

Abstract
In order to manage risk of ochratoxin A (OTA) in foods, we re-evaluated the tolerable daily intake (TDI), derived the negligible cancer risk intake (NCRI), and conducted a probabilistic risk assessment. A new approach was developed to derive ‘usual’ probabilistic exposure in the presence of highly variable occurrence data, such as encountered with low levels of OTA. Canadian occurrence data were used for various raw food commodities or finished foods and were combined with US Department of Agriculture (USDA) food consumption data, which included data on infants and young children. Both variability and uncertainty in input data were considered in the resulting exposure estimates for various age/sex strata. Most people were exposed to OTA on a daily basis. Mean adjusted exposures for all age-sex groups were generally below the NCRI of 4ng OTA kg bw−1, except for 1–4-year-olds as a result of their lower body weight. For children, the major contributors of OTA were wheat-based foods followed by oats, rice, and raisins. Beer, coffee, and wine also contributed to total OTA exposure in older individuals. Predicted exposure to OTA decreased when European Commission maximum limits were applied to the occurrence data. The impact on risk for regular eaters of specific commodities was also examined.
Keywords: ochratoxin A (OTA), mycotoxin, renal cancer, children, soy formula, risk metrics, tolerable daily intake (TDI), negligible cancer risk intake (NCRI), exposure, probabilistic, margin of exposure (MOE), guidelines, risk management
INTRODUCTION
Ochratoxin A (OTA) is a mycotoxin produced by Penicillium verrucosum and Aspergillus ochraceus as well as A. carbonarius and A. niger (JECFA 2001). It occurs naturally in many foods, such as cereal-derived staples as well as other food commodities including grapes, raisins, wine, coffee, beer, corn, and soy. OTA is chemically stable and is not greatly affected by normal processing temperatures (Bullerman and Bianchini 2007).
Based on studies conducted by the National Toxicology Program (NTP) (1989), OTA was found to be the most potent renal carcinogen known to date (Kuiper-Goodman and Scott 1989). Therefore, a good understanding of the foods that contribute to overall OTA exposure is important.
Exposure assessment plays a pivotal role in risk assessment and a critical approach is necessary (for a general review, see Lambe et al. 2002). In the assessment of OTA presented here, we have chosen to use a probabilistic exposure assessment by which distributions of the consumption of the foods of interest by all age groups for two days of recall were combined with distributions of contaminant levels through Monte Carlo simulation (Petersen 2000; Gibney and van der Voet 2003; Guenther et al. 1997; Hoffmann et al. 2002). This approach allows input variability and uncertainty to be taken into account, thus providing a better estimate of exposure.
For natural toxins, occurrence values may show wide fluctuations, with some falling below the limit of detection/quantification (LOD/LOQ). Various approaches, both parametric and non-parametric, have been developed to deal with such left-censored data (Tressou et al. 2004a, 2004b; Counil et al. 2005) and the type of approach used can have a large impact, especially if few values are greater than the limit of detection.
Extremes in food consumption or toxicant occurrence may lead to extreme estimates of exposure in some individuals, but may not be reflective of the true (usual) exposure over a longer time frame. Variance reduction methods to address ‘within person’ variability have been developed for assessing exposure to nutrients. As a result, exposure data obtained through short-term food consumption surveys can readily be adjusted to the ‘usual exposure’ of that population over a longer time frame, such as one year (Nusser et al. 1996; Guenther et al. 1997; Gay 2000); likewise this has been considered for contaminants (Slob 1993). However, the wide occurrence variability and generally skewed nature of the distribution of mycotoxins render these adjustment procedures problematic. For the probabilistic exposure assessment of OTA, new methods were developed to overcome this difficulty.
Risk assessment is an integral step in the iterative process underlying overall risk analysis (Kuiper-Goodman 2004). In the current paper, the assessment of the risks from OTA will be organized along the four components of risk assessment as follows:
Hazard identification: a brief review and re-evaluation of key literature used in the interpretation of the toxicological studies.
Hazard characterization: a critical review of approaches and re-evaluation of risk metrics.
Exposure assessment: method development for probabilistic exposure assessment for all age/ sex strata and results for total exposure of all persons, as well as regular eaters of specific commodities to OTA.
Risk characterization: assessment of the associated risk of adverse health effects from exposure to OTA for all age/sex strata.
To aid in the risk management of OTA in foods, we furthermore modelled the theoretical impact on OTA exposure and risk if the European Commission (EC) maximum limits (MLs) were to apply. The discussion puts the results in context with findings in other jurisdictions.
HAZARD IDENTIFICATION
OTA is a toxic fungal metabolite that may cause nephrotoxic, teratogenic, immunosuppressive, and carcinogenic effects in many species, although species- and sex-related differences in the magnitude of these effects have been noted (Kuiper-Goodman and Scott 1989; O'Brien and Dietrich 2005). OTA causes porcine nephropathy and has also been implicated in the aetiology of Balkan Endemic Nephropathy (BEN), a chronic degenerative kidney disease associated with a high incidence of urinary tract tumours in humans (Kuiper-Goodman and Scott 1989; Stefanovic et al. 2006; Pfohl-Leszkowicz et al. 2007).
Carcinogenicity
OTA is one of the most potent renal carcinogens, inducing cancer in 39% of rats at very low doses (70 µg OTA kg bw−1 per day) (NTP 1989; Kuiper-Goodman and Scott 1989; Kuiper-Goodman 1996; Lock and Hard 2004). Of importance in the weight of evidence (Table 1) is that OTA was carcinogenic via dietary or gavage routes to multiple tissues (i.e. kidney, liver, and mammary glands) in various strains of rats and mice of both sexes (Kanisawa and Suzuki 1978; Bendele et al. 1985; NTP 1989; Castegnaro et al. 1998; Mantle et al. 2005). Consequently, OTA has been classified as a possible human carcinogen (Group 2B) based on sufficient evidence for carcinogenicity in experimental animal studies and inadequate evidence in humans (International Agency for Research on Cancer (IARC) 1993).
Table 1.
Characteristics of induced tumours and other aspects of toxicity typical of threshold and non-threshold chemicals.a
| Threshold | Non-threshold (i.e. OTA) |
|---|---|
| Often single species, site, sex | Two species; several sites; both sexes |
| Low tumorigenic potency; low incidence of tumours | High tumorigenic potency; high incidence of renal tumours Low incidence of renal tumours in (historical) controls |
| Low proportion of carcinomas versus adenomas | High proportion of carcinomas versus adenomas Often bilateral and multiple High cytoplasmic atypia; invasive |
| Mutation frequency similar to spontaneous tumours | Rapid progression |
| Metastases rare | Metastases more common |
| Tumour does not reduce lifespan | Tumour reduces lifespan, large size: many 2.0–6.5 cm; necrosis |
Note: aModified from Kuiper-Goodman (1996); based on Tennant (1994).
The NTP 2-year OTA gavage study (five times/ week) showed that the incidence of tumours (adenomas and carcinomas combined) was dose-related and highest in male rats. At a dose of 210 µg OTA kg bw−1 per day, 72% of male rats had tumours - the highest incidence rate seen in any of the studies by the NTP of the National Cancer Institute (NCI) to date. The incidence in historical controls was less than 1%, indicating that renal tumours are rare in this strain of rat. The aggressive nature of OTA-induced tumours was evident by gross pathology: tumours were often multiple and bilateral with a high ratio of carcinomas to adenomas and many were very large - reaching up to 6.5 cm. There was an early onset of tumours after dosing, and reduced lifespan (Table 1). Although tumour incidence rates in female rats were lower than in males, they were highly significant at the highest dose (16%); here the historical control rate was less than 0.3%.
By light microscopy, the changes in OTA-induced tumours in the NTP study indicated a very aggressive behaviour, as evidenced by a high degree of atypia, rapid progression, and invasiveness (Table 1). In addition, karyomegaly, as evidenced by the presence of large polyploid cells, was observed at the two highest dose levels in males and at all three dose levels in females (no NOAEL). Focal tubular hyperplasia was observed in mid- and high-dose males, and this lesion was suggested as being part of the neoplastic spectrum (NTP 1989). Focal and multifocal tubular cell proliferation was also observed in the mid- and high-dose males. The gross- and histo-pathological aspects of the OTA-induced tumours point to a non-threshold mode of action (Table 1). In a recent 90-day study with a design similar to that of the NTP (1989) study (but including only males), Rached et al. (2007) employed bromodeoxyuridine (BrdU) staining and confirmed the presence of cell proliferation at similar dose levels, a finding that is consistent with OTA being a complete carcinogen. Although it has been suggested that increased cell proliferation following cytotoxicity may play a causative role in OTA-induced carcinogenicity (Rached et al. 2007), cell proliferation is a necessary aspect of carcinogenesis and is also observed with DNA-reactive carcinogens (Lock and Hard 2004). Furthermore, not all cytotoxic chemicals induce uncontrolled growth leading to carcinogenesis (Kuiper-Goodman 2004) and for those chemicals for which cell proliferation is causally implicated as an ‘epigenetic’ mechanism, tumour incidence is generally low and is characterized by a long latency (Lock and Hard 2004).
A significant finding in the NTP (1989) study was the unusually high incidence of metastases — 20% and 36%, respectively, in the mid- and high-dose males with renal cancer, suggestive of a high degree of genomic instability in these renal tumours. Furthermore, based on criteria developed by Tennant (1994), Kuiper-Goodman (1996) determined that the characteristics of the OTA-induced tumours correspond to those typically seen for genotoxic chemicals (Table 1). This position was confirmed by Hard (2000), after a re-examination of the NTP OTA tumours. A further review of all NTP studies in which renal tumours were observed again confirmed this classification (Lock and Hard 2004), suggesting therefore that a non-threshold approach under hazard characterization is appropriate. Thus, based on weight of evidence, the data suggest that OTA in the rat operates as a complete carcinogen (initiator and promoter activity) rather than as a promoter alone.
Non-neoplastic effects in the kidney in rats and pigs
OTA is nephrotoxic in all mammalian species tested to date (Kuiper-Goodman and Scott 1989). In a 90-day feeding study (0, 8, 40 and 160 µg OTA kg bw−1 per day) in female pigs (four to nine pigs per group), Krogh et al. (1974) observed microscopic changes in kidney tubules, reduction or inhibition of kidney enzymes, as well as decreased kidney function in close to 50% (4/9) of pigs exposed to the lowest dose. The most sensitive parameter was a decrease in the maximum tubular excretion (TM) of p-aminohippuric acid (PAH) at all dose levels. In a subsequent 2-year study, six pigs per group were exposed to a single dose level of 40 µg OTA kgbw−1 per day (Krogh et al. 1979) resulting in a more severe nephropathy at 2 years compared with 14 weeks based on histopathology, decreased urine osmolality, and increased glucose excretion. Thus, a NOAEL or LOAEL could not be derived from this single-dose study. Steady-state with regard to OTA residues in several tissues (kidney, liver, adipose tissue) appeared to be achieved after 90 days of dietary administration.
Rats were much less sensitive than pigs, when comparing endpoints for kidney damage such as reduction in phosphoenolpyruvate carboxykinase (PEPCK) enzymatic activity following OTA administration (see reviews Krogh et al. 1988; Kuiper-Goodman and Scott 1989).
In a 90-day Wistar rat study, a dose-related nephropathy was observed in the kidneys of animals fed a 0.2–5.0 µg OTA g−1 diet. Karyomegaly and increased eosinophilia in proximal convoluted tubular cells of the kidney were observed in all treated dose groups (LOAEL calculated as 15 µg OTA kg bw−1 per day). At the end of this study, OTA dosing was stopped, but karyomegaly persisted at higher dose levels for at least another 90 days (Munro et al. 1974); its role in carcinogenicity needs further clarification. Since there were minimal functional changes in the kidneys in the NTP (1989) study, and as recently confirmed in a similar study by Rached et al. (2007), it is unlikely that kidney damage per se (promotor action alone) was responsible for the observed neoplasia (Kuiper-Goodman and Scott 1989).
Genotoxicity
There is an ongoing debate regarding the genotoxic status of OTA, much of it related to the negative results obtained from the majority of microbial mutagenicity assays, including the traditional Ames test (NTP 1989; Kuiper-Goodman and Scott 1989), and because it is at best moderately genotoxic in in vitro or in vivo mammalian test systems (Brambilla and Martelli 2004). The issue of genotoxicity is important, since genotoxic carcinogens tend to be managed more severely than non-genotoxic (threshold carcinogens) (see Hazard Characterization). A recent ILSI-Europe workshop concluded that OTA is genotoxic, albeit the underlying mechanism remains unknown (Fink-Gremmels 2005). Some of the recent developments in this area are discussed below.
