

Abstract
Pancreatic endocrine neoplasms are rare pancreatic tumours that may occur sporadically or as part of inherited syndromes such as multiple endocrine neoplasia-1 syndrome, von Recklinghausen disease, von Hippel–Lindau syndrome and tuberous sclerosis complex. Recent advances in the genetics and pathology of hereditary syndromes have provided valuable insights into the pathophysiology and biology of sporadic pancreatic endocrine neoplasms. Evolving molecular data on the biology of these neoplasms have the potential for diagnostic, therapeutic and prognostic use.
Pancreatic endocrine neoplasms (PENs) are rare neoplasms accounting for less than 3% of all pancreatic neoplasms [1]. PENs may be categorised as functional or non-functional tumours based on the characteristic clinical syndromes they cause owing to specific hormone synthesis and secretion. Non-functional PENs costitue almost half of all PENs. Insulinomas and gastrinomas account for up to 50% of the functional PENs. PENs demonstrate a variable malignant potential; while 90% of insulinomas are benign, 60% of other functional and non-functional PENs show malignant characteristics [2].
Recent advances in pathology and genetics coupled with detailed studies of patients with hereditary syndromes of PENs have led to better understanding of histogenesis, molecular genetics and dominant oncological pathways employed by these tumours. It is now believed that some PENs, despite remarkable similarity to the pancreatic islet cells, originate from pluripotent cells in the pancreatic exocrine (ductal/acinar) system. Continued progress in the field of molecular genetics has provided useful information about the causative genes, their locations and their putative functions, leading to better understanding of inherited syndromes characterised by the development of PENs. All four hereditary PEN syndromes, inherited as autosomal dominant disorders, are characterised by germline mutations leading to inactivation of the tumour suppressor genes. It is interesting to note that a subset of sporadic PENs are associated with variable loss of chromosome fragments or somatic inactivation of tumour suppressor genes involved in the pathogenesis of hereditary PEN syndromes.
In addition to standard cross-sectional modalities (multidetector CT, MRI and ultrasonography), somatostatin receptor CT/single-photon emission CT (SPECT) and endoscopic and intra-operative sonography play important roles in accurate tumour detection and localisation. While surgery is curative with low-stage tumours, a spectrum of treatment modalities (including the use of somatostatin analogues and small-molecule tyrosine kinase inhibitors) are being investigated to treat patients with advanced, symptomatic disease. Cross-sectional imaging also plays a major role in detecting recurrences and monitoring treatment response following targeted therapies.
Pancreatic endocrine neoplasms: epidemiology, taxonomy and natural history
Pancreatic endocrine neoplasms are discovered most commonly in the fourth and fifth decades of life and demonstrate a slight female predominance [3]. The incidence of PENs in unselected autopsy studies is as high as 1.6% and rises to 10% in autopsies at which the whole pancreas is examined both grossly and microscopically [4]. A substantial increase in the incidence of these tumours has been noted over the last 30 years, due in part to increased detection secondary to advances in imaging technologies and techniques [5].
PENs are classified clinically as functional or non-functional depending on the presence or absence of symptoms related to intrinsic hormone release. Functional PENs such as insulinomas, gastrinomas, glucagonomas, somatostatinomas and vasoactive intestinal peptide tumours (VIPomas) present with symptoms arising from hormonal hypersecretion, and thus present early while they are still small (Table 1). Non-functioning PENs (NF-PENs) tend to be larger in size at time of presentation, with the majority detected in asymptomatic individuals or in individuals with symptoms related to mass effect or metastasis [6]. Of note, although NF-PENs produce no clinical signs of hormonal excess, they may produce a precursor hormone that is functionally inert or occurs in amounts too small to be clinically relevant. The relative frequency of PENs varies in surgical or medical series, but most recent studies suggest the following relative order of incidence: NF-PENs, insulinomas, gastrinomas, glucagonomas, VIPomas, somatostatinomas, others [7]. Up to 90% of insulinomas are found in the pancreas, with an even distribution in the head, body and tail. Almost 80% of sporadic gastrinomas are located in the gastrinoma triangle, defined superiorly by the junction of cystic and common bile duct, inferiorly by the junction of second and third part of duodenum, and medially by junction of body and neck of pancreas [3,8].
Table 1. Summary of the most common pancreatic neuroendocrine neoplasms.
| Tumour | Symptoms and clinical findings | Primary location | Malignant (%) |
| Insulinoma | Hypoglycaemia, neuroglycopenic symptoms | Pancreas (equally throughout) | 5–15 |
| Gastrinoma | Abdominal pain, diarrhoea, peptic ulcers | Gastrinoma triangle | >50 |
| Glucagonoma | Necrolytic migratory erythema, hyperglycaemia, venous thrombosis, weight loss | Pancreas (tail) | >60 |
| VIPoma | Watery diarrhoea, hypokalaemia, achlorhydria (WDHA syndrome) | Pancreas (tail>head, body); rarely adrenal and periganglionic tissue | >50 |
| Somatostatinoma | Hyperglycaemia, cholelithiasis, diarrhoea, steatorrhoea | Pancreas, duodenum | >60 |
| Non-functioning tumours | Abdominal pain, weight loss | Pancreas (head>body, tail) | 60–90 |
VIPoma, vasoactive intestinal peptide tumours.
The biological profile and growth pattern exhibited by each subtype of PEN has been found to be independent of the clinical classification. Thus, in 2004, the World Health Organization devised a clinicopathological classification for pancreatic endocrine neoplasms (Table 2).
Table 2. Clinicopathological classification of endocrine tumours of the pancreas.
| Well-differentiated endocrine tumour |
| Benign behaviour: confined to the pancreas, non-angioinvasive, <2 cm in size, ≤2 mitoses and ≤2% Ki-67-positive cells/10 HPF |
| Uncertain behaviour: confined to the pancreas, ≥2 cm in size, >2% Ki-67 positive cells/10 HPF or angioinvasive, perineural invasion |
| Well-differentiated endocrine carcinoma |
| Low grade malignant with gross local invasion and/or metastases |
| Poorly differentiated endocrine carcinoma, small-cell carcinoma, high grade malignant, >10 mitosis/HPF |
HPF, high power fields; Ki-67, antigen/protein that is a proliferation marker.
Most PENs occur sporadically (90%). However, they may occur as part of the following hereditary syndromes: multiple endocrine neoplasia-1 (MEN-1), von Recklinghausen disease (neurofibromatosis-1; NF-1), von Hippel–Landau (VHL) syndrome and tuberous sclerosis complex (TSC) [9]. All these syndromes demonstrate an autosomal dominant inheritance pattern with a specific mutation of the causative genes (MEN VHL, NF-1 and TSC1/2), all of which function as tumour suppressor genes. The molecular and clinical genetics of tumour susceptibility syndromes in which PENs may occur have greatly contributed to the understanding of tumorigenesis (Table 3).
Table 3. Genetic syndromes associated with pancreatic endocrine neoplasm (PEN).
| Syndrome | Gene location | Gene product | PEN frequency and tumour type |
| MEN-1 | 11q3 | Menin | 80–100% (non-functioning, gastrinoma, insulinoma) |
| VHL | 3p25.5 | pVHL | 12–17% (all non-functioning) |
| NF-1 | 17q11.2 | Neurofibromin | 6% somatostatinoma |
| TSC | 9q34 (TSC1) 16p13.3 (TSC2) | Hamartin, tuberin | <5% either functioning or non-functioning |
MEN-1, multiple endocrine neoplasia-1; NF-1, neurofibromatoses-1 (von Recklinghausen disease); TSC, tuberous sclerosis complex; VHL, von Hippel–Landau.
In contrast to ductal adenocarcinoma, the most common pancreatic neoplasm, which shows a 5-year survival rate of less than 5%, PENs (including malignant tumours) portend a better prognosis. Completely resected PENs show an overall median survival of 7 years, and even unresectable tumours, in the absence of widely metastatic disease, are associated with a median survival in the order of 5 years [10].
Pancreatic endocrine neoplasms: histogenesis and histopathology
PENs were originally hypothesised to arise from immature stem cells of the neuroendocrine system referred to as APUD (amine precursor uptake and decarboxylose) cells [11], yet recent studies suggest an exocrine lineage of some PENs. Histologically, however, PENs are similar to endocrine tumours from other primary sites. The growth pattern within the tumour itself can be solid, nested, trabecular, ribbon-like, tubuloacinar or glandular; mixed patterns within a tumour are common [12,13]. The monotonous tumour cells show nuclei with finely stippled chromatin. The cytoplasm varies from pale to moderately eosinophilic. By immunohistochemistry, more than 95% of PENs label for chromogranin and/or synaptophysin, the latter being more consistently expressed (Figure 1) [14]. Barring two exceptions (insulinomas, which show amyloid deposition, and somatostatinomas of the periampullary duodenum, which may show psammoma bodies), the histological pattern is not distinctive enough to determine the functional status of the tumours [13]. Cystic change in PENs histologically has been felt to represent a degenerative alteration within the tumour. In one review on cystic lesions in the pancreas, cystic PENs represented 1.5% of the tumours studied [15]. Calcifications and osseous metaplasia have also been identified within these tumours [16].
Figure 1.
