, et al. Differential diagnosis of lymphocytic interstitial pneumonia and malignant lymphoma on high-resolution CT. AJR Am J Roentgenol 1999;173:71–4 [DOI] [PubMed] [Google Scholar]
- 17.Nicholson AG, Wotherspoon AC, Diss TC, Butcher DN, Sheppard MN, Isaacson PG, et al. Pulmonary B-cell non-Hodgkin's lymphomas: the value of immunohistochemistry and gene analysis in diagnosis. Histopathology 1995;26:395–403 [DOI] [PubMed] [Google Scholar]
- 18.McCulloch GL, Sinnatamby R, Stewart S, Goddard M, Flower CD. High-resolution computed tomographic appearance of MALToma of the lung. Eur Radiol 1998;8:1669–73 [DOI] [PubMed] [Google Scholar]
- 19.Lee DK, Im JG, Lee KS, Lee JS, Seo JB, Goo JM, et al. B-cell lymphoma of bronchus-associated lymphoid tissue (BALT): CT features in 10 patients. J Comput Assist Tomogr 2000;24:30–4 [DOI] [PubMed] [Google Scholar]
- 20.King LJ, Padley SP, Wotherspoon AC, Nicholson AG. Pulmonary MALT lymphoma: imaging findings in 24 cases. Eur Radiol 2000;10:1932–8 [DOI] [PubMed] [Google Scholar]
- 21.Wislez M, Cadranel J, Antoine M, Milleron B, Bazot M, Mayaud C, et al. Lymphoma of pulmonary mucosa-associated lymphoid tissue: CT scan findings and pathological correlations. Eur Respir J 1999;14:423–9 [DOI] [PubMed] [Google Scholar]
- 22.Bae YA, Lee KS, Han J, Ko YH, Kim BT, Chung MJ, et al. Marginal zone B-cell lymphoma of bronchus-associated lymphoid tissue: imaging findings in 21 patients. Chest 2008;133:433–40 [DOI] [PubMed] [Google Scholar]
- 23.Pisani RJ, DeRemee RA. Clinical implications of the histopathologic diagnosis of pulmonary lymphomatoid granulomatosis. Mayo Clin Proc 1990;65:151–63 [DOI] [PubMed] [Google Scholar]
- 24.Hicken P, Dobie JC, Frew E. The radiology of lymphomatoid granulomatosis in the lung. Clin Radiol 1979;30:661–4 [DOI] [PubMed] [Google Scholar]
- 25.Dee PM, Arora NS, Innes DJ., Jr The pulmonary manifestations of lymphomatoid granulomatosis. Radiology 1982;143:613–8 [DOI] [PubMed] [Google Scholar]
- 26.Lee JS, Tuder R, Lynch DA. Lymphomatoid granulomatosis: radiologic features and pathologic correlations. AJR Am J Roentgenol 2000;175:1335–9 [DOI] [PubMed] [Google Scholar]
- 27.Sheehy N, Bird B, O'Briain DS, Daly P, Wilson G. Synchronous regression and progression of pulmonary nodules on chest CT in untreated lymphomatoid granulomatosis. Clin Radiol 2004;59:451–4 [DOI] [PubMed] [Google Scholar]
- 28.Benamore RE, Weisbrod GL, Hwang DM, Bailey DJ, Pierre AF, Lazar NM, et al. Reversed halo sign in lymphomatoid granulomatosis. Br J Radiol 2007;80:e162–6 [DOI] [PubMed] [Google Scholar]
- 29.Heron CW, Husband JE, Williams MP. Hodgkin disease: CT of the thymus. Radiology 1988;167:647–51 [DOI] [PubMed] [Google Scholar]
- 30.Stolberg HO, Patt NL, Macewen KF, Warwick OH, Brown TC. Hodgkin's disease of the lung: roentgenologic–pathologic correlation. Am J Roentgenol Radium Ther Nucl Med 1964;92:96–115 [PubMed] [Google Scholar]
- 31.Mentzer SJ, Reilly JJ, Skarin AT, Sugarbaker DJ. Patterns of lung involvement by malignant lymphoma. Surgery 1993;113:507–14 [PubMed] [Google Scholar]
- 32.Lewis ER, Caskey CI, Fishman EK. Lymphoma of the lung: CT findings in 31 patients. AJR Am J Roentgenol 1991;156:711–4 [DOI] [PubMed] [Google Scholar]
- 33.Ioachim HL, Dorsett B, Cronin W, Maya M, Wahl S. Acquired immunodeficiency syndrome-associated lymphomas: clinical, pathologic, immunologic, and viral characteristics of 111 cases. Hum Pathol 1991;22:659–73 [DOI] [PubMed] [Google Scholar]
- 34.Levine AM, Seneviratne L, Espina BM, Wohl AR, Tulpule A, Nathwani BN, et al. Evolving characteristics of AIDS-related lymphoma. Blood 2000;96:4084–90 [PubMed] [Google Scholar]
- 35.Eisner MD, Kaplan LD, Herndier B, Stulbarg MS. The pulmonary manifestations of AIDS-related non-Hodgkin's lymphoma. Chest 1996;110:729–36 [DOI] [PubMed] [Google Scholar]