The use of hepatic microsomes for metabolic activation in standard mutagenicity tests was suggested as a major factor for not detecting genotoxic carcinogens requiring unusual metabolic activation (Barrett 1992; Brambilla and Martelli 2004). In the presence of microsomes of mouse kidney (i.e. the target tissue), OTA was mutagenic in Salmonella typhimurium strains TA98, TA1535, and TA1538, but not in strains TA100 or TA102 (Obrecht-Pflumio et al. 1999). Using rat kidney microsomes, Zepnik et al. (2001) also demonstrated a lack of mutagenic responses in S. typhimurium strain TA100 and TA2638 - the latter strain genetically related to TA102 (Rydén et al. 2000). However, Zepnik et al. (2001) did not use the S. typhimurium strains that gave positive responses in the previous study (i.e. TA98, TA1535, and TA1538). Thus, while results from both studies are in agreement, Zepnik et al. (2001) stated that their data did not support the positive findings from Obrecht-Pflumio et al. (1999). Surprisingly, others have adopted their opinion (Mally et al. 2005b; Manderville 2005; Turesky 2005), and thus the significance of positive mutagenic responses in the modified Ames test has been overlooked.
The OTA genotoxicity debate is also fuelled by contradictory findings from DNA-binding studies. Most in vitro and in vivo studies that used the 32P-post-labelling approach detected dose- and time-dependent DNA adducts in multiple tissues across several species (Pfohl-Leszkowicz and Castegnaro 2005; Pfohl-Leszkowicz and Manderville 2007). An OTA-DNA standard (C8-OTA-dGMP adduct) has been characterized, and co-migrates with 32P-post-labelling adducts detected in the kidney of rats and pigs following OTA exposure (Faucet et al. 2004). It has been suggested that an electrophilic hydroquinone OTA metabolite may mediate OTA genotoxicity (Manderville 2005; Tozlovanu et al. 2006). Others have not confirmed evidence of DNA adducts (or of an OTA-dGMP adduct) in rats following OTA exposure using 32P-post-labelling (Mally et al. 2004, 2005b) and DNA binding was not demonstrated in a few studies employing 3H- or 14C-OTA (Schlatter et al. 1996; Gautier et al. 2001; Gross-Steinmeyer et al. 2002; Mally et al. 2004). These latter results should be interpreted with caution, since there were notable limitations in the choice of biologically relevant dose levels, the exposure period, as well as methodological aspects such as specific activities of radio-labelled OTA and DNA extraction and purification that may have compromised assay sensitivities. Further studies are needed to resolve these controversial results and methodological issues.
Using the comet assay, in the presence of for-mamido-pyrimidine-DNA-glycosylase (Fpg), a repair enzyme that recognizes oxidized DNA bases such as 8-oxo-7, 8-dihydro-2'deoxyguanosine (8-OH-dG), oxidative DNA damage was detected in several cell lines (Kamp et al. 2005) and in kidney and liver cell suspensions as well as peripheral lymphocytes from rats exposed to increasing levels of OTA (0, 0.25, 0.5, 1, 2 mg OTA kg bw −1 per day) for 2 weeks (Mally et al. 2005b). Based on these findings, these authors speculated that DNA breakage detected by the comet assay is caused by an indirect mechanism involving oxidative stress and implied that DNA adducts detected by the 32P-post-labelling technique may have been endogenously formed either through direct oxidation of DNA or indirectly through lipid peroxidation, a position that has been adopted by others (European Food Safety Authority (EFSA) 2006; Joint Expert Committee on Food Additives (JECFA) 2007). However, results from the same in vivo study do not support the importance of oxidative stress in causing DNA damage. To elaborate, using the comet assay in the presence of Fpg, oxidative DNA damage was observed in the kidney only at a very high dose (2 mg OTA kg bw−1), whereas in the absence of Fpg, more generalized DNA damage was detected in all groups, especially at lower dose levels (0.25 and 0.5 mg) (Mally et al. 2005b). Furthermore, in a concurrent publication by the same authors, the use of specific 32P-post-labelling and chromatography (LC-MS/MS), techniques that can determine the levels of oxidized DNA bases directly, failed to detect increases in the levels of etheno-DNA adducts (associated with lipid peroxidation) and 8-OH-dG (associated with direct oxidation) in the kidney and liver of rats in all dose groups (Mally et al. 2005a). Thus contrary to the hypothesis (stated in Mally et al. 2005b), the authors suggested that oxidative stress does not play a major role in OTA toxicity (Mally et al. 2005b) and OTA-induced carcinogenicity may operate through a unique mechanism. Thus, we conclude that oxidative stress, as an indirect and non-genotoxic agent causing DNA adducts, does not appear to be the major contributing factor in OTA carcinogenicity, a view shared by others (Mally et al. 2005a; Rached et al. 2007). Taken together, the genotoxicity status of OTA remains equivocal.
HAZARD CHARACTERIZATION
Past evaluations by JECFA. EFSA and other organizations
Internationally, several approaches have been used to derive risk metrics for OTA. Examples are the provisional tolerable weekly intake (pTWI) (JECFA 2001), the provisional tolerable daily intake (pTDI) (EFSA 2006), or a negligible cancer risk intake (NCRI) (Kuiper-Goodman 2004).
Based on the carcinogenic properties of OTA, and using both a safety factor approach or modelling (lifetime risk level of 1:100 000), Health Canada estimated pTDIs ranging from 1.2 to 5.7 ng OTA kg bw−1 per day (Kuiper-Goodman and Scott 1989; Kuiper-Goodman 1996). Similarly, the Nordic expert group on food safety based their assessment on the carcinogenic properties of OTA in deriving their TDI of 5 ng OTA kg bw−1 per day (Olsen et al. 1991). The Scientific Committee of Food (SCF) of the European Union concluded in its 1994 assessment that levels of OTA should be minimized owing to its nephrotoxic, carcinogenic and genotoxic properties, and set a pTDI of 5 ng OTA kg bw−1 per day in 1998 (as reviewed in EFSA 2006).
While JECFA had considered the carcinogenic properties of OTA, its final assessment was based on nephrotoxicity in pigs (Krogh et al. 1974), the most sensitive species for this endpoint. By using the lowest dose tested (8 µg OTA kg bw−1 per day) and applying a safety factor of 500, JECFA estimated a pTWI of 112 ng OTA kg bw−1 per week (equivalent to a pTDI of 16 ng OTA kg bw−1 per day (JECFA 1991). These metrics were reduced, through rounding, to 100 ng OTA kg bw−1 per week or 14 ng OTA kg bw−1 per day in 1995 (JECFA 1995) and still stand following more recent evaluations (JECFA 2001; JECFA 2007).
Recently, the European Union (EFSA 2006) adopted a pTWI of 120 ng OTA kg bw−1 per week (equivalent to 17 ng OTA kg bw−1 per day). The EFSA Panel also used nephrotoxicity (Table 2) as the endpoint criterion for adopting these guidelines, since they concluded that OTA was non-genotoxic, based on their interpretation that there was ‘no conclusive evidence’ that OTA binds to DNA, and that there was ‘suggestive evidence’ regarding the role of oxidative processes, such as lipid peroxidation. However, as discussed under Hazard Identification, the genotoxic status of OTA is ‘equivocal'. Recent scientific evidence indicates that at low doses, sufficient to cause cancer, neither oxidative stress involving lipid peroxidation nor direct oxidation of DNA appear to play a primary role in OTA toxicity and carcinogenicity (Mally et al. 2005a; Rached et al. 2007). Thus, the mode of action underlying OTA induced carcinogenicity is still unclear.
Table 2.
Uncertainty factors used in the derivation of risk metrics for OTA from the 90-day pig study.
|
EFSA (2006)a Lowest dose tested: 8 µg kg bw−1 day−1 |
Health Canadaa Derived benchmark dose: BD10 = 1.56 µg kg bw−1 day−1 |
|
|---|---|---|
| Source of uncertainty: | ||
| Intraspecies | 10 | 10 |
| Interspecies | 15b | 25c |
| LOAEL to NOAEL | 3 | |
| 90-Day subchronic to chronic | 2 | |
| Overall uncertainty | 450 | 500 |
| Resulting TDI (ng kg bw−1 day−1) | 17 | 3.0 |
Notes: aUncertainty factors applied to lowest dose tested (8 µg OTA kg bw−1 day−1)orBD10 (data from Krogh et al. 1974).
Toxicodynamics (2.5x); toxicokinetics related to OTA half-life (6x) as estimated by EFSA.
Toxicodynamics (2.5x); toxicokinetics related to OTA half-life (10x) (see Table 3).
An EFSA task force mandated to propose a harmonized approach for the risk assessment of substances that are both genotoxic and carcinogenic stated that:
‘'in the case of a substance that is carcinogenic, but its carcinogenic mode of action has not been identified, it will usually be assumed that genotoxicity is the mode of action. It is important to be aware that this is a default position based on a lack of other information, and is of course not an acknowledgement that genotoxicity is indeed the mode of action.”
(EFSA 2005; also Barlow et al. 2006)
This approach is also shared by other agencies, such as the US Environmental Protection Agency (USEPA) (2005). Since the genotoxic status of OTA is highly controversial, and in light of ongoing studies addressing OTA's mode of action, as well as the overall weight of evidence (discussed under Hazard Identification), we recommend that at present OTA be regulated as a non-threshold carcinogen.
Re-evaluation of the tolerable daily intake (TDI)
Even if OTA were to be regulated as a threshold carcinogen and nephrotoxicity in pigs used as an endpoint, the available 90-day study in pigs (Krogh et al. 1974, 1979, see Hazard Identification) has several limitations. Our review of the recent EFSA Opinion (2006) revealed several methodological issues related to the choice of uncertainty factors applied to the endpoint of nephrotoxicity (Table 2). The EFSA Panel derived a pTWI of 120 ng OTA kg bw−1 per week based on a composite uncertainty factor (UF) of 450 applied to the lowest dose tested (8 µg OTA kg bw−1 per day). This UF was based on intra-species and interspecies factors of 10 and 15, respectively; the latter factor was based on 2.5- and sixfold differences for toxicodynamic and toxicokinetic properties, respectively, as EFSA considered the much longer half-life observed in monkeys and man, compared with pigs (Table 3). However, the factor of 6 was based on a comparison of half-lives following intravenous administration in pigs (sex not specified) and oral exposure in a human. When the same route of administration (i.e. oral) is used for both human (male) and pigs (all males), this factor increases to 10 (Table 3), resulting in an overall interspecies factor of 25 (Table 2).
Table 3.
Species-and route-specific half-life of OTA.a
| Half-life (h) |
||
|---|---|---|
| Species | Intravenous | Oral |
| Mouse | 48 | 39 |
| Rat | 170 | 120 |
| Pig | 150 | 89b |
| Monkey | 840 | 510 |
| Human | 1400c | 853d |
Notes: aHagelberg et al. (1989).
Estimated; from Petzinger and Ziegler (2000).
An additional UF of 3 accounted for the use of an LOAEL rather than an NOAEL. Here the Panel referred to WHO-IPCS (1999) recommendations, which state that in the absence of an NOAEL, an UF of 3 may be used if the LOAEL is of sufficient quality. However, considering the small number of animals per group in these studies and the fact that, in the lowest dose group, four out of nine pigs showed microscopic and functional kidney changes, it is our opinion that this LOAEL is not of sufficient quality. Thus it is reasonable to apply a more conservative UF of up to 10 (WHO-IPCS 1999). As a substitute for the NOAEL of the most sensitive endpoint (TM renal clearance), a hybrid benchmark dose response of 10% above background (BD10) was derived (Figure 1) using the Fortran program BENCH_C (Crump 1995; Crump and Van Landingham 1996). BD10s of 1.20 and 1.92 µg OTA kg bw−1 per day were estimated for decreases in TM renal clearance, expressed relative to body weight or to inulin clearance, respectively. Both of these metrics are lower than the putative NOAEL of 2.7µg OTA kg bw−1 per day resulting from the use of a factor of 3 by EFSA. Thus, the BD10 approach would suggest that an UF of at least 5 would be more appropriate for going from the LOAEL to the NOAEL. In addition, since the pig study was a sub-chronic rather than a chronic study, an additional UF should be used (WHO-IPCS 1999). Taking into account that on average the differences in NOAELs between sub-chronic and chronic studies are two- to three-fold, with a small proportion of chemicals exceeding ten-fold ratios (Dourson et al. 1996), a sub-chronic to chronic UF of at least 2 would need to be applied (Table 2). Taken together, this would result in an overall UF in the order of at least 2500 applied to the lowest dose tested (8 µg OTA kg bw−1 per day), or an UF of 500 applied to the average BD10 of 1.56 µg OTA kg bw−1 per day, thereby reducing the estimated pTDI (after rounding) to 3.0 ng OTA kg bw−1 per day (Table 2 and Figure 1).
Figure 1.
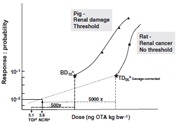
Derivation of risk metrics for OTA.
While the pig is a more sensitive species with regard to kidney damage, the available 90-day study in pigs should be considered inadequate for the estimation of a TDI, as evidenced by the high uncertainty factor. Furthermore, the endpoint nephrotoxicity is less appropriate, since in the chronic rat study (NTP 1989) kidney damage was not a prominent factor and is unlikely to be the causal factor for the observed carcinogenicity in the rat.