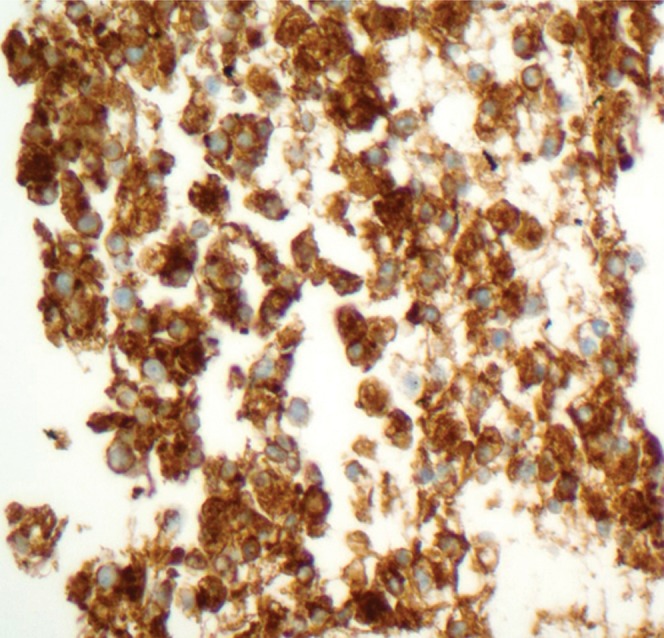
High-power photomicrograph using the chromogranin stain reveals diffusely positive cytoplasmic staining of the tumour cells.
Multiple endocrine neoplasia-1: loss of menin, a tumour suppressor gene product
MEN-1 syndrome is an autosomal dominant disorder that is characterised by multifocal endocrine tumours involving the anterior pituitary, parathyroid, stomach, duodenum, pancreas, adrenal cortex and lungs. In addition, various uncommon tumours may occur in the skin, central nervous system and soft tissues such as angiofibromas and collagenoma [17,18]. Typically, patients with MEN-1 syndrome present with hyperparathyroidism in the third decade of life, and develop clinical symptoms related to PEN between the ages of 35 and 50 years (in 20–70% of cases) [18]. In a large pathology series, almost all patients with MEN-1 were found to harbour a small, non-functioning PEN at autopsy [19]. A variety of PEN subtypes are found in MEN-1 patients, including NF-PENs (80–100%), gastrinomas (20–61%), insulinomas (7–31%) and other functioning tumours (<5%). A strong association of NF-PENs and gastrinomas is seen with MEN-1, whereas insulinomas, VIPomas and glucagonomas have been shown less frequently with the MEN-1 syndrome [20]. Overall, about 10% of PENs are found to be associated with MEN-1. MEN-1 syndrome is also diagnosed in 20–25% of all patients with gastrinomas and 4% of patients with insulinomas [21].
The pancreatic tumours associated with MEN-1 syndrome are typically multiple and widespread (Figures 2 and 3). The most characteristic MEN-1 pancreatic lesion is pancreatic microadenomatosis, defined as the presence of numerous pancreatic microadenomas (up to 5 mm in diameter) [22]. Gastrinomas seen with MEN-1 may also arise in the duodenum. In contrast to sporadic PENs, the tumours associated with MEN-1 tend to present at an earlier age (30–50 years), have a higher rate of post-operative recurrence and are a common cause of death in these patients [19].
Figure 2.

48-year-old male with multiple endocrine neoplasia-1 and elevated gastrin levels. (a, b) Axial contrast-enhanced CT in the arterial phase shows two hypervascular lesions (white arrows) in the pancreas head and body. At time of surgery the entire pancreas was found to be studded with multiple gastrinomas. Total pancreatectomy was performed. Incidental note is made of bilateral adrenal adenomas.
Figure 3.

31-year-old male with multiple endocrine neoplasia-1 and history of subtotal thyroidectomy for medullary cancer. (a, b) Axial contrast-enhanced CT in the arterial phase shows multiple hypervascular lesions (white arrows) in the pancreatic head and tail.
MEN-1 syndrome is the result of an inactivating mutation of the MEN-1 gene. The MEN-1 gene is a tumour supressor gene that is located on chromosome 11q13 (gene product: menin). Genetic mapping studies show somatic loss of heterozygosity (LOH), suggesting the two-hit hypothesis. Initially, a germline mutation affects the MEN-1 gene, making the carrier of the inherited defective gene heterozygous and predisposed to tumour development (first hit), and then a somatic inactivation of the unaffected allele by LOH occurs (second hit), resulting in the development of MEN-1-associated lesions [23,24].
Menin is a cell cycle-regulated nuclear protein, and is assumed to play an important role in pathways controlling cell growth and differentiation during embryogenesis and post-natal life. Menin has been shown to interact with numerous proteins involved in regulation of transcription, DNA replication, mitosis, apoptosis, genome integrity, growth factor signalling pathways and extracellular matrix organisation [18,25]. Menin binds to activating protein-1 transcription factor JunD and neuronal factor κB (NF-κB), resulting in inhibition of transcription caused by each of these proteins. Menin also interacts with SMA/mothers against decapentaplegic (MAD) Drosophila homologue protein 3 (SMAD3), and the SMAD1/5 complex results in enhanced transcriptional activation and transforming growth factor β (TGFβ) signalling. Studies of menin in irradiated cells have indicated that menin may be important for repairing DNA damage.
Multiple allelic deletions involving chromosomes 6, 8, 10, 11, 18 and 21 have also been demonstrated in PEN arising in MEN-1 patients [26]. Inter- and intratumoral genetic heterogeneity or variation is noted, suggesting that there is chromosomal instability in these tumours [26].
It has been noted that 5% to 93% of PENs from patients with sporadic PENs (not caused by MEN-1) show LOH at the MEN-1 locus (11q13), and from 27% to 39% of the sporadic tumours had a mutation in the MEN-1 gene [27-30]. The most common sporadic PENs containing somatic MEN-1 mutations are gastrinomas and NF-PENs [29-31]. These observations suggest that the MEN-1 gene may be involved in the pathogenesis of both familial and sporadic PENs, and also suggest a possibility of additional tumour suppressor genes located on the distal part of 11q that may co-operate with the MEN-1 gene in the pathogenesis of PENs.
von Hippel–Lindau disease: dysregulation of hypoxia-inducible factor pathway due to von Hippel–Lindau gene inactivation
VHL syndrome is characterised by retinal or central nervous system haemangioblastomas, clear cell renal carcinomas, and a phaeochromocytomas, as well as pancreatic cystic tumours and PENs [32]. Pancreatic lesions are noted in 20–75% of patients with VHL [33]; however, the majority of these pancreatic lesions are reported to be true cysts (91%). Other pancreatic lesions associated with VHL include cystadenomas (12%), PENs (10–17%), haemangioblastomas (<1%) and adenocarcinomas (<1%) [34].
Endocrine pancreatic tumours in VHL patients are being increasingly recognised and emerging as life-threatening lesions because other serious associated diseases (i.e. renal carcinomas, symptomatic central nervous system haemangioblastomas and phaeochromocytomas) are now well recognised and better managed [35]. Almost all VHL-related pancreatic PENs reveal LOH of the VHL gene locus on chromosome 3p25 [36]. The gene product pVHL is a tumour suppressor protein and is involved in oxygen-dependent, proteosomal degradation of the alpha subunit of hypoxia-inducible factor (HIF-α), a key regulator of the tissue hypoxia response mechanism. Inactivation of VHL thus leads to inappropriate up-regulation of HIF and its downstream hypoxia response genes, including vascular and somatic growth factors, resulting in angiogenesis and cell proliferation. Investigations in a kindred with VHL PEN revealed LOH in other sites of chromosome 3p, suggesting that apart from the VHL gene, other suppressor genes, not yet identified, may cause these tumours [37].
PENs in the setting of VHL syndrome have been seen to occur in young patients, may be multiple (up to five), can be located anywhere in the pancreas, are functionally inactive (immunohistochemistry may however demonstrate focal positivity for pancreatic polypeptide, somatostatin, glucagon and/or insulin in 30–40% of cases) and are not associated with either microadenomas or nesidioblastosis [38-40]. VHL-related pancreatic PENs also grow slowly. Although VHL-related tumours are often multifocal, precursor lesions have not been identified in a systematic analysis of 14 patients [36]. The distinguishing feature of VHL PEN on histology is clear cell morphology, seen in up to 60% of tumours [36].
The risk of malignancy in PEN in association with VHL syndrome seems to be directly proportional to the diameter of the tumour, as with sporadic PEN. In the largest reported series of 30 patients, the median size of the tumour in patients without metastases was 2 cm (n=25) compared with 5 cm for those with metastases (n=5) [39].
Allelic loss on chromosome 3p has also been described in one-third of sporadic PENs; however, the region of allelic loss did not involve the VHL locus [41]. A novel gene located close to the VHL locus is thus suspected to be associated with development of sporadic PENs. It has also been noted that the tumours with 3p allelic loss were associated with metastatic disease, whereas PENs showing an intact 3p region were more likely to be benign [41].
Neurofibromatosis-1: loss of neurofibromin, a tumour suppressor protein
NF-1, a neurocutaneous phakomatosis, is clinically characterised by the presence of café au lait spots on the skin, cutaneous or subcutaneous neurofibromas, optic gliomas, benign iris hamartomas and specific dysplastic bone lesions. The prevalence of NF-1 has been estimated to be 1 in 4500 newborns [42]. Duodenal somatostatinomas are the most frequent endocrine tumours in NF-1, and uncommonly pancreatic somatostatinomas (16 times less common than duodenal somatostinomas), insulinomas or pancreatic NF-PEN and gastrointestinal stromal tumours are also noted. Characteristically these tumours occur in the periampullary region, cause biliary dilatatio and present with jaundice and/or pancreatitis. NF-1 and sporadic duodenal somatostatinomas differ from sporadic pancreatic somatostatinomas in the frequency of clinical somatostatinoma syndrome (1–2 vs 66%), mean tumour size (2.8 vs 5.9 cm), psammoma body production (61% vs 0%) and the presence of metastases (30% vs 71%) [43,44].