- 36.Hamilton-Dutoit SJ, Pallesen G, Franzmann MB, Karkov J, Black F, Skinhoj P, et al. AIDS-related lymphoma. Histopathology, immunophenotype, and association with Epstein-Barr virus as demonstrated by in situ nucleic acid hybridization. Am J Pathol 1991;138:149–63 [PMC free article] [PubMed] [Google Scholar]
- 37.Blunt DM, Padley SP. Radiographic manifestations of AIDS related lymphoma in the thorax. Clin Radiol 1995;50:607–12 [DOI] [PubMed] [Google Scholar]
- 38.Sider L, Weiss AJ, Smith MD, VonRoenn JH, Glassroth J. Varied appearance of AIDS-related lymphoma in the chest. Radiology 1989;171:629–32 [DOI] [PubMed] [Google Scholar]
- 39.Knowles DM, Inghirami G, Ubriaco A, Dalla-Favera R. Molecular genetic analysis of three AIDS-associated neoplasms of uncertain lineage demonstrates their B-cell derivation and the possible pathogenetic role of the Epstein-Barr virus. Blood 1989;73:792–9 [PubMed] [Google Scholar]
- 40.Simonelli C, Spina M, Cinelli R, Talamini R, Tedeschi R, Gloghini A, et al. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single-institution study. J Clin Oncol 2003;21:3948–54 [DOI] [PubMed] [Google Scholar]
- 41.Dotti G, Fiocchi R, Motta T, Facchinetti B, Chiodini B, Borleri GM, et al. Primary effusion lymphoma after heart transplantation: a new entity associated with human herpesvirus-8. Leukemia 1999;13:664–70 [DOI] [PubMed] [Google Scholar]
- 42.Teruya-Feldstein J, Zauber P, Setsuda JE, Berman EL, Sorbara L, Raffeld M, et al. Expression of human herpesvirus-8 oncogene and cytokine homologues in an HIV-seronegative patient with multicentric Castleman's disease and primary effusion lymphoma. Lab Invest 1998;78:1637–42 [PubMed] [Google Scholar]
- 43.Nalesnik MA. Clinicopathologic characteristics of post-transplant lymphoproliferative disorders. Recent Results Cancer Res 2002;159:9–18 [DOI] [PubMed] [Google Scholar]
- 44.Scarsbrook AF, Warakaulle DR, Dattani M, Traill Z. Post-transplantation lymphoproliferative disorder: the spectrum of imaging appearances. Clin Radiol 2005;60:47–55 [DOI] [PubMed] [Google Scholar]
- 45.Aris RM, Maia DM, Neuringer IP, Gott K, Kiley S, Gertis K, et al. Post-transplantation lymphoproliferative disorder in the Epstein-Barr virus-naive lung transplant recipient. Am J Respir Crit Care Med 1996;154:1712–17 [DOI] [PubMed] [Google Scholar]
- 46.Bates WD, Gray DW, Dada MA, Chetty R, Gatter KC, Davies DR, et al. Lymphoproliferative disorders in Oxford renal transplant recipients. J Clin Pathol 2003;56:439–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nalesnik MA, Makowka L, Starzl TE. The diagnosis and treatment of posttransplant lymphoproliferative disorders. Curr Probl Surg 1988;25:367–472 [DOI] [PubMed] [Google Scholar]
- 48.Harris NL, Ferry JA, Swerdlow SH. Posttransplant lymphoproliferative disorders: summary of Society for Hematopathology Workshop. Semin Diagn Pathol 1997;14:8–14 [PubMed] [Google Scholar]
- 49.Carignan S, Staples CA, Muller NL. Intrathoracic lymphoproliferative disorders in the immunocompromized patient: CT findings. Radiology 1995;197:53–8 [DOI] [PubMed] [Google Scholar]
- 50.Dodd GD, 3rd, Ledesma-Medina J, Baron RL, Fuhrman CR. Posttransplant lymphoproliferative disorder: intrathoracic manifestations. Radiology 1992;184:65–9 [DOI] [PubMed] [Google Scholar]
- 51.Harris KM, Schwartz ML, Slasky BS, Nalesnik M, Makowka L. Posttransplantation cyclosporine-induced lymphoproliferative disorders: clinical and radiologic manifestations. Radiology 1987;162:697–700 [DOI] [PubMed] [Google Scholar]
- 52.Collins J, Muller NL, Leung AN, McGuinness G, Mergo PJ, Flint JD, et al. Epstein-Barr-virus-associated lymphoproliferative disease of the lung: CT and histologic findings. Radiology 1998;208:749–59 [DOI] [PubMed] [Google Scholar]