Derivation of the negligible cancer risk intake (NCRI)
We have continued to use both a safety factor (threshold) and a model-based (non-threshold) approach and currently the two estimates are combined into one metric, the negligible cancer risk intake (NCRI). The NCRI is defined as the exposure associated with a risk level of 1:100 000 and is equivalent in units to the TDI. To derive the NCRI, we have used the tumorigenic dose at which 5% of the animals are likely to have tumours (TD05), as a point of departure on the dose response curve. The TD05 was derived through modelling the tumour incidence data in the observable range of the dose response curve using the multi-stage method (THRESH; Howe 1995) and then corrected downward from 27.4 to 19.6 µg OTA kg bw−1 to adjust for the fact that animals were dosed by gavage for only 5 days per week (Figure 1). The TD05 is slightly below the lowest dose level (21 µg OTA kg bw−1, unadjusted for gavage) tested in the NTP study. Dividing the TD05 (5/100) by 5000, equivalent to linear extrapolation to zero exposure, gives the exposure (NCRI = 3.9 ng OTA kg bw−1 per day, rounded to 4 ng OTA kg bw−1 per day) associated with a risk level of 1:100 000 (Kuiper-Goodman 2004). The risk level here pertains to the test species (rats) developing tumours.
While both approaches discussed above have merit, depending on the underlying mode of action, there is considerable convergence in the derived TDI and NCRI estimates (Figure 1). But, in light of the limitations with the pig study, the equivocal genotoxicity status, the associated recommended default position, and the pathological characteristics of the tumours, a non-threshold approach is recommended. Under Risk Characterization, the risk metrics derived here will be compared with estimates of exposure. In addition, and as an alternative to using the NCRI, we have also used a margin of exposure (MOE) approach (see Risk Characterization).
EXPOSURE ASSESSMENT
Methods and approaches
The objective of the exposure assessment is to obtain a realistic view of the factors affecting the exposure of different age sex strata to OTA, paying particular attention to subgroups of the population that are likely to have a higher level of exposure.
In a typical exposure assessment, means (Docc, where D is deterministic) or distributions (Pocc, where P is probabilistic) of contaminant concentrations in foods, are combined with the distributions (Pcon)of the consumption of the foods of interest (Figure 2). Throughout this paper we avoid the use of the word ‘intake', as this term has been variously used to indicate either food consumption or exposure, and may lead to misinterpretation if not qualified. Details related to the need for specific corrections and modifications of occurrence data and consumption data, as well as approaches used in assessing exposure, are described below.
Figure 2.
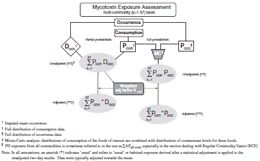
Multi-commodity exposure assessment for OTA.
Occurrence data
In this probabilistic assessment, we have used Canadian occurrence data for raw food commodities and various finished foods gathered over the last decade (Table 4). For details on analytical methods, the reader is referred to the original source data.
Table 4.
Sources of OTA occurrence data for Canada.
| Project | Survey years | Limit of detection (LOD) (ng OTA g−1) | Limit of quantification (LOQ) (ng OTA g−1) | Recovery (%)a | Reference |
| Canadian Grain Commission (CGC) | 1994–2005 | – | 1 | 88.4 | CGC – TW Nowicki, Personal Communication |
| HC – rice | 1993–1995 | −−− | 0.5 | 97.0 | HC – Dr. P Pantazopoulos, Personal Communication |
| HC – breakfast cereals | 1999/2000–2001/2002 | 0.05 | 0.2 | 83.3 | Roscoe et al. (2008) |
| HC – bran cereals | 2004/2005–2005/2006 | 0.05, 0.2 | 0.2, 0.5 | 93.0 | HC – Dr. P Pantazopoulos, Personal Communication |
| HC – infant cereals | 1997/1998–1999/2000 | – | 0.2, 0.5 | 92.0 | Lombaert et al. (2003) |
| HC – infant cereals | 1998–1999, 2000–2002 | 0.15 | 0.5 | 96.1 | HC – Ms V Roscoe, Personal Communication |
| HC – soy-based infant formula | 2000–2001 | 0.05 | 0.2 | 85.4 | HC – Ms V Roscoe, Personal Communication |
| HC – beer | 1995 | 0.05, 0.1 | – | 95.3 | Scott and Kanhere (1995) |
| HC – wine and grape juice | 1999/2000–2001/2002 | 0.008b | 0.04b | 87.4 | Ng et al. (2004) |
| HC – coffee | 1997–1998 | – | 0.1 | 78.5 | Lombaert et al. (2002) |
| HC – dried fruit | 1998/1999–2000/01 | 0.03 | 0.1 | 81.0 | Lombaert et al. (2004) |
| HC – pork kidney | 1990–1997 | – | 0.5, 1.0 | 91.7 | HC – Kuiper-Goodman et al, 1993; Mr G Lombaert, Personal Communication |
| HC – pasta | 2004–2006 | 0.2, 0.05 | 0.5, 0.2 | 91.0 | Ng et al. (2009) |
Notes: aRecovery is the mean of all recoveries.
White wine had an LOD = 0.004 ng OTA g−1 and an LOQ = 0.02 ng OTA g−1 compared with 0.008 and 0.04 ng OTA g−1 for red wine and grape juice, respectively, indicated above.
Processing factors
It is known that a redistribution of OTA occurs during the milling process, resulting in differences in OTA concentrations in the various fractions (flour, bran, etc.). To account for this redistribution, processing factors are used to convert the concentration of OTA in raw grain to the expected concentrations in ‘surrogate’ flour, bran and germ. These factors may also depend on the degree of fungal infection (including penetration into grain kernels) in a particular load of grain. Although definitive processing factors are currently not available for OTA, we chose to use a likely range of processing factors. As an upper bound for flour, we applied a conservative processing factor of 0.82 for the three classes of wheat: hard, soft and durum. As a lower bound, we used a processing factor of 0.64 for hard and soft wheat, and available finished-food pasta data for durum wheat. The surrogate flour values were then applied to foods such as bread and cookies, according to the amount of flour these foods contain. Under both of these scenarios, processing factors of 1.75 and 0.82 were used to convert the grain occurrence data to bran and germ, respectively.
Due to the reported stability of OTA during the baking of bread (Scudamore et al. 2003), no further processing adjustments were included. For foods that were analysed on a finished food basis (breakfast cereals, infant cereals, soy formula, raisins, wine, coffee, beer and others), no processing factor was required.
Handling of censored data
Values that fall below the limit of detection/quantification (censored values) may not reflect true zeros, and need to be considered in the distribution of occurrence data. To deal with such censored values, an imputing program was developed in-house, by which a parametric (lognormal) distribution was fitted to both censored and non-censored data. In the analysis, imputed values from the left-tail of the fitted distribution of occurrence values were used to replace censored values; observed values were used for the non-censored data. Together these data represented a more realistic distribution of occurrence values (Pocc) to be used for further Monte Carlo analysis, and from which an imputed mean (Docc) could be derived as required for certain calculations (Figure 2). Using this imputing procedure, data sets with different detection limits could be combined, as validated in house. For comparison, we also estimated mean occurrence after replacing censored values with values equal to 1/2LOD or 1/2LOQ, as appropriate, since many scientists have used this non-parametric method for handling censored data (Counil et al. 2005). This latter method tends to overestimate the mean when the number of positives is small.
Modelling with European Commission guidelines (maximum levels, MLs)
In Europe, maximum limits have been set for OTA in many food commodities (Table 5). Although Canada currently does not have such limits, we modelled the possible impact that EC maximum limits could have on OTA exposure in Canada. Using a conservative approach, OTA occurrence values above these limits were assigned values equal to these limits. These ML-modified distributions of occurrence data were then used in further analyses.
Table 5.
Mean occurrence data for OTA in various food commodities in Canada.
| Not corrected for recovery |
Corrected for recoverya |
|||||||
|---|---|---|---|---|---|---|---|---|
| Commodity assessed | EC ML | Number of samples | Number of positives | 1/2 LODb, mean (ng OTA g-1) | Imputed, mean (ng OTA g−1) | Number > ML | No ML (ng OTA g−1) | With ML (ng OTA g−1)c |
| Health Canada (HC) | ||||||||
| Rice | 3 | 17 | 4 | 0.90 | 0.78 | 0 | 0.80 | 0.68 |
| Corn based breakfast cereal | 3 | 34 | 6 | 0.04 | 0.04 | 0 | 0.04 | 0.04 |
| Multigrain based breakfast cereal | 3 | 83 | 46 | 0.25 | 0.25 | 0 | 0.27 | 0.27 |
| Oat based breakfast cereal | 3 | 61 | 33 | 0.39 | 0.38 | 1 | 0.43 | 0.40 |
| Rice based breakfast cereal | 3 | 29 | 3 | 0.04 | 0.03 | 0 | 0.03 | 0.03 |
| Wheat-based breakfast cereal | 3 | 132 | 54 | 0.30 | 0.28 | 4 | 0.31 | 0.30 |
| Infant cereal | 0.5 | 296 | 101 | 0.24 | 0.23 | 26 | 0.25 | 0.17 |
| Soy-based infant formulad | 0.5 | 108 | 16 | 0.04 | 0.03 | 0 | 0.04 | 0.04 |
| Pasta | 3 | 274 | 205 | 0.47 | 0.48 | 1 | 0.53 | 0.52 |
| Beer | – | 41 | 26 | 0.05 | 0.05 | – | 0.05 | – |
| Wine | 2 | 180 | 63 | 0.10 | 0.10 | 3 | 0.11 | 0.09 |
| Grape juice | 2 | 71 | 9 | 0.01 | 0.01 | 0 | 0.01 | 0.01 |
| Raisins | 10 | 151 | 118 | 1.83 | 1.83 | 9 | 2.27 | 1.68 |
| Coffee – ground regular | 5 | 59 | 38 | 0.42 | 0.42 | 0 | 0.53 | 0.53 |
| Coffee – ground decaffeinated | 5 | 12 | 4 | 0.22 | 0.21 | 0 | 0.26 | 0.26 |
| Coffee – instant regular | 10 | 21 | 15 | 0.81 | 0.81 | 0 | 1.07 | 1.07 |
| Coffee – instant decaffeinated | 10 | 9 | 5 | 0.52 | 0.51 | 0 | 0.68 | 0.68 |
| Pork | – | 90 | 19 | 0.42 | 0.32 | – | 0.35 | – |
| Canadian Grain Commission (CGC) | ||||||||
| Hard wheat | 5 | 521 | 92 | 0.97 | 0.78 | 15 | 0.88 | 0.70 |
| Soft wheat | 5 | 7(1 | 6 | 0.67 | 0.40 | 1 | 0.45 | 0.38 |
| Durum wheat | 5 | 235 | 60 | 1.18 | 1.05 | 13 | 1.19 | 0.98 |
| Oats | 3 | 54 | 18 | 2.41e | 2.24e | 8 | 2.54e | 0.92 |
| Barley | 3 | 136 | 13 | 0.75 | 0.46 | 7 | 0.52 | 0.41 |
| Peas | 3 | 49 | 12 | 1.35 | 1.17 | 6 | 1.33 | 0.84 |
Notes: aCorrection for recovery was based on the imputed data.
Values below the limit of detection (LOD) were set to 1/2 LOD.
All values above the European maximum limits (MLs) were set to the ML.
Due to the lack of positives and higher detection limit in 1997, imputing was not possible for 40 of the samples, Thus samples were not included in the analysis.
The high mean values for oats are due to a single sample that was over ten times higher than the other occurrence values.
Food consumption data
Because of limitations in available Canadian food consumption data, we used the 24-h recall survey ‘Continuing Survey of Food Intakes by Individuals’ (CSFII), encompassing the years 1994–1996 and 1998, conducted on 2 non-consecutive days by the US Department of Agriculture (USDA 2000). This large data set (over 20 000 individuals) also included a large number of infants and young children. Validation procedures were run to ensure that these data were appropriate for Canada.
In collaboration with Dr. Barbara Petersen (Exponent, Washington, DC, USA), we used the Food Analysis and Residue Evaluation (FARETM) software (Copyrighted Durango Software LLC, Bethesda, MD, USA, and licensed through Exponent, Inc) as an interface to the CSFII survey. This software provides information on food composition through several large recipe databases. FARETMwas used to assess frequency of consumption and amounts consumed of multiple food commodities/ categories (total = 37) known to sometimes contain OTA; some of these are shown in Table 5. Food consumption for certain foods (breads, cookies, pasta etc.) was expressed on a raw ingredient (i.e. flour and bran) basis. As a result, we obtained distributions (Pcon) of the amounts of relevant foods or food ingredients consumed for various age sex strata (Figure 2). Measures were taken to ensure that double counting of raw ingredients and finished foods did not occur.