The NF-1 gene is a tumour suppressor gene that is located on 17q11.2 and encodes a protein called neurofibromin. Neurofibromin affects cell proliferation/growth and signalling by regulating the activation of p21 Ras by its Ras guanosine triphosphatase (GTPase) protein-activating activity, binding microtubules, modulating adenylate cyclase activity and interacting with the cellular cytoskeleton [45]. This is also linked with the genes responsible for TSC, regulating especially TSC2 through the mammalian target of rapamycin (mTOR) pathway. The mTOR is a serine threonine kinase that participates in the regulation of apoptosis, proliferation and cell growth through modulation of cell cycle progression. It has been shown that the NF-1 gene acts as a negative regulator of mTOR; therefore LOH of the NF-1 gene results in loss of neurofibromin expression, mTOR activation and hence tumour development [46]. These observations suggest that therapy targeting mTOR could be of benefit for NF-1-associated PEN.
Tuberous sclerosis complex
TSC is a multisystem disorder exhibiting a wide range of manifestations characterised by hamartomatous lesions in the brain, skin, eyes, heart, lungs and kidneys. TSC has a prevalence of 1∶10 000 and demonstrates autosomal dominant mode of transmission with a penetrance of up to 100%. Two-thirds of the cases result from new dominant mutations. The diagnosis of TSC is usually based on both the clinical and radiological findings.
TSC is caused by inactivating mutations in either the TSC1 gene at 9q34 or the TSC2 gene at 16p13 [34]. The products of the TSC1 and TSC2 genes, hamartin and tuberin, respectively, form a dimer that mediates a key step in the phosphoinositide 3-kinase signalling pathway. They are also important for regulation of the small GTPase, rheb, which, in turn, is involved in regulating the activity of mTOR, a master controller of protein translation, integrating information on growth stimuli, cellular energy levels, nutrient availability, hypoxia and cell growth [47].
Both functional PENs and NF-PENs are reported in a small percentage of patients with TSC and usually occur in patients with TSC2. Gastrinomas and insulinomas are the most common functional PENs described in TSC, and some of these may be malignant [48,49].
Pancreatic endocrine neoplasms: role of imaging
Non-invasive imaging techniques for localising pancreatic endocrine tumours include ultrasound, contrast-enhanced imaging, CT, MRI and somatostatin receptor scintigraphy with SPECT/CT. Conventionally, angiography and venous sampling have also been used; however, they are seldom performed now. More recently, endoscopic ultrasound (EUS) and EUS-guided five-needle aspiration (EUS-FNA) have become preferred modalities for diagnosing PENs, with excellent sensitivity for detection of tumours that are too small to be detected with conventional cross-sectional imaging [50].
Contrast-enhanced ultrasound (CEUS) can be performed in conjunction with greyscale and Doppler examination by using a wide array of transabdominal, intra-operative or endoscopic transducers [51]. The contrast agents consist of stabilised gas microbubbles (measuring >10 μm), which act as blood pool tracers and produce harmonic signals at low acoustic powers [52]. The contrast agents are non-embolising and non-toxic, and only mild adverse reactions have been recorded [52]. CEUS involves acquisition of dynamic images (arterial, portal venous and delayed phases) following IV injection of a second-generation contrast agent such as sulphur hexafluoride. CEUS allows real-time depiction of tumour angioarchitecture with high contrast and spatial resolution. Studies on detection of PENs by CEUS have reported high sensitivity, specificity, positive predictive value and negative predictive value of 94%, 96%, 75% and 99%, respectively [53].
PENs, on account of rich vascularity, usually show rapid and marked enhancement, appearing hyperechoic in comparison with surrounding parenchyma in the arterial phase, with washout giving a slight hypoechoic appearance on the delayed phase (Figure 4). Uncommonly, the tumours may show persistent enhancement, becoming more obvious during the portal venous phase. An advantage of CEUS over CT or MR techniques is the ability to constantly monitor the lesion for evaluation of peak enhancement, in contrast to CT/MR, where timing of imaging is far more critical and lesions may be missed if imaging is performed early or late [54]. Other advantages of CEUS include its non-invasive nature, cost-effectiveness and potential use in patients with renal failure.
Figure 4.

(a) Greyscale ultrasound image shows a solid mass (arrow) with internal anechoic areas/debris in the region of pancreatic body (star). (b) Contrast-enhanced ultrasound image obtained in the arterial phase (20 s) shows marked enhancement of solid elements (arrow) leading to a hyperechoic appearance. Central cystic/necrotic elements are non-enhancing (star). Figure courtesy of Dr R Sinha, FRCR, FICR, MD, Warwick Hospital and Medical School, Warwick, UK.
A dedicated multiphase contrast-enhanced CT or dynamic gadolinium-enhanced MRI is a prerequisite for optimal assessment of pancreatic endocrine neoplasms. A study comparing multiphasic CT and MRI found similar effectiveness of both techniques in identification and localisation of these tumours [55]. Although PENs are classically considered hypervascular in the arterial phase, the degree of enhancement in the arterial phase and timing of enhancement may be variable. In fact, some of the PENs may remain isodense to the pancreas on arterial phase and are best identified on the unenhanced and/or venous phase (Figure 5). Multiple studies have thus concluded that multiphasic scanning leads to improved detection of pancreatic islet cell tumours [56,57]. As the conspicuousness of the tumour, as well as the metastasis lesions, depends on density difference between tumour and surrounding parenchyma, low-density oral intraluminal agents are used to improve the identification of tumours and also facilitate three-dimensional (3D) volumn-rendering technology. A commonly used protocol involves 500 ml of water given orally over 30 min with an additional 250 ml of water immediately prior to the study to achieve adequate distention of the stomach and duodenum. Small tumours are also easier to detect on reformatted and 3D images than axial images alone (Figure 6). A common pitfall is mistaking vascular structure for small arterially enhancing lesions on the axial images; however, the vessels are apparent on the reformatted and 3D images.
Figure 5.

(a) Axial T2 weighted MRI shows a lesion with high T2 signal in the pancreatic head (arrow). The lesion remained isointense to the rest of the pancreatic parenchyma on all phases following contrast administration. (b) Axial post-gadolinium image in the portal venous phase shows no definite lesion. The arrow in (b) points to the expected location of the tumour. Surgical pathology revealed a moderately differentiated neuroendocrine tumour.
Figure 6.
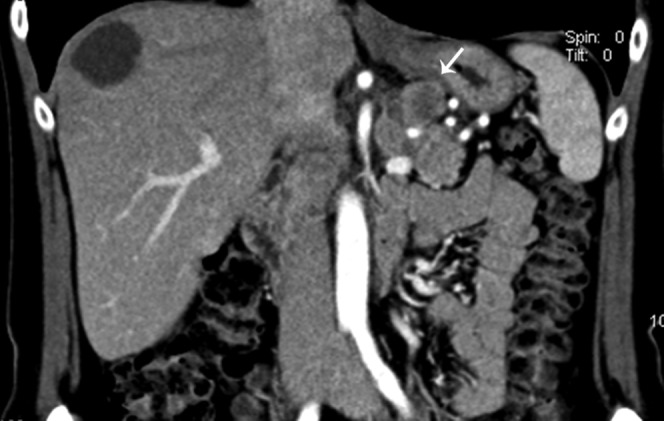
47-year-old female with history of unintentional weight loss with elevated pancreas polypeptide. Coronal reformatted contrast-enhanced CT image in the arterial phase shows a 2.4-cm exophytic pancreatic body mass (arrow). Pathology revealed a well-differentiated low grade pancreatic endocrine neoplasm.
Tumour enhancement also depends on the tumour size. While small lesions commonly demonstrate homogeneous enhancement, larger lesions often exhibit a heterogeneous enhancement pattern (Figures 7 and 8) [57,58]. A ring-like peripheral enhancement is also considered characteristic both within the primary tumour and its metastasis (Figures 9 and 10). Most PENs are solid; however, cystic variants have been reported. Cystic PENs are more likely to be associated with MEN-1 and more likely to be non-functional (Figures 11 and 12) [59].
Figure 7.

Coronal reformatted image from the arterial phase showing a partly exophytic endocrine tumour (arrow) arising from the proximal pancreatic body infiltrating the hepatoduodenal ligament. Multiple arterially enhancing lesions are seen throughout the liver, consistent with metastasis, proven by core biopsy.
Figure 8.
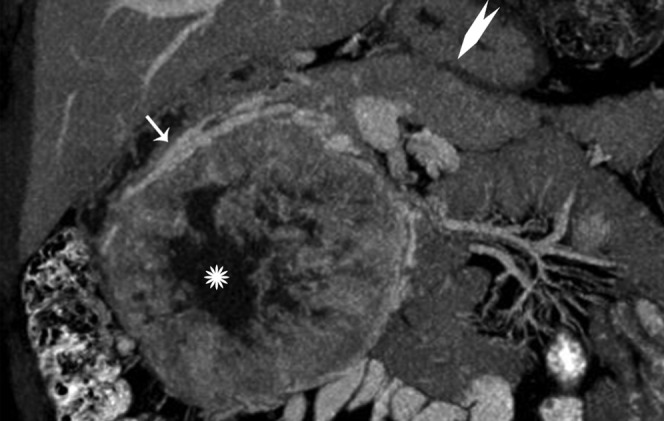
33-year-old female with a large solitary hypervascular neoplasm in the head of the pancreas (white arrow) with central necrosis (star). The body and tail of pancreas is normal (chevron).
Figure 9.
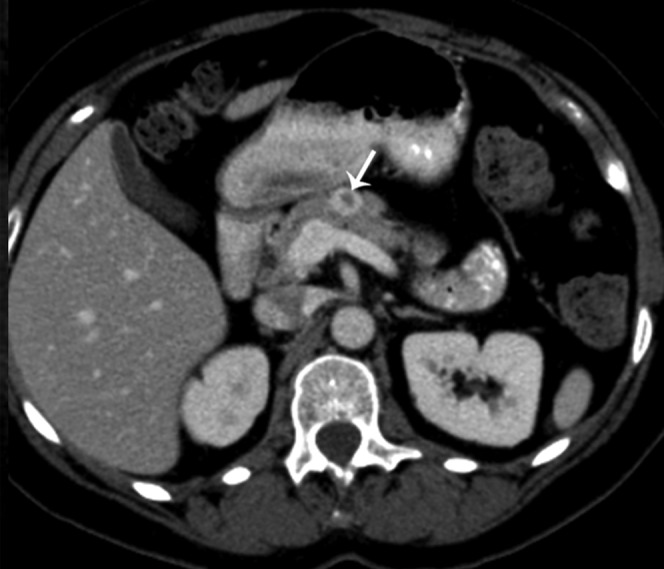
51-year-old female with heartburn and nausea. Axial contrast-enhanced CT shows a peripherally enhancing lesion (arrow) in the pancreatic body. Pathology following surgical enucleation revealed a gastrinoma.
Figure 10.
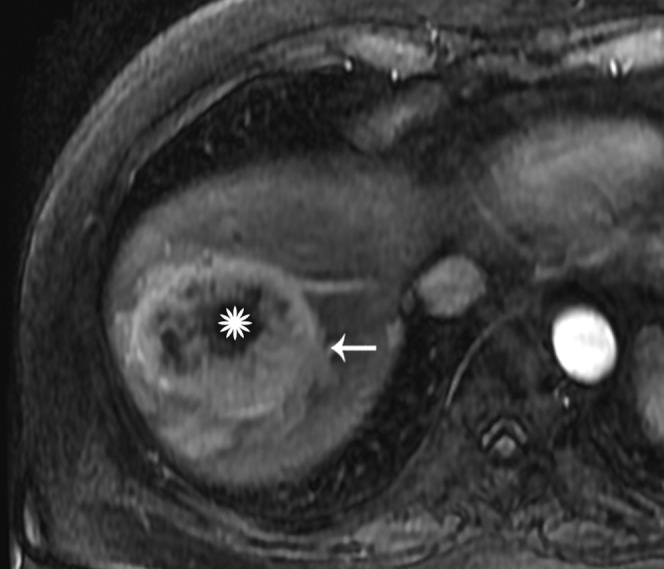
Axial gadolinium-enhanced arterial phase image showing the targetoid appearance with intense peripheral arterial enhancement (arrow) and central necrosis (star).
Figure 11.
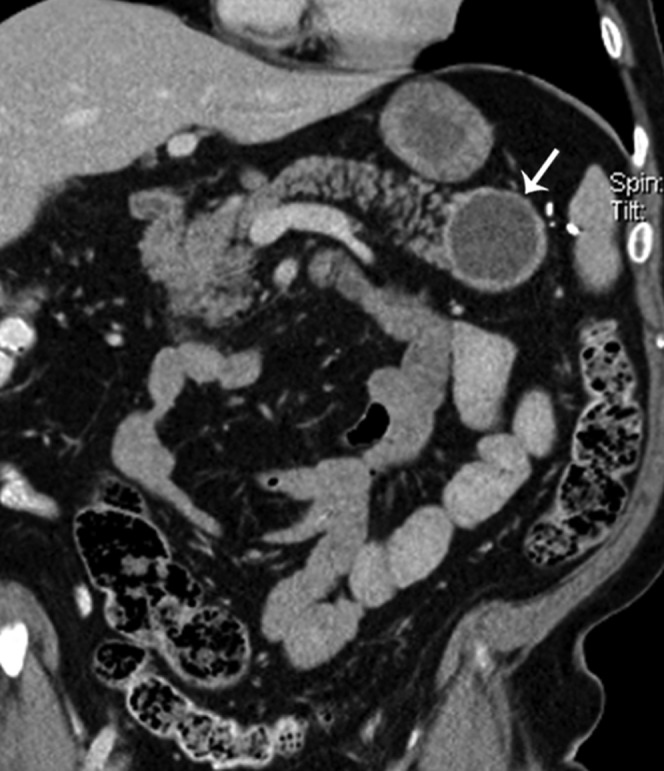
67-year-old female with incidental detection of a pancreatic tail mass. Reformatted coronal contrast-enhanced CT shows a round cystic mass in the pancreatic tail with a thick enhancing wall (arrow). Laparoscopic splenic preserving distal pancreatectomy showed a well-differentiated neuroendocrine tumour with cystic change.
Figure 12.

(a) Axial contrast-enhanced CT (CECT) image shows a thick-walled cystic lesion (thick arrow) with a suspicious mural nodule (thin arrow) in the pancreatic tail. The patient was lost to follow-up and a repeat CECT was performed 1 year later. (b) Axial CECT image shows significant increase in size of pancreatic mass (thick arrow) with eccentric solid component, also larger. The patient also developed hepatic metastasis in the interval (star).
An optimal MRI protocol for detection of PENs should include breath-hold T2 weighted sequences, axial in-phase and opposed-phase T1 weighted images, and breath-hold T1 weighted fat-suppressed sequences acquired before and after contrast administration. The T1 fat-suppressed sequence shows excellent contrast between the low-signal intensity (SI) tumour and normally bright pancreatic parenchyma secondary to acinar proteins [60]. The tumours usually exhibit high SI on T2 weighted fat-suppressed images (Figure 13) [61].
Figure 13.

34-year-old female with family history of multiple endocrine neoplasia-1 and hyperparathyroidism. (a) Axial contrast-enhanced CT from the arterial phase shows mild/vague enlargement of pancreatic head/uncinate process (arrow) without a discrete lesion and mild extrahepatic biliary dilatation. (b) Coronal half-Fourier acquisition single-shot turbo spin-echo image shows a lobulated T2 intermediate–high signal intesnity mass (arrow) causing extrinsic mass effect on the distal common bile duct, proven to represent a pancreatic endocrine neoplasm.
Pancreatic endocrine neoplasms: clinical and imaging features of specific subtypes
Functioning pancreatic endocrine neoplasms
Insulinoma, gastrinoma, glucagonoma and VIPomas are the most common functional PENs.
Insulinoma
Insulinomas are usually benign, small, solitary tumours within the pancreas, although MEN-1-related insulinomas may be multicentric. Most insulinomas are under 2 cm in size with an equal distribution in the head, body and tail of the pancreas. The highest incidence is found in the fifth and sixth decades, and females are more frequently affected (male-to-female ratio, 4:6) [62]. Insulinomas may present as diffuse hyperplasia or microadenomatosis in 2% of cases [63]. Tumours that produce a hypoglycaemic syndrome are usually larger than 1 cm; microadenomas are typically functionally silent.
Two recent studies employing multidetector, multiphase contrast-enhanced thin section CT have reported sensitivities of 83% and 94% for detection of insulinomas [57,64]. Typically, insulinomas are hypervascular, and show intense enhancement during the arterial and portal phases following contrast administration (Figure 14). The enhancement is usually uniform, but may also be targetoid [65]. Because small tumours are non-contour-deforming, detection of the vascular blush is essential for the diagnosis of small tumours. Many small lesions are easier to detect in the arterial phase [66]. Multiplanar reconstructions can improve detection and localisation of small tumours. In a recent retrospective series of 30 patients with post-operative diagnosis of insulinoma, 63% of the tumours had been identified prospectively and 83% retrospectively on CT. The study reported the following causes for the false-negative studies: proximity of the lesions to the vessels, cystic/pedunculated mass and non-inclusion of 3D images for interpretation [57]. On MRI, insulinomas show low SI on T1 weighted images and high SI on T2 weighted images. They are especially well visualised on T1 and T2 weighted images with fat suppression. On dynamic contrast-enhanced T1 weighted images, the tumours show typical enhancement pattern as on CT scan.
Figure 14.
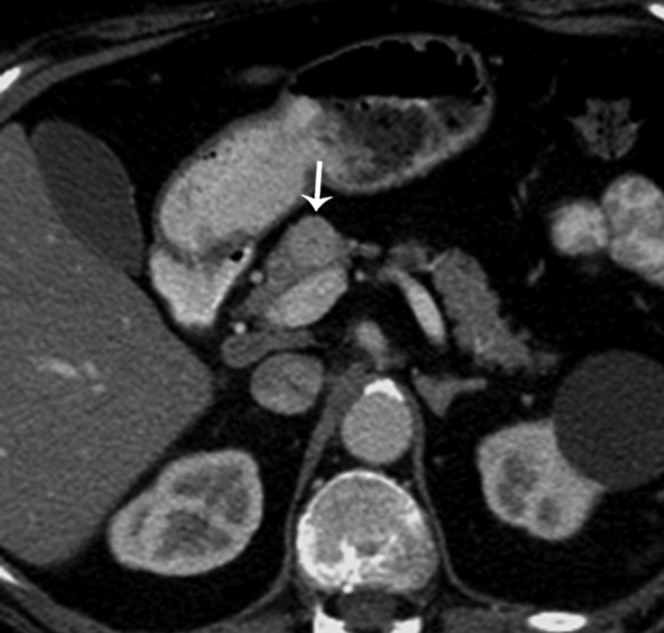
35-year-old with hypoglycaemic attacks, elevated proinsulin blood level (640 pmol l−1) and very low fasting blood sugar. Axial contrast-enhanced CT in the arterial phase reveals an intensely enhancing mass (arrow) in the pancreatic neck, consistent with an insulinoma.
The reported sensitivity of EUS for the detection of insulinoma ranges between 82 and 94% (Figure 15). The combination of thin section multislice CT and endoscopic ultrasound was demonstrated to have a combined sensitivity for pre-operative detection of insulinomas of 100% [57,64]. Somatostatin-receptor scintigraphy (SRS), which has a major role in imaging other PENs, is less useful in locating insulinomas as these tumours have a lower density of somatostatin receptors and generally do not express the somatostatin subtype-2 cell-surface receptor.