Partial (PD) probabilistic exposure assessment
A number of approaches were developed to derive exposure estimates for various age sex strata. The simplest of these, ‘partial probabilistic exposure,’ is generally used as an initial step, and specifically for analyses of individual food commodities and their contribution to total exposure. Partial probabilistic (PD) exposure is derived by multiplying distributions of ‘all person’ food consumption data (Pcon)by the (imputed) mean of occurrence data (Docc) for each commodity assessed and summing across commodities for each person and day to give ∑k=137 Pcon Docc (PD, Figure 2), also expressed as ∑k=137 APall com or ∑APall com in the section involving regular commodity eaters (RCEs).
OTA exposure for regular eaters of specific commodities
Because ‘all person’ exposure is representative of the overall population, it is expected that subgroups, which regularly consume specific commodities known to potentially contain OTA, may have higher exposure to OTA than indicated in the ‘all person’ PD exposure derived above, and thus be at greater risk. While most people consume wheat, not everyone consumes, on a regular basis, coffee, wine, beer, hot oatmeal, or in the case of infants, soy formula. Therefore, for the subpopulation of regular eaters of specific commodity k (k = 1,…, 37), their ‘actual’ exposure to OTA from this specific commodity (aRCEcom) was used, and their ‘total’ exposure to OTA (tRCEcom) from all commodities in the presence or absence of EC guidelines (MLs) (Table 5) was estimated as follows:
| (1) |
where APcom is the average PD ‘all person’ exposure component for the specific food commodity being investigated, based on mean occurrence of OTA in that food. Estimates of tRCEcom±ML exposure were compared with ‘all person’ (∑APall com) exposure, to determine the increase in exposures for these subpopulations of regular commodity eaters.
Full probabilistic (PP) exposure and Monte Carlo simulation
Full probabilistic exposure assessment combines full distributions of occurrence of OTA in foods with distributions of consumptions of those foods. The full probabilistic approach takes account of the fact that the level of contamination is not constant, but varies from individual to individual and from one day of consumption to the next (Counil et al. 2005). Health Canada scientists developed and modified statistical programs utilizing SAS (Statistical Analysis Software, version 9.1.3 SP4 for SunOS, SAS Institute, Inc., Cary, NC, USA) to conduct probabilistic exposure assessments through Monte Carlo (MC) simulations (Figure 2). In our simulations, we chose a time window of ‘one day’ implying complete independence of OTA concentrations within food groups between the two non-consecutive days of the food consumption survey, but complete dependence within the same day. This approach seems to reflect best the real life scenario, especially for young children, and is more conservative than the ‘occasion’ approach (Counil et al. 2005), which assigns different OTA concentrations for each eating occasion within a day. The model for assessing the exposure for each of the two survey days is as follows:
| (2) |
where Xij is the exposure of individual i (i = 1, …, 20 607) in ng OTA kg bw−1 per day on food consumption survey day j (j = 1, 2); Qijk is the quantity of food group k (k = 1,…,37) consumed by individual i on day j, expressed in g kg−1bw; and Cijk is the concentration of OTA (ng OTA g−1) for individual i encountered on day j in food group k.
For each of the 2 survey days in each of the 1000 iterations of the exposure simulation, sampling of OTA values proceeded as follows: for each food k (or ingredient thereof) that each individual reported consuming, they received one OTA concentration value Cijk drawn randomly, with replacement, from the OTA occurrence distribution for the corresponding food (Pocc). This value was then combined with the quantity of this food (Qijk) consumed by this individual to obtain their exposure from that particular food. Summation across all food commodities consumed by that individual gave their total OTA exposure for that iteration on that survey day. Repeating this process for the entire population resulted in a distribution of exposure for that iteration and day, which was then broken down into age sex strata, and described by calculating certain distribution parameters (mean, 50th, 75th, 90th and other percentiles of exposure).
To summarize the results from the 1000 iterations, overall distribution parameters were obtained for each survey day by calculating the median of the 1000 individual parameters (e.g. the overall 90th percentile is calculated as the median of the 1000 different 90th percentiles from the individual iterations). In addition to the median, the 5th and 95th percentiles of these parameters were taken to give a measure of the variance between iterations. The 2-day average ∑k=137 Pcon Pocc (or PP) of these overall results gave a rough estimate of chronic exposure (Figure 2).
Adjustment to ‘usual’ exposure
When assessing long-term health effects, an important concept is ‘usual’ or habitual exposure, sometimes defined as the long-term average daily exposure over a period of at least one year. To estimate usual exposure, one must consider both the ‘within’ and ‘between’ individual components of variance. Since people, for the most part, do not eat the same foods every day, there tends to be a large ‘within person’ variation in the foods consumed, which needs to be removed as it interferes with the measurement of ‘between person’ variation that is of interest (Slob 1993).
The method of adjusting the raw exposure distribution to a ‘usual’ one is an adaptation of variance reduction methods developed for nutrients by the US National Research Council (NRC) (1986) and subsequently modified at Health Canada by Karpinski and Nargundkar (1992) and elsewhere (Nusser et al. 1996; Guenther et al. 1997; Hoffman et al. 2002). Since there is little variability in nutrient concentrations for a particular food commodity, these scientists used constant occurrence values (food composition data) and were effectively adjusting ∑PD exposure (Figure 2). On the other hand, because mycotoxin concentrations within a particular food commodity can vary by several orders of magnitude, we use a Monte Carlo simulation to capture the impact of this occurrence variability on OTA exposure.
Since the Monte Carlo analysis captures the two types of ‘within person’ variability (food consumption and mycotoxin concentration) the adjustment for nutrients could not be used. Thus, a new two-stage hybrid procedure was developed. For stage one, we first derived unadjusted PD exposures. In this way, consumption values for each of the 37 different food commodities were essentially weighted in proportion to the average amount of OTA they contained, using the mean occurrence data for each food (Docc). We then applied the variance reduction procedure, as developed for nutrients, to the PD exposures, and derived adjustment factors (Figure 2). In stage two, these factors were used, to adjust both the single-iteration PD exposure and the 1000-iteration PP exposure, yielding ‘adjusted’ partial probabilistic (∑k=137 P * D) exposure and ‘adjusted’ full probabilistic (∑k=137 P * P) exposure, respectively (Figure 2), thus avoiding the computational problems and excessive reduction of desirable variance that occurred when adjustment factors were calculated separately for each iteration of the Monte Carlo simulation. As part of our nomenclature, an asterisk ‘*’ is used to indicate that an adjustment to usual exposure has been made. The steps used to conduct this adjustment are detailed below.
First, the distribution of unadjusted PD exposures was normalized through a quarter root transformation (other data sets could require different transformations to normalize), and an ANOVA model with fixed main effects for population strata (i.e. age-sex group, region, race and origin) was fitted to the transformed exposure data in order to calculate estimates of the variance components. To eliminate their influence on the estimation of these components, observations with large studentized residuals and large standardized differences (>3.090, the critical value on the normal distribution indicating a significance of α = 0.001) between the two repeat recalls were flagged as ‘outliers’ and removed. For each age-sex group h, estimates, s2intra(h) and s2inter(h), of the within and between subject components of variance, σ2intra(h) and σ2intra(h), were generated using the method of moments (Kempthorne 1952). An adjustment factor ![]() was then derived from the variance component estimates as follows:
was then derived from the variance component estimates as follows:
| (3) |
Thus far, all the steps have been performed on the PD data - the occurrence variability introduced by the Monte Carlo simulation has not yet been included, so it does not interfere with the adjustment process.
Subsequently, the adjustment factors ![]() were applied to both the partial probabilistic (PD) exposure, and the individual iterations of the full probabilistic (PP) exposure to adjust towards the median on the original scale (Figure 2), as follows:
were applied to both the partial probabilistic (PD) exposure, and the individual iterations of the full probabilistic (PP) exposure to adjust towards the median on the original scale (Figure 2), as follows:
| (4) |
where g is fixed main effects other than age (region, etc.); h is the age-sex group; i is the individual in the gh-th sampling stratum; j is the day of recall (day 1 or 2); yghij is the original exposure value for a particular individual on a given day of recall on the transformed scale (y = x0:25); ![]() is the Windsorized mean of exposures for that stratum on the transformed scale (Windsorization involves setting values outside the 5th and 95th percentiles to equal the 5th and 95th percentiles to produce a mean that is more robust to the effects of extreme values); and Zmedianghij is the exposure adjusted toward the distribution median on the original scale. With this procedure, all observations are adjusted, including the outliers removed for the purpose of estimating the variance components. This adjustment preserves the median on the original scale after back transformation (mean of the distribution on the normalized scale). As a result, the adjusted mean has shifted from the mean in the original observed distribution (Carriquiry and Camañio-Garcia 2006). In order to preserve means rather than medians on the original scale, the data are transformed as follows:
is the Windsorized mean of exposures for that stratum on the transformed scale (Windsorization involves setting values outside the 5th and 95th percentiles to equal the 5th and 95th percentiles to produce a mean that is more robust to the effects of extreme values); and Zmedianghij is the exposure adjusted toward the distribution median on the original scale. With this procedure, all observations are adjusted, including the outliers removed for the purpose of estimating the variance components. This adjustment preserves the median on the original scale after back transformation (mean of the distribution on the normalized scale). As a result, the adjusted mean has shifted from the mean in the original observed distribution (Carriquiry and Camañio-Garcia 2006). In order to preserve means rather than medians on the original scale, the data are transformed as follows:
| (5) |
where ![]() and
and ![]() are the respective Windsorized means of the median-adjusted and unadjusted exposures for the gh-th sampling stratum on the original scale; and Zmeanghij is the exposure for individual i adjusted to the mean on the original scale.
are the respective Windsorized means of the median-adjusted and unadjusted exposures for the gh-th sampling stratum on the original scale; and Zmeanghij is the exposure for individual i adjusted to the mean on the original scale.
As was done for the original unadjusted data, repeating this process for the entire population resulted in a distribution of adjusted exposures for that iteration, broken down into age-sex strata, and described by calculating certain distribution parameters (mean, 50th, 7th, 90th and other percentiles of exposure); overall distribution parameters were then calculated to describe these adjusted results (∑k=137 P * P) of the 1000 iterations (Figure 2). Due to the possibility of systematic bias on the second day of reporting, the adjusted first day exposure is taken as usual exposure.
Results
Occurrence data
A summary of the occurrence data for various food commodities is shown in Table 5. This table shows the impact on mean occurrence of using for censored data either 1/2LOD (or 1/2LOQ, where applicable) or the HC imputing method, our preferred approach. Approximately 9% of infant cereal data exceeded the EC ML, whereas none of the soy based infant formula exceeded the EC ML, and 6% of the raisin data exceeded the EC ML. With regard to wheat grain, approximately 3% of hard wheat, 1.4% of soft wheat and 5.5% of durum wheat exceeded the EC ML. In addition, recent HC results on OTA in finished pasta products indicate that only one of 274 samples was above the EC ML of 3 ng OTA g−1 (Ng et al. 2009). Furthermore, OTA levels in finished pasta were lower than ‘surrogate’ durum flour derived from grain data using processing factors of 0.82 or 0.64.
As indicated under Methods and approaches, we modelled the data with two processing factor scenarios. For most of the following assessment, and unless otherwise indicated, we show the results using the conservative processing factor of 0.82 for all wheat (grain to flour). Furthermore, throughout this communication ‘all person’ exposure is implied, unless indicated otherwise.
Partial probabilistic (PD) unadjusted exposure
Most individuals from 6 to 9 months of age and older consumed OTA-containing food on both days of the survey (Table 6), confirming earlier HC findings that OTA was present in all serum samples of the Canadian population sampled (Scott et al. 1998).
Table 6.
Frequency of consumption of OTA-containing food commodities by age-sex strata.a
| Age group | All persons | Total eatersc | One day onlyd | Both dayse | Eaters/all persons (%) | Both days/ eaters (%) |
|---|---|---|---|---|---|---|
| 0–2 months | 344 | 148 | 23 | 125 | 43 | 84.5 |
| 3–5 months | 428 | 319 | 49 | 270 | 74.5 | 84.6 |
| 6–8 months | 365 | 354 | 25 | 329 | 97 | 92.9 |
| 9–11 months | 349 | 347 | 10 | 337 | 99.4 | 97.1 |
| 1 year | 1040 | 1035 | 14 | 1021 | 99.5 | 98.6 |
| 2 years | 1056 | 1054 | 4 | 1050 | 99.8 | 99.6 |
| 3 years | 1759 | 1759 | 3 | 1756 | 100 | 99.8 |
| 4 years | 1782 | 1781 | 6 | 1775 | 99.9 | 99.7 |
| 5–6 years | 1420 | 1420 | 2 | 1418 | 100 | 99.9 |
| 7–11 years | 1343 | 1343 | 3 | 1340 | 100 | 99.8 |
| 12–18 years, male | 629 | 629 | 6 | 623 | 100 | 99 |
| 19–30 years, male | 854 | 853 | 14 | 839 | 99.9 | 98.4 |
| 31–50 years, male | 1684 | 1684 | 19 | 1665 | 100 | 98.9 |
| 51–70 years, male | 1606 | 1605 | 9 | 1596 | 99.9 | 99.4 |
| 71+ years, male | 674 | 674 | 3 | 671 | 100 | 99.6 |
| 12–18 years, female | 632 | 632 | 11 | 621 | 100 | 98.3 |
| 19–30 years, female | 827 | 827 | 14 | 813 | 100 | 98.3 |
| 31–50 years, female | 1653 | 1652 | 25 | 1627 | 99.9 | 98.5 |
| 51–70 years, female | 1539 | 1539 | 11 | 1528 | 100 | 99.3 |
| 71 + years, female | 623 | 623 | 2 | 621 | 100 | 99.7 |
Notes: aBased on USDA food intake surveys (2 non-consecutive survey days for >20 000 persons, collected between 1994 and 1998).