Figure 15.
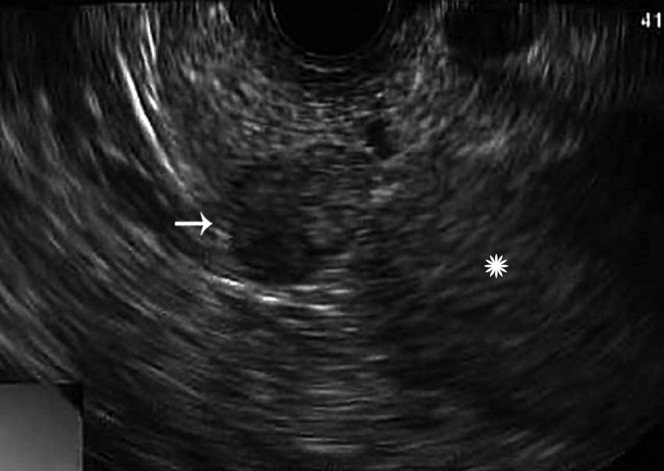
Endoscopic ultrasound image shows a well-defined hypoechoic mass (arrow) in the distal pancreatic body/tail. The spleen (star) is noted on the right of the image.
Gastrinoma
Gastrinoma is the second most common endocrine tumour of the pancreas. Gastrinomas cause hypersecretion of gastrin, resulting in hyperacidity, Zollinger–Ellison syndrome, abdominal pain and diarrhoea. They are more often extrapancreatic and multiple compared with insulinoma. 90% of the gastrinomas occur in the gastrinoma triangle and are frequently small (3–5 mm in diameter; Figure 16) [67]. Associated gastric rugal hypertrophy and wall thickening, indicating hyperplastic gastritis and peptic ulcer disease, are often useful in the diagnosis. Metastases to the liver or lymph nodes are seen in up to 70–80% of cases at the time of diagnosis. Gastrinomas tend to be less vascular than insulinomas [63]. EUS has a sensitivity of 80–94% for detection of intrapancreatic tumours and 70% for detection of extrapancreatic tumours [68]. SRS also has high sensitivity, ranging between 74 and 87%, for detection of gastrinomas (Figure 17) [68].
Figure 16.
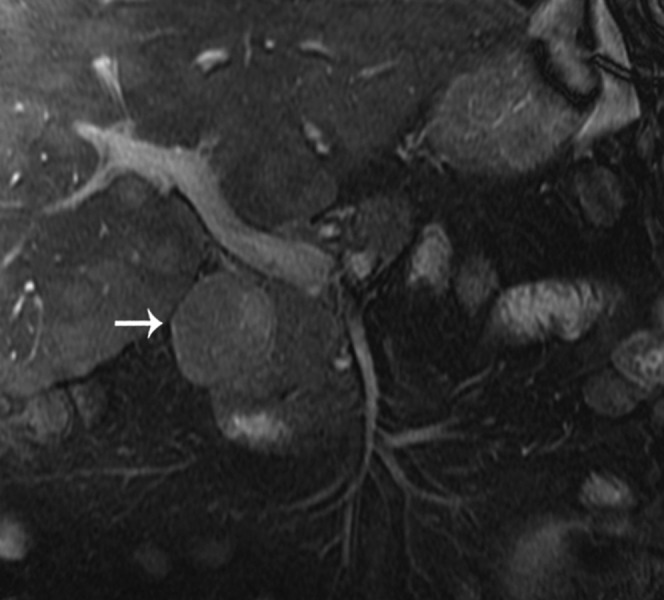
Coronal fast imaging employing steady-state acquisition image shows an intermediate signal mass (arrow) consistent with a gastrinoma in the typical boundaries of the gastrinoma triangle.
Figure 17.
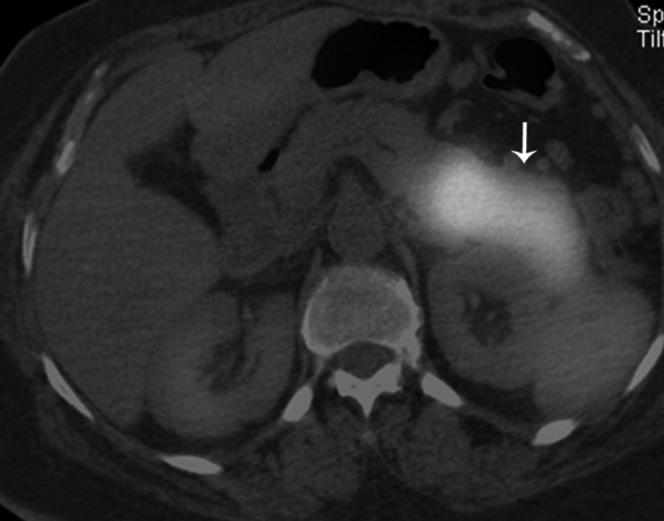
Fused axial image from CT/single-photon emission CT using octreotide showing intense uptake (arrow) in the distal body and tail of the pancreas consistent with presence of a neuroendocrine tumour.
Glucagonoma and VIPoma
Glucagonomas and VIPomas usually tend to be large solitary tumours, commonly occurring in the tail of the pancreas (Figure 18) [69]. Most of these are solid and enhancing but may show cystic change. Approximately 60–70% of glucagonomas and 50% of VIPomas present with metastasis at the time of diagnosis [70]; however, glucagonomas tend to grow slowly and patients survive for many years [69-71]. SRS is the imaging of choice for detection of VIPomas, with a sensitivity of up to 88% for detection.
Figure 18.
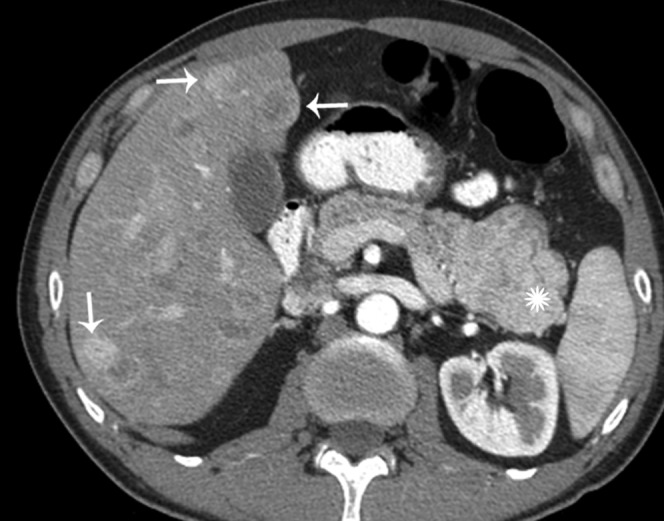
42-year-old male with incidental finding of a pancreatic mass. Axial contrast-enhanced CT in the arterial phase shows a hypervascular mass in the pancreatic tail (star) as well as hypervascular liver metastasis (arrows).
Non-functioning pancreatic endocrine neoplasms
90% of non-functioning tumours are malignant at the time of presentation. They are almost always very large, ranging from 3 to 24 cm in diameter (Figures 19 and 20) [72]. Recent studies have shown that NF-PENs are smaller than previously reported, probably because of increasing detection of incidental lesions through widespread use of cross-sectional imaging (Figure 21) [73]. Calcifications can be seen in approximately 20% of cases and are usually discrete, nodular or shell-like [74]. Calcification has also been reported more frequently in malignant than in benign neoplasms. On MRI, NF-PENs show high SI on T2 weighted image with homogeneous enhancement [12]. Imaging findings that are suggestive of malignancy include necrosis, invasion of retroperitoneal structures and discrete nodular calcification. The liver and lymph nodes are the most common sites for metastasis. Like the primary tumour, liver metastases tend to be hypervascular to the normal hepatic parenchyma or show uniform peripheral ring enhancement [3].
Figure 19.
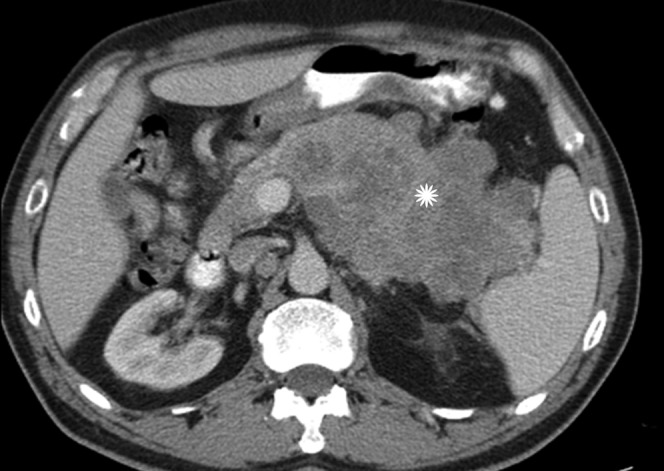
Axial contrast-enhanced CT in the portal venous phase shows an infiltrative mass in the body and tail of pancreas (star). Biopsy revealed intermediate-grade neuroendocrine carcinoma.
Figure 20.
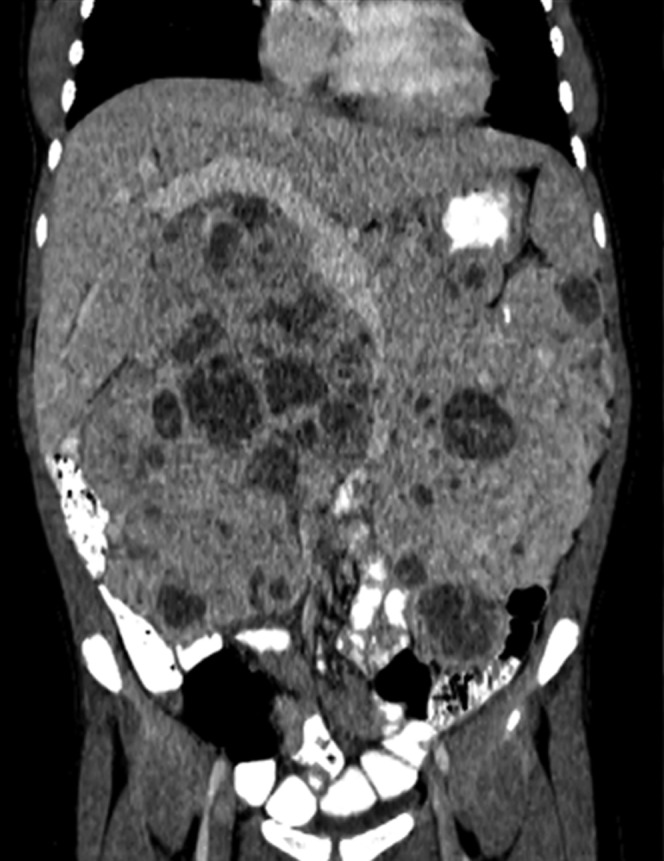
18-year-old female with a large non-functioning neuroendocrine tumour. Coronal contrast-enhanced CT showing a large mass with areas of necrosis in the retroperitoneum.
Figure 21.
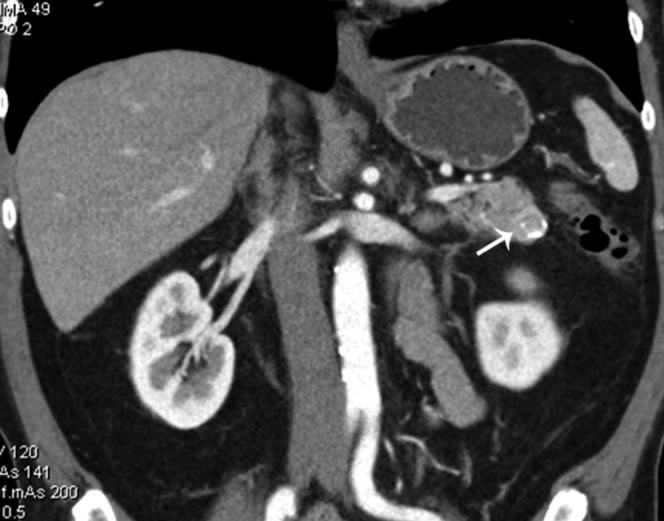
60-year-old male with incidentally detected 1.5 cm peripherally calcified mass in the pancreatic tail, during workup for back and left flank pain, with 40 lb of unintentional weight loss. Coronal reformatted contrast-enhanced CT from arterial phase demonstrates minimal enhancement within the tumour (arrow). Surgical pathology from distal pancreatectomy showed well-differentiated endocrine neoplasm of benign behaviour.
Malignant non-functioning tumours often present similarly to adenocarcinoma. Pre-operative accurate differentiation between PENs and exocrine pancreatic tumours is very important because PENs reveal more indolent behaviour, higher resectability, better response to chemotherapy and better prognosis compared with adenocarcinomas [3]. Imaging findings that are useful in the differentiation of endocrine tumours from ductal adenocarcinomas include the high SI on T2 weighted images, hypervascular primary tumour and liver metastases, presence of calcification lack of vascular encasement, lack of ductal obstruction, and lack of desmoplastic reaction [75]. In contrast to the frequent occurrence of venous thrombosis in pancreatic ductal adenocarcinoma, thrombosis is rare in the setting of pancreatic endocrine neoplasms. Peritoneal metastasis and/or regional lymph node enlargement, characteristic features of pancreatic ductal adenocarcinoma, are generally not present in pancreatic endocrine tumours.
Management and prognosis
In contrast to patients with adenocarcinoma of the pancreas, those who have pancreatic endocrine carcinomas can achieve long-term survival even if their disease is advanced. Strategies for management of neuroendocrine tumours include complete surgical resection, as well as medical management. For functional tumours, biotherapy with secretory inhibitors such as somatostatin analogues and interferon-α can be used. Treatment for metastatic tumours may include systemic chemotherapy, hepatic artery embolisation or radiofrequency ablation for hepatic lesions. In general, well-differentiated tumours do not demonstrate a high degree of sensitivity to chemotherapy which has been explained by their low mitotic rates, high levels of antiapoptotic protein bcl-2 and increased expression of the multidrug resistance gene [76].
Recent advances in delineation of molecular pathogenesis in the genetic PEN syndromes has provided insights into the pathogenesis of sporadic tumours and demonstrated potential molecular targets where targeted disruption may prevent tumour progression. PENs frequently produce multiple growth factors including vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), insulin-like growth factor (IGF-1), basic fibroblast growth factor and transforming growth factor, as well as expressing receptors for these (VEGFR, PDGFR, IGF-1R) and other growth factors (epidermal growth factor receptor, EGFR) [77]. These are potential molecular targets for directed therapy (Figure 22). Multiple such novel targeted therapies are currently under investigation, directed at various growth factors and receptors on the gastrointestinal PEN surface, including a monoclonal antibody to VEGF (bevacizumab), as well as small-molecule inhibitors of the intracellular tyrosine kinase domain of VEGFR or other growth factor receptors: sunitinib, sorafenib, vatalanib, imatinib (gleevac), gefitinib [78,79].
Figure 22.

Diagram demonstrating the sites and mechanism of action of novel agents for the management of pancreatic endocrine neoplasm. The growth factor receptors when occupied by their respective growth factors (in an autocrine or paracrine manner) lead to autophosphorylation of the intracellular tyrosine kinase component of the receptor. This activates the mammalian target of rapamycin (mTOR) pathway (among others), ultimately promoting protein synthesis, cell cycle progression and cell survival. The pathway can be inhibited by monoclonal antibodies to growth factor receptors, tyrosine kinase inhibitors or downstream mTOR inhibitors.
In the preliminary report of a Phase II trial evaluating gefitinib, a small-molecule inhibitor of the EGFR tyrosine kinase domain, a 6-month progression-free survival was noted in 7 out of 24 PEN patients. Prior to therapy all patients had radiographic evidence of progression [80,81]. Sunatinib maleate (SU-11248) is a selective inhibitor of tyrosine kinases such as VEGFR-1 to -3, PDGFR, fibromyalg syndrome-like tyrosine kinase 3, c-KIT and RET with antitumour activity caused by inhibition of angiogenesis and its antiproliferative effects (Figure 23) [82]. In a Phase 2 multicentric trial in patients with advanced neuroendocrine tumours, overall objective response rate achieved was 16.7%, and 68% of patients had stable disease in pancreatic tumours [83]. Sirolimus (rapamycin) and its derivatives block mTOR and yield antiproliferative activity. Temsirolimus and everolimus have also been evaluated in endocrine tumours and significant clinical benefits have been found in Phase 2 trials of both these drugs [84,85].
Figure 23.

Axial contrast-enhanced images of the liver from a patient on sunatinib therapy for metastatic neuroendocrine tumour. (a) Pre-treatment axial contrast-enhanced CT (CECT) image shows a well-defined enhancing lesion (arrow) in the liver, consistent with a metastatic lesion. (b) Post-treatment axial CECT image at the same level reveals evidence of necrosis within the lesion (arrow), suggesting treatment response. Multiple other low-density areas correspond to other metastatic lesions, not well visualised on the pre-treatment CT.
Conclusion
There is a wide spectrum of non-functional and functional PENs with characteristic histogenesis, pathology, natural history and tumour biology. Functional PENs are characterised by specific hormone excess syndromes. A variety of imaging modalities permit detection and staging of PENs that allows optimal management. Surveillance imaging studies assist in detecting recurrences as well as documenting treatment response. The malignant PENs demonstrate variable biological behaviour, with prognosis being better than that of pancreatic cancers even when the disease is advanced. The emerging knowledge of the molecular biology of PENs has facilitated the development of new targeted therapies. It is hoped that these novel agents may play a greater role in the future to improve quality of life and prolong disease-free survival for patients with PENs.