Total number of persons in each age-sex stratum.
Number of consumers of any potentially OTA-containing commodity in each age-sex stratum.
Persons consuming on only of the 2 survey days.
Persons consuming on both survey days.
As indicated under Methods and approaches, the Partial probabilistic (PD) exposure (∑APall com) uses mean imputed occurrence values (Figure 2). For 1-year-olds and 31–50-year-old males PD exposure was 4.42 and 1.62 ng OTA kg bw−1, respectively), with the greater value in children attributed to a higher food intake relative to their body weight. Accordingly, much of the subsequent risk assessment focused on this vulnerable age group.
Overall contribution of various foods to total ‘all person’ exposure to OTA
To determine the contribution of various foods to total ‘all person’ exposure, we used PD exposure. For different age groups, differences in food consumption patterns will result in differences in the average contributions of specific grouped food commodities to total exposure. Comparing 1-year-olds with 31- 0-year-old males clearly shows that for both age groups the largest contributing commodity was wheat, with hard > durum > soft wheat (Figure 3).
Figure 3.
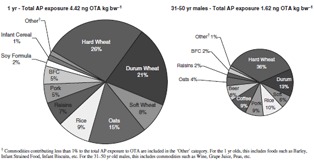
Percent contribution of food commodities to total ‘all person’ OTA exposure (PD, unadjusted 2d average).
Using raisins as an example, the ‘all person’ consumption of raisins, from all sources, for 1-year-olds is about 0.13 g raisins kg bw−1 (Figure 4c). Since the mean occurrence of OTA in raisins is 2.27 ng OTA g−1 (Table 4), the ‘all person’ exposure from raisins (APraisins) is 0.3 ng OTA kg bw−1. Thus, for this age group, the ‘all person’ raisin contribution to total exposure is 0.3* 100/4.42 = 6.8%, rounded to 7%, compared with 2% for older age groups (Figure 3). The contribution of oats (processing factor of 1.0) to exposure was approximately twice that of soft wheat or rice. Overall, the OTA contribution from pasta (expressed here as durum wheat) decreased with age, whereas the OTA contributions from rice and soft wheat were similar across various age-sex groups (Figure 3).
Figure 4.
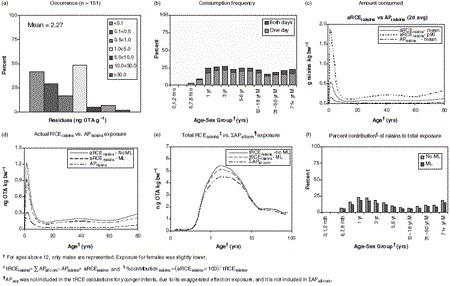
OTA data on occurrence, consumption, and exposure for regular eaters of raisins for various age groups.
Regular eaters of specific commodities (RCEcom)
Whereas most people consumed wheat each day, the ‘all person’ exposures for other individual commodities (APcom) used in the average total ‘all person’ OTA exposure (∑k=137 APall com) (Figure 3) included many non-consumers. Thus, subpopulations which regularly consume foods known to potentially contain OTA, such as coffee, wine, or raisins, may have a greater than average exposure to OTA. To assess the total exposure of regular commodity eaters, (tRCEcom) we used PD exposure (see equation, methods), and in the first example discussed below, we show again detailed data for raisin eaters. Data for other specific commodities, discussed below, were modelled in a similar manner, but no detailed data are provided.
Raisins. Approximately 23% of 1-year-old children consume raisins, and about one-third of them consume raisins on both survey days (Figure 4b). The mean and 90th percentile of the amount of raisins (2-day average) consumed by this subpopulation of children are 0.52 and 1.8 g OTA kg bw−1, respectively (Figure 4c). With a mean occurrence of 2.27 ng OTA g−1 raisins, their mean and 90th percentile actual exposure from raisins (aRCEraisins) is 1.2 and 4.1 ng OTA kg bw−1, respectively, compared with the ‘all person’ exposure from raisins (APraisins) of 0.3 ng OTA kg bw−1 (Figure 4d) derived in the previous section. Using equation 1 (see Methods and approaches) ∑APall com – APraisins + aRCEraisins = tRCEraisins, the mean exposure to OTA for regular raisin eaters (tRCEraisins) of this age group is 4.42-0.3 + 1.2 = 5.35 ng OTA kg bw−1 (Figure 4e) and for the 90th percentile of raisin eaters, exposure is 8.28 ng OTA kg bw−1, compared with the ‘all person’ exposure of 4.42 ng OTA kg bw−1 for this age group (Table 7). For raisins, nine out of 151 values were above the EC ML of 10 ng OTA g−1 (Table 5). Modelling with this ML showed that the above exposures would have decreased to 5.04 and 7.21 ng OTA kg bw−1, respectively (Table 7). The contribution for the exposure of actual raisin eaters (aRCErasin) to total exposure (tRCEraisin) = 1.2* 100/ 5.3 = 23%. (Figure 4f), compared with the ‘all person’ raisin contribution to total ‘all person’ exposure of 7%, estimated in the previous section (Figure 3).
Table 7.
Partial probabilistic (PD) exposure to OTA for ‘regular commodity eaters’ (tRCE) compared with ‘all persons’ (AP) for select age sex strata.
| Exposure (ng OTA kg bw-1 per day) |
|||||
|---|---|---|---|---|---|
| No ML |
ML |
||||
| Age group | Commodity | Mean | p90 | Mean | p90 |
| 0–2 months | Soy-based formulaa | 6.30 | 10.22 | b | b |
| 1 year | ∑AP all comc | 4.42 | – | 3.25 | – |
| Hard wheat | 4.51 | 5.74 | 4.21 | 5.13 | |
| Soft wheat | 4.50 | 4.98 | 4.42 | 4.79 | |
| Durum wheat | 5.06 | 6.89 | 4.65 | 6.01 | |
| Hot oatmeal | 8.94 | 14.98d | 5.65 | 7.83 | |
| Breakfast cereal | 4.56 | 4.91 | 4.54 | 4.88 | |
| Raisins | 5.35 | 8.28 | 5.04 | 7.21 | |
| Rice | 4.93 | 6.12 | 4.93 | 6.12 | |
| 31–50 years, male | ∑APall comc | 1.62 | – | 1.33 | – |
| Hard wheat | 1.63 | 2.10 | 1.49 | 1.84 | |
| Soft wheat | 1.65 | 1.80 | 1.63 | 1.74 | |
| Durum wheat | 1.99 | 2.63 | 1.84 | 2.31 | |
| Hot oatmeal | 2.75 | 3.72d | 1.99 | 2.34 | |
| Beer | 1.99 | 2.53 | e | e | |
| Wine | 1.73 | 1.93 | 1.71 | 1.89 | |
| Coffee | 1.71 | 1.92 | b | b | |
| Breakfast cereal | 1.72 | 1.86 | 1.71 | 1.85 | |
| Raisins | 1.77 | 2.06 | 1.72 | 1.93 | |
| Rice | 1.92 | 2.52 | 1.92 | 2.52 | |
Notes: aInfant formula and breast milk are usually the only foods consumed in this age group, so ∑APall com is not shown.
All occurrence values were below the EC ML for infant formula, cereal and coffee.
ML values for ∑APall com were modelled using all the EC MLs; those for regular eaters of a specific commodity were modelled using only that commodity's EC ML.
The high mean values for oats are due to a single sample that was over ten times higher than the other occurrence values.
There is presently no EC ML for beer.
Hot oatmeal. Only a small proportion of the population regularly consume hot oatmeal. Thus, regular consumers of this food are under-represented in the ‘all person’ distribution of exposure. The mean and 90th percentiles of exposure for 1-year-old regular consumers of hot oatmeal (tRCEoatmeal) were 8.94 and 15.0 ng OTA kg bw−1, respectively. These values were two (mean) to nearly four (p90) times higher than the ‘all person’ (∑APall com) exposure of the average 1-year-old (4.42 ng OTA kg bw−1). Modelling, using the EC ML for oats, showed that these values would decrease to 5.65 and 7.83 ng OTA kg bw−1 (Table 7).
Cold breakfast cereals. Approximately two-thirds of 1-year-olds consume some form of breakfast cereal (BFC), and half of these consumers do so on both survey days (data not shown). The exposure to OTA for 1-year-old regular breakfast cereal consumers (tRCEBFC) was only about 3% higher than the mean exposure for their age group (Table 7). Considering that a person who consumes BFC is likely to consume less of other cereal-derived foods such as bread, the overall impact on OTA exposure from being a BFC consumer is probably negligible. There was very little impact on exposure when EC MLs were applied to BFC occurrence data, as most values were below the ML.
Coffee. In the food consumption survey, about 60% of adults consumed coffee (brewed by percolation from ground coffee), and three-quarters of them did so on both days. While other types of coffee were considered, the number of consumers of these was small. In the subpopulation of regular coffee drinkers (tRCEcoffee), the 31–50-year-old males had a mean total daily exposure of 1.71 ng OTA kg bw−1 (Table 7), compared with 1.62 ng OTA kg bw−1 for the general population (∑APall com). For heavy coffee drinkers, represented as the 90th percentile of tRCEcoffee, exposure was 1.92 ng OTA kg bw−1. No occurrence values for coffee were above the EC ML (Table 5); the mean occurrence was 0.53 ng OTA per g, compared with 0.72 ng OTA per g reported in Europe (SCOOP 2002).
Beer. Beer drinkers comprised at most 30% of the adult population, with almost a third consuming beer on both days of the survey (data not shown). Again taking the 31–50-year-old males as an example, the exposures of regular (mean), heavy (p90), and very heavy (p95) beer drinkers (tRCEbeer) were 1.99, 2.53, and 2.88 ng OTA kg bw−1, respectively compared with 1.62 ng OTA kg bw−1 for the general population (∑APall com) (Table 7). Currently there are no EC MLs for OTA in beer (Table 5).
Wine. Wine was consumed by only 10% of the population, and rarely on both survey days (data not shown). After modelling for differences in OTA levels between wines of different countries and market share of domestic versus imported wine, exposures for regular consumers of wine (tRCEwine) for 31–50-year-old males were 1.73 (mean) and 1.93 (p90) ng OTA kg bw−1 compared with 1.62 ng OTA kg bw−1for the average 31–50-year-old male (∑APall com) (Table 7). Again, EC MLs had only a slight impact on OTA exposure from this source, as few occurrence values were above the EC limits.
Infant cereals (IFC). The consumption of infant cereals peaks at the age of 6–8 months, when 80% of infants consume them. This is followed by a gradual transition to ready-to-eat breakfast cereals. Three quarters of these consumers eat infant cereals on both days of the survey (data not shown). OTA exposure (tRCEIFC) for 6–11-month-old consumers was 2.69–3.77 ng OTA kg bw−1 compared with ‘all person’ (∑APall com) exposure of 2.593.58 ng OTA kg bw−1. Modelling with the EC ML had only a slight impact on OTA exposure from this source.
Soy infant formula. One commodity of particular interest was soy-based infant formula, a significant contributor of OTA for infant (zero to 6 months) consumers of this product. Other than human breast milk or substitutes thereof, this age group consumes very few foods. In Canada, soy formula is consumed by about 10% of young infants (Christine Zehaluk, Health Canada, personal communication), compared with 30% of infants in the US survey, and almost always on both days. Although all soy formula samples tested were below the EC limits (Table 5), as one of the sole foods consistently consumed by some infants, the mean and 90th percentile exposure estimates (tRCEsoy) were 6.30 and 10.22 ng OTA kg bw−1, respectively (zero to 2 months old, Table 7), compared with essentially zero exposure for non-consumers of soy formula. Consequently, the APsoy component had an exaggerated effect on ‘all person’ OTA (∑APall foods) exposure and for infants zero to 5 months old was excluded from the tRCE estimates.
Ochratoxin exposure for regular eaters of various classes of wheat. As indicated above, wheat was the major contributor to OTA exposure for all age groups. Summary data on OTA occurrence in hard, soft and durum wheat are shown in Table 5. The highest frequency of consumption was for foods based on hard wheat such as bread and similar foods. Most people consumed these foods on both survey days. Using recipes to calculate the amount of hard wheat in each food, 1-year-olds consumed about 2 g hard wheat kg bw−1. Partly because of their lower body weight, this was about twice as much as their older counterparts (data not shown). As hard wheat was part of the diet for almost all individuals in the survey, the difference between the total exposure of the average hard wheat consumer (tRCEhard) and the average person ('all person’ or ∑APall com) was minimal for both 1-year-olds and 31–50-year-old males (Table 7).