References
- 1.Hoffmann AC, Mori R, Vallbohmer D, Brabender J, Drebber U, Baldus SE, et al. High expression of heparanase is significantly associated with dedifferentiation and lymph node metastasis in patients with pancreatic ductal adenocarcinomas and correlated to PDGFA and via HIF1a to HB-EGF and bFGF. J Gastrointest Surg 2008;12:1674–81 (discussion: 81–2) [DOI] [PubMed] [Google Scholar]
- 2.Metz DC, Jensen RT. Gastrointestinal neuroendocrine tumors: pancreatic endocrine tumors. Gastroenterology 2008;135:1469–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kloppel G, Heitz PU. Pancreatic endocrine tumors. Pathol Res Pract 1988;183:155–68 [DOI] [PubMed] [Google Scholar]
- 4.Kimura W, Kuroda A, Morioka Y. Clinical pathology of endocrine tumors of the pancreas. Analysis of autopsy cases. Dig Dis Sci 1991;36:933–42 [DOI] [PubMed] [Google Scholar]
- 5.Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol 2008;9:61–72 [DOI] [PubMed] [Google Scholar]
- 6.Ferrone CR, Tang LH, Tomlinson J, Gonen M, Hochwald SN, Brennan MF, et al. Determining prognosis in patients with pancreatic endocrine neoplasms: can the WHO classification system be simplified? J Clin Oncol 2007;25:5609–15 [DOI] [PubMed] [Google Scholar]
- 7.Yamada T, AD, Kaplowitz N, Owyang C, Powell DW, Kalloo AN. Endocrine neoplasms of the pancreas. 5th edn Oxford, UK: Blackwell; 2008 [Google Scholar]
- 8.Norton JA, Doppman JL, Collen MJ, Harmon JW, Maton PN, Gardner JD, et al. Prospective study of gastrinoma localization and resection in patients with Zollinger–Ellison syndrome. Ann Surg 1986;204:468–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Calender A. Genetics of neuroendocrine tumours. Caplin M, Kvols L, Handbook of neuroendocrine tumours. Bristol, UK: BioScientifica; 2006. pp. 53–79 [Google Scholar]
- 10.Sutliff VE, Doppman JL, Gibril F, Venzon DJ, Yu F, Serrano J, et al. Growth of newly diagnosed, untreated metastatic gastrinomas and predictors of growth patterns. J Clin Oncol 1997;15:2420–31 [DOI] [PubMed] [Google Scholar]
- 11.Klöppel G, Schröder S, Heitz PU. Histopathology and immunopathology of pancreatic endocrine tumors. Mignon M, Jensen RT, Endocrine tumors of the pancreas: recent advances in research and management. Basel, Switzerland: Karger; 1995. pp. 99–120 [Google Scholar]
- 12.Kloppel G, Heitz PU. Tumors of the endocrine pancreas. Fletcher CD, Diagnostic histopathology of tumors. 2nd edn London, UK: Churchill Livingstone; 2000. pp. 1083–98 [Google Scholar]
- 13.Heitz PU, Komminoth P, Perren A, Klimstra DS, Dayal Y, Bordi C, et al. Pancreatic endocrine tumors: introduction. DeLellis DA, Lloyd RV, Heitz PU, Eng C, Pathology and genetics of tumours of endocrine organs. World Health Organization classification of tumours. Lyon, France: IARC Press; 2004. pp. 177–82 [Google Scholar]
- 14.Lloyd RV, Mervak T, Schmidt K, Warner TF, Wilson BS. Immunohistochemical detection of chromogranin and neuron-specific enolase in pancreatic endocrine neoplasms. Am J Surg Pathol 1984;8:607–14 [DOI] [PubMed] [Google Scholar]
- 15.Fernandez-del Castillo C, Warshaw AL. Cystic tumors of the pancreas. Surg Clin North Am 1995;75:1001–16 [DOI] [PubMed] [Google Scholar]
- 16.Perez-Montiel MD, Frankel WL, Suster S. Neuroendocrine carcinomas of the pancreas with ‘rhabdoid’ features. Am J Surg Pathol 2003;27:642–9 [DOI] [PubMed] [Google Scholar]
- 17.Lemmens I, Van deVen WJ, Kas K, Zhang CX, Giraud S, Wautot V, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet 1997;6:1177–83 [DOI] [PubMed] [Google Scholar]
- 18.Calender AMC, Komminoth P, Scoazec JY, Sweet KM, Teh BT, Multiple endocrine neoplasia type 1. Lyon, France: IARC; 2004 [Google Scholar]
- 19.Jensen RT. Pancreatic endocrine tumors: recent advances. Ann Oncol 1999;10(Suppl. 4):170–6 [PubMed] [Google Scholar]
- 20.Akerstrom G, Hessman O, Hellman P, Skogseid B. Pancreatic tumours as part of the MEN-1 syndrome. Best Pract Res Clin Gastroenterol 2005;19:819–30 [DOI] [PubMed] [Google Scholar]
- 21.Jensen RT, Berna MJ, Bingham DB, Norton JA. Inherited pancreatic endocrine tumor syndromes: advances in molecular pathogenesis, diagnosis, management, and controversies. Cancer 2008;113(Suppl. 7):1807–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Anlauf M, Schlenger R, Perren A, Bauersfeld J, Koch CA, Dralle H, et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am J Surg Pathol 2006;30:560–74 [DOI] [PubMed] [Google Scholar]
- 23.Rindi G, Bordi C. Endocrine tumours of the gastrointestinal tract: aetiology, molecular pathogenesis and genetics. Best Pract Res Clin Gastroenterol 2005;19:519–34 [DOI] [PubMed] [Google Scholar]
- 24.Knudson AG. Hereditary cancer: two hits revisited. J Cancer Res Clin Oncol 1996;122:135–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marx SJ, Simonds WF. Hereditary hormone excess: genes, molecular pathways, and syndromes. Endocr Rev 2005;26:615–61 [DOI] [PubMed] [Google Scholar]
- 26.Hessman O, Skogseid B, Westin G, Akerstrom G. Multiple allelic deletions and intratumoral genetic heterogeneity in men1 pancreatic tumors. J Clin Endocrinol Metab 2001;86:1355–61 [DOI] [PubMed] [Google Scholar]
- 27.D'Adda T, Keller G, Bordi C, Hofler H. Loss of heterozygosity in 11q13–14 regions in gastric neuroendocrine tumors not associated with multiple endocrine neoplasia type 1 syndrome. Lab Invest 1999;79:671–7 [PubMed] [Google Scholar]
- 28.Debelenko LV, Emmert-Buck MR, Manickam P, Kester M, Guru SC, DiFranco EM, et al. Haplotype analysis defines a minimal interval for the multiple endocrine neoplasia type 1 (MEN1) gene. Cancer Res 1997;57:1039–42 [PubMed] [Google Scholar]
- 29.Zhuang Z, Vortmeyer AO, Pack S, Huang S, Pham TA, Wang C, et al. Somatic mutations of the MEN1 tumor suppressor gene in sporadic gastrinomas and insulinomas. Cancer Res 1997;57:4682–6 [PubMed] [Google Scholar]
- 30.Goebel SU, Heppner C, Burns AL, Marx SJ, Spiegel AM, Zhuang Z, et al. Genotype/phenotype correlation of multiple endocrine neoplasia type 1 gene mutations in sporadic gastrinomas. J Clin Endocrinol Metab 2000;85:116–23 [DOI] [PubMed] [Google Scholar]
- 31.Moore PS, Missiaglia E, Antonello D, Zamo A, Zamboni G, Corleto V, et al. Role of disease-causing genes in sporadic pancreatic endocrine tumors: MEN1 and VHL. Genes Chromosomes Cancer 2001;32:177–81 [DOI] [PubMed] [Google Scholar]
- 32.Woodward ER, Maher ER. Von Hippel-Lindau disease and endocrine tumour susceptibility. Endocr Relat Cancer 2006;13:415–25 [DOI] [PubMed] [Google Scholar]
- 33.Iliopoulos O. von Hippel-Lindau disease: genetic and clinical observations. Front Horm Res 2001;28:131–66 [DOI] [PubMed] [Google Scholar]
- 34.Hammel PR, Vilgrain V, Terris B, Penfornis A, Sauvanet A, Correas JM, et al. Pancreatic involvement in von Hippel-Lindau disease. The Groupe Francophone d'Etude de la Maladie de von Hippel-Lindau. Gastroenterology 2000;119:1087–95 [DOI] [PubMed] [Google Scholar]
- 35.Lonser RR, Glenn GM, Walther M, Chew EY, Libutti SK, Linehan WM, et al. von Hippel-Lindau disease. Lancet 2003;361:2059–67 [DOI] [PubMed] [Google Scholar]
- 36.Lubensky IA, Pack S, Ault D, Vortmeyer AO, Libutti SK, Choyke PL, et al. Multiple neuroendocrine tumors of the pancreas in von Hippel-Lindau disease patients: histopathological and molecular genetic analysis. Am J Pathol 1998;153:223–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lott ST, Chandler DS, Curley SA, Foster CJ, El-Naggar A, Frazier M, et al. High frequency loss of heterozygosity in von Hippel-Lindau (VHL)-associated and sporadic pancreatic islet cell tumors: evidence for a stepwise mechanism for malignant conversion in VHL tumorigenesis. Cancer Res 2002;62:1952–5 [PubMed] [Google Scholar]
- 38.Alexakis N, Connor S, Ghaneh P, Lombard M, Smart HL, Evans J, et al. Hereditary pancreatic endocrine tumours. Pancreatology 2004;4:417–33 (discussion: 34–5) [DOI] [PubMed] [Google Scholar]
- 39.Libutti SK, Choyke PL, Alexander HR, Glenn G, Bartlett DL, Zbar B, et al. Clinical and genetic analysis of patients with pancreatic neuroendocrine tumors associated with von Hippel-Lindau disease. Surgery 2000;128:1022–7 (discussion: 7–8) [DOI] [PubMed] [Google Scholar]
- 40.Neumann HP, Dinkel E, Brambs H, Wimmer B, Friedburg H, Volk B, et al. Pancreatic lesions in the von Hippel-Lindau syndrome. Gastroenterology 1991;101:465–71 [DOI] [PubMed] [Google Scholar]
- 41.Chung DC, Smith AP, Louis DN, Graeme-Cook F, Warshaw AL, Arnold A. A novel pancreatic endocrine tumor suppressor gene locus on chromosome 3p with clinical prognostic implications. J Clin Invest 1997;100:404–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, et al. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 1997;278:51–7 [PubMed] [Google Scholar]
- 43.Mao C, Shah A, Hanson DJ, Howard JM. Von Recklinghausen's disease associated with duodenal somatostatinoma: contrast of duodenal versus pancreatic somatostatinomas. J Surg Oncol 1995;59:67–73 [DOI] [PubMed] [Google Scholar]
- 44.Nesi G, Marcucci T, Rubio CA, Brandi ML, Tonelli F. Somatostatinoma: clinico-pathological features of three cases and literature reviewed. J Gastroenterol Hepatol 2008;23:521–6 [DOI] [PubMed] [Google Scholar]
- 45.Trovo-Marqui AB, Tajara EH. Neurofibromin: a general outlook. Clin Genet 2006;70:1–13 [DOI] [PubMed] [Google Scholar]
- 46.Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA 2005;102:8573–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Slegtenhorst M, Nellist M, Nagelkerken B, Cheadle J, Snell R, van denOuweland A, et al. Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 1998;7:1053–7 [DOI] [PubMed] [Google Scholar]
- 48.Francalanci P, Diomedi-Camassei F, Purificato C, Santorelli FM, Giannotti A, Dominici C, et al. Malignant pancreatic endocrine tumor in a child with tuberous sclerosis. Am J Surg Pathol 2003;27:1386–9 [DOI] [PubMed] [Google Scholar]
- 49.Eledrisi MS, Stuart CA, Alshanti M. Insulinoma in a patient with tuberous sclerosis: is there an association? Endocr Pract 2002;8:109–12 [DOI] [PubMed] [Google Scholar]
- 50.McLean A. Endoscopic ultrasound in the detection of pancreatic islet cell tumours. Cancer Imaging 2004;4:84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Giovannini M. Contrast-enhanced and 3-dimensional endoscopic ultrasonography. Gastroenterol Clin North Am 2010;39:845–58 [DOI] [PubMed] [Google Scholar]
- 52.Badea R, Seicean A, Diaconu B, Stan-Iuga R, Sparchez Z, Tantau M, et al. Contrast-enhanced ultrasound of the pancreas—a method beyond its potential or a new diagnostic standard? J Gastrointestin Liver Dis 2009;18:237–42 [PubMed] [Google Scholar]
- 53.Rickes S, Unkrodt K, Ocran K, Neye H, Wermke W. Differentiation of neuroendocrine tumors from other pancreatic lesions by echo-enhanced power Doppler sonography and somatostatin receptor scintigraphy. Pancreas 2003;26:76–81 [DOI] [PubMed] [Google Scholar]
- 54.D'Onofrio M, Mansueto G, Falconi M, Procacci C. Neuroendocrine pancreatic tumor: value of contrast enhanced ultrasonography. Abdom Imaging 2004;29:246–58 [DOI] [PubMed] [Google Scholar]
- 55.Ichikawa T, Peterson MS, Federle MP, Baron RL, Haradome H, Kawamori Y, et al. Islet cell tumor of the pancreas: biphasic CT versus MR imaging in tumor detection. Radiology 2000;216:163–71 [DOI] [PubMed] [Google Scholar]
- 56.Van Hoe L, Gryspeerdt S, Marchal G, Baert AL, Mertens L. Helical CT for the preoperative localization of islet cell tumors of the pancreas: value of arterial and parenchymal phase images. AJR Am J Roentgenol 1995;165:1437–9 [DOI] [PubMed] [Google Scholar]
- 57.Fidler JL, Fletcher JG, Reading CC, Andrews JC, Thompson GB, Grant CS, et al. Preoperative detection of pancreatic insulinomas on multiphasic helical CT. AJR Am J Roentgenol 2003;181:775–80 [DOI] [PubMed] [Google Scholar]
- 58.Buetow PC, Parrino TV, Buck JL, Pantongrag-Brown L, Ros PR, Dachman AH, et al. Islet cell tumors of the pancreas: pathologic-imaging correlation among size, necrosis and cysts, calcification, malignant behavior, and functional status. AJR Am J Roentgenol 1995;165:1175–9 [DOI] [PubMed] [Google Scholar]
- 59.Bordeianou L, Vagefi PA, Sahani D, Deshpande V, Rakhlin E, Warshaw AL, et al. Cystic pancreatic endocrine neoplasms: a distinct tumor type? J Am Coll Surg 2008;206:1154–8 [DOI] [PubMed] [Google Scholar]
- 60.Herwick S, Miller FH, Keppke AL. MRI of islet cell tumors of the pancreas. AJR Am J Roentgenol 2006;187:W472–80 [DOI] [PubMed] [Google Scholar]
- 61.Semelka RC, Custodio CM, Cem Balci N, Woosley JT. Neuroendocrine tumors of the pancreas: spectrum of appearances on MRI. J Magn Reson Imaging 2000;11:141–8 [DOI] [PubMed] [Google Scholar]
- 62.Service FJ, McMahon MM, O'Brien PC, Ballard DJ. Functioning insulinoma—incidence, recurrence, and long-term survival of patients: a 60-year study. Mayo Clin Proc 1991;66:711–9 [DOI] [PubMed] [Google Scholar]
- 63.Simon P, Spilcke-Liss E, Wallaschofski H. Endocrine tumors of the pancreas. Endocrinol Metab Clin North Am 2006;35:xii, 431–47 [DOI] [PubMed] [Google Scholar]
- 64.Gouya H, Vignaux O, Augui J, Dousset B, Palazzo L, Louvel A, et al. CT, endoscopic sonography, and a combined protocol for preoperative evaluation of pancreatic insulinomas. AJR Am J Roentgenol 2003;181:987–92 [DOI] [PubMed] [Google Scholar]
- 65.Power N, Reznek RH. Imaging pancreatic islet cell tumours. Imaging 2002;14:147–59 [Google Scholar]
- 66.Sheth S, Hruban RK, Fishman EK. Helical CT of islet cell tumors of the pancreas: typical and atypical manifestations. AJR Am J Roentgenol 2002;179:725–30 [DOI] [PubMed] [Google Scholar]
- 67.Howard TJ, Stabile BE, Zinner MJ, Chang S, Bhagavan BS, Passaro E., Jr Anatomic distribution of pancreatic endocrine tumors. Am J Surg 1990;159:258–64 [DOI] [PubMed] [Google Scholar]
- 68.Zimmer T, Scherubl H, Faiss S, Stolzel U, Riecken EO, Wiedenmann B. Endoscopic ultrasonography of neuroendocrine tumours. Digestion 2000;62(Suppl. 1):45–50 [DOI] [PubMed] [Google Scholar]
- 69.Ruttman E, Kloppel G, Bommer G, Kiehn M, Heitz PU. Pancreatic glucagonoma with and without syndrome. Immunocytochemical study of 5 tumour cases and review of the literature. Virchows Arch A Pathol Anat Histol 1980;388:51–67 [DOI] [PubMed] [Google Scholar]
- 70.Perry RR, Vinik AI. Clinical review 72: diagnosis and management of functioning islet cell tumors. J Clin Endocrinol Metab 1995;80:2273–8 [DOI] [PubMed] [Google Scholar]
- 71.Prinz RA, Dorsch TR, Lawrence AM. Clinical aspects of glucagon-producing islet cell tumors. Am J Gastroenterol 1981;76:125–31 [PubMed] [Google Scholar]
- 72.Eelkema EA, Stephens DH, Ward EM, Sheedy PF., 2nd CT features of nonfunctioning islet cell carcinoma. AJR Am J Roentgenol 1984;143:943–8 [DOI] [PubMed] [Google Scholar]
- 73.Jani N, Khalid A, Kaushik N, Brody D, Bauer K, Schoedel K, et al. EUS-guided FNA diagnosis of pancreatic endocrine tumors: new trends identified. Gastrointest Endosc 2008;67:44–50 [DOI] [PubMed] [Google Scholar]
- 74.Imhof H, Frank P. Pancreatic calcifications in malignant islet cell tumors. Radiology 1977;122:333–7 [DOI] [PubMed] [Google Scholar]
- 75.Semelka RC, Cumming MJ, Shoenut JP, Magro CM, Yaffe CS, Kroeker MA, et al. Islet cell tumors: comparison of dynamic contrast-enhanced CT and MR imaging with dynamic gadolinium enhancement and fat suppression. Radiology 1993;186:799–802 [DOI] [PubMed] [Google Scholar]
- 76.Eriksson B. New drugs in neuroendocrine tumors: rising of new therapeutic philosophies? Curr Opin Oncol 2010;22:381–6 [DOI] [PubMed] [Google Scholar]
- 77.Fjallskog ML, Lejonklou MH, Oberg KE, Eriksson BK, Janson ET. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin Cancer Res 2003;9:1469–73 [PubMed] [Google Scholar]
- 78.Strosberg JR, Kvols LK. A review of the current clinical trials for gastroenteropancreatic neuroendocrine tumours. Expert Opin Investig Drugs 2007;16:219–24 [DOI] [PubMed] [Google Scholar]
- 79.Chan JA, Kulke MH. Emerging therapies for the treatment of patients with advanced neuroendocrine tumors. Expert Opin Emerg Drugs 2007;12:253–70 [DOI] [PubMed] [Google Scholar]
- 80.Hobday TJ, Mahoney M, Erlichman C, Lloyd R, Kim G, Mulkerin D, Picus J, et al. Preliminary results of a phase II trial of Gefitinib in progressive metastatic neuroendocrine tumors (NET): a phase II Consortium (PC2) study. Proc Am Soc Clin Oncol 2005;23:328s [Google Scholar]
- 81.Yao JC. Neuroendocrine tumors. Molecular targeted therapy for carcinoid and islet-cell carcinoma. Best Pract Res Clin Endocrinol Metab 2007;21:163–72 [DOI] [PubMed] [Google Scholar]
- 82.Durán I, Salazar R, Casanovas O, Arrazubi V, Vilar E, Siu LL, et al. New drug development in digestive neuroendocrine tumors. Ann Oncol 2007;18:1307–13 [DOI] [PubMed] [Google Scholar]
- 83.Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol 2008;26:3403–10 [DOI] [PubMed] [Google Scholar]
- 84.Yao JC, Phan AT, Chang DZ, Jacobs C, Mares JE, Rashid A, et al. Phase II study of RAD001 (everolimus) and depot octreotide (Sandostatin LAR) in patients with advanced low grade neuroendocrine carcinoma (LGNET). J Clin Oncol 2006;24:18S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Duran I, Le L, Saltman D, Singh D, Kocha W, Cheiken R, A phase II trial of temsirolimus in metastatic neuroendocrine tumors. Proceedings of 2005 Gastrointestinal Cancers Symposium. Chicago, IL: American Society of Clinical Oncology; 2005. [Google Scholar]