The consumption of foods containing soft wheat (cookies, cakes, biscuits, etc.) was slightly less frequent, with about 80% of individuals in most age groups consuming soft wheat, and about half of these on both days (data not shown). For both age groups, the exposure of consumers of soft wheat (tRCEsoft) was only slightly higher than that of the average Canadian (Table 7).
However, for foods derived from durum wheat (i.e. pasta), the situation was somewhat different. Durum wheat was consumed by less than half the population in most age groups, and rarely on both days of the survey (data not shown). The exposure for 1-year-old durum wheat consumers (tRCEdurum) was 5.06 ng OTA kg bw−1, compared with the average exposure of 4.42 ng OTA kg bw−1 for this age group.
When the data were modelled with EC flour MLs applied to the OTA occurrence data for wheat grain-derived ‘surrogate flour', total OTA exposure (tRCEwheat) decreased for all three classes of wheat, with the greatest impact on hard and durum wheat (Table 7). Thus, total OTA exposure for 1-year-old durum wheat consumers (tRCEdurum) decreased from 5.06 to 4.65 ng OTA kg bw−1 for the PF = 0.82 run.
Full probabilistic exposure assessment and adjustment to usual exposure
Exposure modelling in the absence of guidelines.
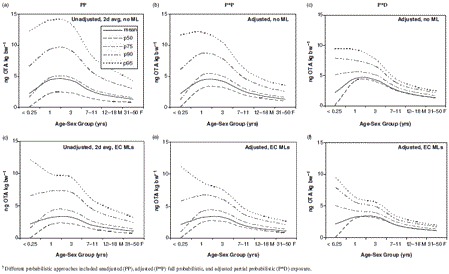
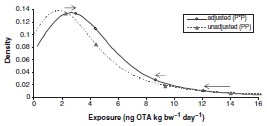
With the exception of infants, there were minimal differences in mean exposure between unadjusted 2-day average ‘all person’ exposure (PP) (Figure 5a), ‘eaters only’ exposure (data not shown), adjusted usual exposure (P*P) (Figure 5b and Table 8), and partially probabilistic (P*D) exposure (Figure 5c). Figure 6 compares the distribution of unadjusted (PP) and adjusted (P*P) exposure for 1-year-olds.
Figure 5.
OTA exposure modeling as a function of age for select percentiles using three approaches†, and with or without the EC MLs.
Table 8.
Full probabilistic usual (P*P) exposure to ochratoxin A (ng OTA kg bw−1 per day) for ‘all persons’ (AP) and each age-sex stratum.
| Age | Mean | SD | p50 | p75 | p90 | p95 | p97.5 | ||||||||||||||
| 0–2 months | 1.62a | 2.22 | 2.91 | 4.54 | 6.57 | 8.89 | 0.00 | 0.00 | 0.00 | 0.80 | 1.11 | 1.63 | 4.26 | 6.07 | 8.29 | 8.21 | 11.59 | 17.74 | 12.38 | 20.89 | 33.25 |
| 3–5 months | 1.53 | 1.93 | 2.44 | 3.67 | 5.06 | 7.25 | 0.15 | 0.20 | 0.24 | 1.10 | 1.41 | 1.75 | 3.93 | 5.18 | 6.61 | 7.03 | 9.42 | 14.14 | 10.62 | 17.26 | 26.18 |
| 6–8 months | 2.05 | 2.45 | 3.12 | 3.09 | 4.18 | 9.32 | 0.88 | 1.04 | 1.22 | 2.24 | 2.64 | 3.15 | 4.74 | 5.88 | 7.39 | 7.15 | 9.53 | 13.28 | 10.49 | 15.13 | 21.18 |
| 9–11 months | 2.86 | 3.45 | 4.64 | 2.93 | 4.20 | 13.99 | 1.86 | 2.16 | 2.56 | 3.45 | 4.11 | 4.90 | 5.95 | 7.43 | 9.45 | 8.30 | 10.78 | 14.42 | 10.94 | 14.74 | 19.73 |
| 1 year | 3.79 | 4.38 | 5.29 | 3.67 | 5.60 | 10.35 | 2.66 | 2.99 | 3.41 | 4.49 | 5.05 | 5.82 | 7.63 | 8.66 | 10.04 | 10.37 | 12.08 | 14.31 | 13.45 | 16.18 | 19.86 |
| 2 years | 3.31 | 4.36 | 5.10 | 2.77 | 3.71 | 5.25 | 3.04 | 3.42 | 3.94 | 4.55 | 5.17 | 5.96 | 6.87 | 7.88 | 9.16 | 8.79 | 10.30 | 12.13 | 10.96 | 12.99 | 15.74 |
| 3 years | 3.79 | 4.22 | 4.78 | 3.01 | 3.95 | 6.70 | 2.92 | 3.19 | 3.54 | 4.48 | 4.94 | 5.49 | 7.00 | 7.81 | 8.75 | 9.24 | 10.43 | 11.84 | 11.67 | 13.33 | 15.49 |
| 4 years | 3.62 | 3.96 | 4.47 | 2.67 | 3.31 | 5.10 | 2.87 | 3.09 | 3.42 | 4.30 | 4.66 | 5.18 | 6.50 | 7.16 | 8.07 | 8.45 | 9.48 | 10.78 | 10.57 | 11.99 | 14.00 |
| 5–6 years | 3.2S | 3.66 | 4.25 | 2.53 | 3.28 | 5.61 | 2.55 | 2.80 | 3.14 | 3.91 | 4.29 | 4.83 | 6.02 | 6.77 | 7.67 | 7.88 | 8.97 | 10.23 | 9.67 | 11.39 | 13.45 |
| 7–11 years | 2.31 | 2.60 | 2.95 | 1.71 | 2.18 | 3.19 | 1.32 | 2.01 | 2.23 | 2.75 | 3.04 | 3.38 | 4.18 | 4.72 | 5.34 | 5.45 | 6.27 | 7.21 | 6.86 | 8.05 | 9.56 |
| 12–18 years, male | 1.52 | 1.76 | 2.07 | 1.14 | 1.50 | 2.22 | 1.17 | 1.34 | 1.54 | 1.81 | 2.08 | 2.41 | 2.77 | 3.25 | 3.92 | 3.62 | 4.35 | 5.36 | 4.62 | 5.73 | 7.28 |
| 19–30 years, male | 1.56 | 1.76 | 2.04 | 1.23 | 1.65 | 2.44 | 1.20 | 1.32 | 1.48 | 1.93 | 2.14 | 2.41 | 2.96 | 3.40 | 3.95 | 3.86 | 4.54 | 5.39 | 4.32 | 5.85 | 7.08 |
| 31–50 years, male | 1.49 | 1.62 | 1.79 | 1.23 | 1.50 | 1.95 | 1.15 | 1.23 | 1.33 | 1.80 | 1.95 | 2.11 | 2.80 | 3.06 | 3.36 | 3.64 | 4.04 | 4.56 | 4.62 | 5.23 | 6.03 |
| 51–70 years, male | 1.29 | 1.43 | 1.58 | 1.13 | 1.46 | 2.02 | 0.97 | 1.05 | 1.14 | 1.56 | 1.69 | 1.84 | 2.48 | 2.73 | 3.01 | 3.29 | 3.67 | 4.11 | 4.18 | 4.78 | 5.52 |
| 71+ years, male | 1.14 | 1.33 | 1.61 | 1.03 | 1.68 | 2.73 | 0.80 | 0.91 | 1.03 | 1.36 | 1.54 | 1.75 | 2.23 | 2.58 | 3.05 | 3.02 | 3.61 | 4.40 | 3.86 | 4.90 | 6.44 |
| 12–18 years, female | 1.19 | 1.41 | 1.74 | 0.82 | 1.11 | 1.33 | 0.95 | 1.11 | 1.35 | 1.42 | 1.66 | 2.02 | 2.09 | 2.53 | 3.14 | 2.71 | 3.30 | 4.18 | 3.31 | 4.24 | 5.48 |
| 19–30 years, female | 1.17 | 1.33 | 1.56 | 0.93 | 1.18 | 1.80 | 0.90 | 1.01 | 1.14 | 1.42 | 1.60 | 1.82 | 2.20 | 2.54 | 2.94 | 2.90 | 3.42 | 4.06 | 3.62 | 4.39 | 5.34 |
| 31–50 years, female | 1.21 | 1.33 | 1.47 | 1.02 | 1.25 | 1.61 | 0.92 | 0.99 | 1.07 | 1.47 | 1.59 | 1.73 | 2.32 | 2.54 | 2.31 | 3.02 | 3.42 | 3.86 | 3.37 | 4.44 | 5.17 |
| 51–70 years, female | 1.11 | 1.23 | 1.33 | 0.99 | 1.28 | 1.77 | 0.83 | 0.90 | 0.98 | 1.33 | 1.45 | 1.59 | 2.12 | 2.35 | 2.62 | 2.83 | 3.16 | 3.59 | 3.54 | 4.12 | 4.81 |
| 71+ years, female | 0.96 | 1.15 | 1.40 | 0.87 | 1.36 | 2.53 | 0.70 | 0.80 | 0.92 | 1.15 | 1.33 | 1.54 | 1.88 | 2.23 | 2.65 | 2.54 | 3.07 | 3.73 | 3.26 | 4.07 | 5.30 |
Notes: a95% Confidence intervals are shown in lower font size and italic font.
Figure 6.
Distribution of unadjusted and adjusted exposure to OTA for 1 year olds.
Figures 5a-c and 6 clearly show how the adjustment to usual exposure decreased the higher percentiles of the OTA exposure distribution and increased the lower percentiles, such as the median, especially with the (P*D) exposure. This latter approach uses mean OTA occurrence in various foods and thus variability resulting from the occurrence distribution is not captured in the exposure estimates. Specific P*P exposure values for 1-year-olds, were 4.38, 8.66 and 12.08 ng OTA kg bw−1 for the mean, 90th and 95th percentiles, respectively (Figure 6 and Table 8). For this age group, P*D exposures were 4.53, 7.43, and 9.28 ng OTA kg bw−1 for the same percentiles, respectively (Figure 5c).
Table 8 also shows that for 31–50-year-old males, usual exposures were 1.62, 3.06, and 4.04 ng OTA kg bw−1 for the mean, 90th and 95th percentiles, respectively. For comparison, P*D exposure estimates for 31–50-year-old males were 1.65, 2.53, and 2.98 ng OTA kg bw−1, respectively. Over time, true exposure would be expected to fall between the P*P and P*D estimates (Figures 5b and c). Exposures for females were lower than for males of the same age group (Table 8).
Overall impact of lower processing factors.
Differences in estimated processing factors introduce a systematic bias, with lower processing factors resulting in lower estimates of exposure. Using the lower bound processing factor of 0.64 for hard and soft wheat, together with actual pasta occurrence data, gave a mean exposure (P*P) of 3.68 ng OTA kg bw−1 per day for 1-year-olds compared with 4.38 ng OTA kg bw−1 per day (Table 8), when the higher processing factor of 0.82 was used.
Overall impact of EC MLs.
Modelling showed that an overall introduction of EC MLs for all affected commodities would have considerably lowered estimates of exposure. Removal of high occurrence values also reduces exposure variability (Figures 5d-f), and would have a pronounced effect on the higher exposure percentiles. For example, the mean and 90th percentile P*P exposure to OTA for 1-year-olds, would have decreased from 4.38 to 3.27 and from 8.66 to 6.65 ng OTA kg bw−1, respectively (Figure 5e).
RISK CHARACTERIZATION
Margin of exposure (MOE) estimates
Estimates of the margin of exposure (MOE) with respect to the cancer endpoint (Kuiper-Goodman 2004; O'Brien et al. 2006) were made for selected age groups by dividing the TD05 (19.6 µg OTA kg bw−1)by the mean or 90th percentile of exposure. When this ratio equals 5000, the risk of developing renal cancer was taken as 1:100 000. For MOE values <5000, there is a high priority for risk reduction, especially when one also considers the longer half-life of OTA in humans as compared to rats. Regardless of which mean exposure estimate was used (P*P, P*D or PD), in scenarios that involved a PF of 0.82, a high priority for risk reduction was demonstrated for younger age groups. Thus, the MOE for mean and 90th percentile ‘all person’ PD (∑APall com) OTA exposure for 1-year-olds was 4426 and 2446, respectively (Table 9a). For most older age groups the MOEs were >5000. Modelling with EC MLs showed that the MOEs for the mean and 90th percentile of the 1-year age group would have increased to 6026 and 3289, respectively.
Table 9a.
Margin of exposure (MoEa) for regular specific commodity eaters (tRCEcom) for select age sex strata and various exposure scenarios.
| No ML | ML | ||||||||||
| PD exposureb | Age (years): Sex: |
1 M + F |
7–11 M + F |
12–18 M |
19–30 M |
31–50 F |
1 M + F |
7–11 M + F |
12–18 M |
19–30 M |
31–50 F |
| ∑APall com | Mean: | 4426 | 7552 | 10856 | 11358 | 14836 | 6026 | 9778 | 13626 | 13859 | 18223 |
| p90: | 2446 | 4360 | 6306 | 6399 | 8230 | 3289 | 5723 | 7854 | 7462 | 10228 | |
| tRCEcom | Means | ||||||||||
| Durum wheatb Means | 3867 | 6108 | 8528 | 9076 | 12013 | 4205 | 6682 | 9304 | 9910 | 13062 | |
| Durumb PF = 0.64 & pasta data | 4735 | 7790 | 10892 | 11355 | 14841 | 4741 | 7800 | 10905 | 11368 | 14858 | |
| Rice | 3972 | 6588 | 8767 | 9611 | 12384 | c | c | c | c | c | |
| Hot oatmeal | 2188 | 3918 | 5633 | 5821 | 8563 | 3464 | 5815 | 8231 | 8570 | 11935 | |
| Breakfast cereal | 4298 | 7344 | 10292 | 10762 | 13857 | 4314 | 7369 | 10332 | 10791 | 13903 | |
| Raisins | 3658 | 7152 | 10286 | 10558 | 13578 | 3887 | 7284 | 10468 | 10792 | 13961 | |
| Beer | 9043 | 11817 | d | d | |||||||
| Coffee | 10349 | 10464 | 13729 | c | c | c | |||||
| Wine | 10860 | 13486 | 10927 | 13668 | |||||||
Notes: aMoE = TD05 (19.6 µg OTA kg bw−1 per day adjusted for 5–7-day gavage) divided by total RCE mean exposure to ochratoxin A (ng OTA kg bw−1 per day). MoE < 5000 (in bold) points to need for risk reduction.
Using a processing factor of 0.82 or 0.64 plus pasta occurrence data where indicated.
All occurrence values were below the EC ML for rice and coffee.
There is presently no EC ML for beer.
As expected, the MOEs for exposure to OTA of regular eaters of specific commodities (tRCEspec com) were lower than MOEs for ‘all person’ exposure from all commodities (∑APall com) (Table 9a). Thus, MOEs for OTA exposure of regular consumers from foods such as pasta and rice (tRCEpasta, etc.) were about 10–12% lower (Table 9a). While MOEs were about the same for regular consumers of breakfast cereals, the MOE was about 50% lower for 1-year-old regular consumers of hot oatmeal. Here the EC ML would have had a large impact, as one of the occurrence values was particularly high. For 1-year-old raisin consumers, a lower MOE of 3658 was observed, but here the current EC ML would not have had much of an impact. The MOEs for coffee, wine and beer consumers were all much greater than 5000. Heavy beer consumers (90th percentile of 19–30 yr M, data not shown) would have an MOE in the order of 6800. When exposure was modelled with the less conservative processing factor of 0.64 combined with actual pasta occurrence data, MOEs for pasta were higher, but for 1-year-olds, these remained <5000. For soy formula, the MOE was well below 5000 for the two youngest groups of infants, and since all data were below the EC ML, there would have been no impact of this ML for this commodity (Table 9b). On the other hand, the MOEs for infant cereal were >5000 (Table 9b).
Table 9b.
Margin of Exposure (MoEa) for regular commodity eaters of infant foods.
| No ML |
ML |
|||||||
|---|---|---|---|---|---|---|---|---|
| Age (months) | 0–2 | 3–5 | 6–8 | 9–11 | 0–2 | 3–5 | 6–8 | 9–11 |
| Soy-based formula | 3107 | 3799 | Few eaters | b | b | Few eaters | ||
| Infant cereal | Few eaters | 7263 | 5198 | Few eaters | 7740 | 5427 | ||
Notes: aMoE = TD05 (19.6 µg OTA kg bw−1 per day adjusted for 5–7-day gavage) divided by total RCE mean exposure to ochratoxin A (ng OTA kg bw−1 per day). MoE < 5000 (in bold) points to need for risk reduction.
All occurrence values were below the EC ML for this commodity.
Comparison of mean and percentiles of full probabilistic exposure to the NCRI
There are several risks associated with exposure to OTA. Of these, the risk of nephropathy and the risk of cancer were considered the most important. As was seen under hazard characterization, there is a convergence in the value of risk metrics associated with these two endpoints, the NCRI (cancer, 4 ng kg bw −1 per day) and the TDI (nephropathy, 3 ng kg bw−1 per day). As these metrics are close in value, we have used the NCRI, to compare with the mean or higher percentiles of exposure.
The usual OTA exposure estimates (P*P) were compared with risk metrics such as the NCRI or multiples thereof (8, 12, or 16 ng OTA kg bw−1 per day). With the exception of 1 to 3-year-olds, mean estimates were below 4 ng OTA kg bw−1, whereas the 90th and 95th percentiles were generally below 12 ng OTA kg bw−1 (Table 8 and Figure 5b). Modelling with EC MLs showed that all mean P*P estimates would have been below the NCRI (Figure 5e).
Frequency and magnitude of excursions above the NCRI
Occasionally exceeding the NCRI may not be a cause of concern, provided that the duration is brief and exposure is not excessive. To examine this, we made a plot of adjusted (P*P) exposure percentiles of 1-year-olds (p5, p10, p50, etc.) for 1000 Monte Carlo iterations sorted by exposure (Figure 7). In the absence of EC MLs the lower 10th percentile segment of the population of 1-year-olds rarely (10.8% of iterations) exceeded the NCRI, and the 90th percentile segment did so frequently (66.2% of iterations) (Figure 7 left, Table 10). It appears that an individual's food consumption pattern, reflecting the number of potentially OTA containing foods and amounts thereof consumed, determines the exposure percentile to which one belongs. At the lower exposure iterations, exposures for this age group ranged from <0.1 to about 3 ng OTA kg bw−1 per day. But, for the highest exposure iterations, most people exceeded the NCRI with P*P exposures ranging from 5.1 to over 100 ng OTA kg bw−1 per day (Figure 7 left, Table 10).
Figure 7.
Usual exposure, of select percentiles (5, 10, 50, 90, 95)† of the population of 1-year-olds for the 1000 Monte Carlo iterations, sorted to show the relative impact of consumption pattern and occurrence variability, modelled without and with the EC MLs.
Table 10.
Per cent of iterations above the NCRIa and multiples thereof for usual exposure (P*P) to OTA of various population percentiles of select age sex groups.
| Per cent of iterations out of 1000 where exposure is above the indicated value | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| >4 ng OTA kg bw1 | >8 ng OTA kg bw−1 | >12ng OTA kg bw1 | > 16 ng OTA kg bw−1 | ||||||
| Age group | Population percentiles | No ML | ML | No ML | ML | No ML | ML | No ML | ML |
| 1 year | p5 | 1.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| p10 | 10.8 | 11.3 | 1.3 | 0.0 | 0.3 | 0.0 | 0.0 | 0.0 | |
| p50 | 33.7 | 20.7 | 9.5 | 0.6 | 3.3 | 0.0 | 1.3 | 0.0 | |
| p90 | 66.2 | 55.7 | 27.7 | 18.0 | 15.3 | 7.6 | 8.2 | 0.9 | |
| p95 | 79.5 | 70.6 | 37.4 | 34.1 | 21.6 | 18.4 | 13.2 | 6.4 | |
| 7–11 years | p5 | 0.9 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| p10 | 3.8 | 0.0 | 0.5 | 0.0 | 0.2 | 0.0 | 0.0 | 0.0 | |
| p50 | 10.5 | 4.7 | 2.5 | 0.0 | 0.6 | 0.0 | 0.3 | 0.0 | |
| p90 | 29.2 | 20.2 | 5.9 | 0.1 | 2.5 | 0.0 | 0.8 | 0.0 | |
| p95 | 36.3 | 28.0 | 9.3 | 0.9 | 2.9 | 0.0 | 0.9 | 0.0 | |
| 31–50 years, male | p5 | 0.1 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 |
| p10 | 0.2 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| p50 | 3.7 | 0.0 | 0.3 | 0.0 | 0.1 | 0.0 | 0.0 | 0.0 | |
| p90 | 11.6 | 1.7 | 1.7 | 0.0 | 0.5 | 0.0 | 0.2 | 0.0 | |
| p95 | 21.1 | 6.7 | 4.9 | 0.0 | 1.8 | 0.0 | 0.6 | 0.0 | |
Note: aNegligible cancer risk intake (4 ng OTA kg bw−1 per day) associated with a risk level of 1:100 000−1.
As expected, the proportion of individuals that exceed the NCRI and the frequency with which they did so decreased with age (Table 10). Modelling shows that the introduction of EC MLs, through removal of the higher occurrence values, would reduce the probability of exceeding the NCRI, with the greatest impact seen at the higher multiples of the NCRI, especially for 1-year-olds (Figure 7 right, Table 10).
DISCUSSION
The many challenges associated with estimating possible health risks from OTA exposure relate to uncertainties in estimating both hazard and exposure. First, while there is no disagreement that OTA is a nephrotoxic agent and very potent ‘complete’ renal carcinogen, a clear understanding of the underlying mechanism and mode of action for carcinogenicity is lacking. Various hypotheses have been proposed but remain unproven and there is a lack of consensus on appropriate risk metrics for OTA. As we have shown, the two approaches converge whether one assumes a threshold mode of action (Krogh et al. 1974 pig study and modified uncertainty factors), or a non-threshold mode of action (NTP 1989 rat study), with both leading to a TDI or NCRI in the order of 4ngkgbw−1 per day. The uncertainties regarding OTA's mode of action for the initiation of renal tumours can only be resolved through further studies.
Secondly, there are always difficulties in obtaining estimates of true exposure within a population or in the same individual over a long time. For OTA, these can be due to an insufficient number of samples, lack of representative sampling of all relevant foods, or difficulties in obtaining representative long-term food consumption estimates. Nevertheless, our results have shown that it is important to reduce the risk even if there is some uncertainty inherent in its estimation.
Since OTA has a long half-life and tends to bioaccumulate over time in most individuals, as evidenced by its presence at low levels in all Canadian serum samples analysed for OTA (Scott et al. 1998), we mainly considered chronic exposure.
Though probabilistic assessments (PP) and adjusted partial probabilistic assessments (PD) have been conducted by other groups in the past, to our knowledge, this is the first time that a full probabilistic ‘usual’ assessment (P*P) has been conducted, taking into account both variability in food consumption patterns as well as variability in the concentrations of a contaminant, and we consider this approach as the most appropriate for assessing overall risk from OTA.
Exposure for various age-sex strata
The ‘all person’ probabilistic exposure assessment for OTA showed that OTA is ubiquitous and consumed at low levels on a daily basis by most Canadians. Whereas other exposure assessments for OTA have mainly considered older age groups, we examined all age groups and found that exposure to OTA was highest in young children (aged 1 4 years). OTA exposure for infants was generally lower than for 1-year-olds, except for infants consuming soy formula. No Canadian data are available yet for human breast milk, which, based on limited data from other countries, is likely to be a source of OTA as well (Scott 2005). Above the age of 6 months, infant cereals were a lesser source of OTA exposure. A gradual shift to regular breakfast cereals was observed for older infants, but these foods were not a major source of OTA.
For young children, the main sources of contributing foods are wheat, oats, rice and raisins. While wheat and rice were major contributing foods for older individuals as well, coffee and beer also made important contributions.
Employing a ‘usual’ partial probabilistic approach, similar to our P*D approach, Counil et al. (2006) estimated that the mean and 95th percentile exposure to OTA for the adult French population were 1.69 and 3.28 ng OTA kg bw−1 per day, respectively. Her results are similar to our P*D results of 1.65 and 2.53 ng OTA kg bw−1 for 31–50-year-old males, respectively. Their data set, however, did not include children under the age of 15.
Compared with Canada, exposure results for the Swedish population, based on deterministic (DconDoccor DD) estimates using mean consumption and occurrence values, were lower, with exposures of 1.2ng OTA kg bw−1 per day for children aged 7–14 years and 0.7 ng OTA kg bw−1 per day for adults (Thuvander et al. 2001). Based on deterministic estimates, ‘all person’ exposures for adults for most European Union countries ranged from 1.09 to 1.71 ng OTA kgbw−1, whereas estimates for the UK, Portugal, and Greece were below this, ranging from 0.23 to 0.81 ng OTA kg bw−1 (SCOOP 2002). In Germany and the UK, exposure estimates for young children were almost three-fold higher than for adults, in line with the results reported here. The SCOOP report indicated that since most European Union countries did not provide information on all food products potentially affected by OTA contamination, the total dietary intake by country could be underestimated (SCOOP 2002).
Previous deterministic (DD) estimates of exposure to OTA in Canada, stratified by age groups, and based only on limited grain, breakfast cereal, and pork data showed ‘all person’ estimates of 1.5 and 1.1 ng OTA kg bw−1 for 1–4- and 12–19-year-olds, respectively (Kuiper-Goodman et al. 1993). Those ‘all person’ estimates were again lower than the results for the much larger probabilistic data set reported here, which included many more foods.
For comparison, OTA blood plasma samples for adults (aged 19–68 years) collected from 16 ‘city locations’ in Canada had an overall mean OTA concentration of 0.88 (range = 0.29–2.37) ng OTA ml−1 serum (Scott et al. 1998). Mean calculated exposures of 1.22 and 1.74 ng OTA kg bw−1, respectively, were derived from this value using either the relationship of dietary exposure = 1.34* Cp (where Cp is plasma concentration) (Hagelberg et al. 1989) based on inulin clearance, or the relationship of dietary exposure = 1.97* Cp (Schlatter et al. 1996) based on clearance of radio-labelled OTA in one person. The higher value of 1.74 ng OTA kg bw−1 corresponds well with our current OTA exposure data. Gilbert et al. (2001) found OTA serum concentrations from adult control subjects in the UK to be similar to those reported for Canada (Scott et al. 1998), and slightly higher when these volunteers were participating in a duplicate diet study. Estimated dietary exposure levels were 1.46 and 2.15 ng OTA kg bw−1, using the same two respective calculation methods shown above, placing them in the same range as our study. However, the analysis of the actual duplicate diet indicated an exposure of 0.94 ng OTA kg bw−1, thus lower than indicated above. It is possible that this lower-than-expected value was due to the 30-day duplicate diet study not truly reflecting all the various foods that may contain OTA. Furthermore, the lack of a close agreement between individual plasma levels and the amount of OTA consumed from the diet by the volunteers may be attributed to the long half life of OTA in humans and the steady-state serum concentrations of OTA, which would not change significantly when such a diet contains OTA at normal concentration levels.
Impact of different processing factors (PF) on OTA exposure
Currently, there are no definitive studies that can be used to derive processing factors for OTA in naturally contaminated wheat for commercial milling of grain to flour and products, such as bran. While Scudamore et al. (2003) addressed the effect of milling on OTA levels, it was felt that their data could not be applied to general commercial milling, as their results were based on wheat grain purposely inoculated with Penicillium verrucosum, yielding ochratoxin levels in bran that were much higher than those typically encountered. As shown in results, the uncertainties regarding an appropriate processing factor were thus addressed by using two scenarios: a higher processing factor of 0.82 or a lower processing factor of 0.64 for hard and soft wheat, together with actual pasta occurrence data. The second scenario resulted in approximately 16–19% lower mean exposure estimates for those less than 30 years of age. These two sets of estimates were seen as delimiting the window of uncertainty attributed to processing factors; true exposure falls somewhere between these two bounds. Moreover, the processing factor may be a dynamic variable that changes with the degree of infection in the grain kernel.
Comparing ‘all person’ OTA exposure with that of regular commodity eaters (tRCE)
Although ‘all person’ exposure could be considered to reflect typical day-to-day background exposure, we showed that it was underestimating total OTA exposure for regular (or habitual) eaters of specific commodities. Thus, even more useful for characterizing risk were the higher estimates of total OTA exposures for such regular consumers (tRCEcom) compared with background ‘all person’ exposure (∑APall com, Table 7).
Thus, for young children, higher total OTA exposure (tRCEcom) was seen for regular consumers of hot oatmeal and raisins. Although soy formula had low concentrations of OTA, infants drinking soy formula tend to consume this as their main food source for the first 6 months of life, resulting in mean exposures exceeding 4 ng OTA kg bw−1.
For adults, regular consumers of pasta, rice, raisins, coffee, and wine did not have much higher exposure (tRCEcom) than the average Canadian (∑APall com). The portion of exposure obtained from coffee, for both the average Canadian (APcoffee = about 0.14 ng OTA kg bw−1) and regular coffee drinkers (aRCEcoffee = about 0.22 ng OTA kg bw−1) were in the range of those reported for Europe (SCOOP 2002) where ‘all person’ coffee estimates ranged from 0.06 ng OTA kg bw−1 in Italy to 0.42 ng OTA kg bw−1in Finland. Coffee was reported to have contributed 9% to the mean European total OTA exposure, the same as seen in our exposure assessment.
It is known that wines from Southern Europe have a higher incidence and higher concentrations of OTA compared with those from Northern Europe (SCOOP 2002). Similarly, occurrence data for wines available in Canada varied depending on the country of origin. Canadian ‘all person’ OTA exposure from wine for 31 50-year-old females was 0.02 ng OTA kg bw−1 compared with 0.15 ng OTA kg bw−1 for regular consumers of wine. These estimates fell in the lower ranges of estimates reported for Europe (SCOOP 2002). Since wine is consumed less frequently in North America than in Europe and OTA levels are typically low in oft-consumed domestic wine, wine in Canada contributes less than 1% to total ‘all person’ OTA exposure and is included in the ‘other’ category in Figure 3. In contrast, in Europe wine was reported to have contributed 10% of the mean total OTA exposure (SCOOP 2002).
Risk characterization
The ‘usual’ full probabilistic (P*P) exposure assessment provided the best overall approach for comparison with available risk metrics and assessing the impact of introducing MLs. Here, the tails of the distribution gave a better understanding of the risk at higher percentiles of exposure. While this assessment focused on risks for various age groups, the impact of socio-economic factors or geography could also be examined.
For the MOE estimates, we have typically used a point estimate such as the TD05 (19.6 µg OTA kg bw−1 per day) rather than its lower confidence estimate, the TDL10. The latter value tends to be statistically less robust. MOE estimates were useful as an estimator of risk for various age groups, for percentiles of ‘all person’ exposure, and for regular eaters of specific commodities (RCEs). Such estimates bypass the need for applying uncertainty factors to the experimental risk metric, regardless of the endpoint used. Yet data quality and concerns about the significance of the endpoint should influence what is an acceptable ratio. For mean ‘all person’ exposure in 1-year-olds, MOEs indicating a high priority risk (<5000) became slightly greater than 5000 when a less conservative processing factor together with substitution of lower pasta occurrence data were modelled (Table 9a). On the other hand, 90th percentile MOE ratios for young children all indicated a high priority risk, regardless of the processing factor used. Furthermore, for 1-year-olds, most of the MOEs for regular consumers of specific commodities (Table 9a), with the exception of hot oatmeal, were greater than the MOEs for the ‘all person’ 90th percentiles, indicating that this higher percentile can be useful as a consideration of an upper bound on risk in a market economy. Thus examining risk for regular consumers helps in deciding on priorities for risk management.
Our approach of determining the frequency (number of iterations) and magnitude of excursions above the NCRI was another way of examining risk, and again the younger age groups were at higher risk (see section on Risk Characterization). The long-term health implications of the higher exposures to OTA for young children are not known and require further investigation.
Uncertainties in the exposure estimates
In undertaking this exposure assessment, which involved multiple food commodities, recipe sets, and occurrence data over a number of years, we encountered many uncertainties, which we attempted to resolve within the limits of the available data.
Although we did have OTA occurrence data for most of the major foods known to be susceptible to OTA contamination, for some commodities the data set was considered weak due to small sample size (e.g. rice, beer), while for others, the data set was excluded from the assessment due to very small sample size (e.g. corn, rye) or lack of data (e.g. human breast milk, cocoa products, spices). In the case of spices, food consumption is small and there may be little impact on overall exposure. Future sampling and inclusion of these other commodities could slightly change the overall estimates of exposure as well as the relative contribution of various commodities to total exposure.
Most commodities were sampled randomly by Health Canada over several years, and grain data from the CGC represented about 10 years of data and a large sample size. While CGC data provided information on OTA occurrence in grain intended for export, rather than domestic grain, it was assumed, after analysing distribution patterns of various grain classes in domestic versus export grain, that the occurrence data are likely representative of the levels of OTA in food on the Canadian market.
To deal with the uncertainty regarding the limit of detection, we developed an imputing program for nondetects (censored data) similar to Bakker and Pieters (2002), and modified it to accommodate changes in detection limits over time. This approach allowed for the development of simulated distributions of censored occurrence data. Means derived from such overall distributions were considered more appropriate than those using 1/2LOD/LOQ for censored data, especially when the number of positives is small.
CONCLUSION
Currently, the extensive data presented herein are the most appropriate for the risk assessment of OTA. The approach used is suitable not only for Canada, but probably for many other jurisdictions as well. We have presented the results for all person ‘usual’ exposure, as well as the higher exposure of subpopulations known to consume foods such as coffee, beer, oats, raisins, and soy formula on a regular basis. Such comparisons are important when considering risk management options for specific foods. The frequency and magnitude of excursions above the NCRI, modelled using the iterations of the Monte Carlo simulation, and MOE estimates for all age-sex strata, for both ‘all persons’ (AP) and regular commodity eaters (RCEs), helped to assess the potential risks from the presence of OTA in foods and to set priorities for risk reduction. The impact of possible risk reduction strategies was assessed through modelling exposure with or without the current European MLs in place. As a result of this probabilistic risk assessment, Health Canada is currently considering upper limits for the presence of OTA in various food commodities, as well as guidelines based on Hazard Analysis of Critical Control Points (HACCP) in food production, to reduce the levels of OTA in the Canadian diet.
ACKNOWLEDGEMENTS
The authors gratefully acknowledge Dr Tom Nowicki of the Canadian Grain Commission for sharing with us unpublished results on OTA; and Mike Walker, Biostatistics, Healthy Environments and Consumer Safety, for deriving the TD05 and BD10. The authors also thank Dr Peter Scott for helpful discussions on processing factors for wheat; and Dr Barbara Petersen and the staff of Exponent for very helpful discussions on the use of the licensed software FARETM. This work was made possible through financial support, in the form of 3-year funding under the Agricultural Policy Framework, from Agriculture Agri-Foods Canada. Part of this work was first presented at the 12th International IUPAC Symposium on Mycotoxins and Phycotoxins, Istanbul, Turkey, 21–25 May 2007. A visiting scientist travel grant from Tubitak made this possible.
References
- Bakker M. Pieters MN. Risk assessment of ochratoxin A in the Netherlands. 2002. Report Number 388802025/2002. RIVM, Bilthoven (the Netherlands).
- Barlow S. Renwick AG. Kleiner J. Bridges JW. Busk L. Dybing E. Edler L. Eisenbrand G. Fink-Gremmels J. Knaap A. Kroes R. Liem D. Müller DJG. Page S. Rolland V. Schlatter J. Tritscher A. Tueting W. Würtzen G. Risk assessment of substances that are both genotoxic and carcinogenic: report of an International Conference organized by EFSA and WHO with support of ILSI Europe. Food Chem Toxicol. 2006;44:1636–1650. doi: 10.1016/j.fct.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Barrett JC. Mechanisms of action of known human carcinogens. In: Vainio H, Magee PN, McGregor DB, McMichael AJ, editors. Mechanisms of carcinogenesis in risk identification. Scientific Publications Number 116. Lyon (France): International Agency for Research on Cancer (IARC); 1992. pp. 115–134. [Google Scholar]
- Bendele AM. Carlton WW. Krogh P. Lillehoj EB. Ochratoxin A carcinogenesis in the (C57BL/6J × C3H)F1mouse. J Natl Canc Inst. 1985;75:733–742. [PubMed] [Google Scholar]
- Brambilla G. Martelli A. Failure of the standard battery of short-term tests in detecting some rodent and human genotoxic carcinogens. Toxicology. 2004;196:1–19. doi: 10.1016/j.tox.2003.11.003. [DOI] [PubMed] [Google Scholar]
- Bullerman LB. Bianchini A. Stability of mycotoxins during food processing. Int J Food Microbiol. 2007;119:140–146. doi: 10.1016/j.ijfoodmicro.2007.07.035. [DOI] [PubMed] [Google Scholar]
- Carriquiry AL. Camañio-Garcia G. Evaluation of dietary intake data using the tolerable upper intake levels. J Nutr. 2006;136:507S–513S. doi: 10.1093/jn/136.2.507S. [DOI] [PubMed] [Google Scholar]
- Castegnaro M. Mohr U. Pfohl-Leszkowicz A. Estève J. Steinmann J. Tillmann T. Michelon J. Bartsch H. Sex- and strain-specific induction of renal tumors by ochratoxin A in rats correlates with DNA adduction. Int J Cancer. 1998;77:70–75. doi: 10.1002/(sici)1097-0215(19980703)77:1<70::aid-ijc12>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Counil É. Verger P. Volatier JL. Handling of contamination variability in exposure assessment: a case study with ochratoxin A. Food Chem Toxicol. 2005;43:1541–1555. doi: 10.1016/j.fct.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Counil E. Verger P. Volatier JL. Fitness-for-purpose of dietary survey duration: a case-study with the assessment of exposure to ochratoxin A. Food Chem Toxicol. 2006;44:499–509. doi: 10.1016/j.fct.2005.08.024. [DOI] [PubMed] [Google Scholar]
- Crump KS. Calculation of benchmark doses from continuous data. Risk Analys. 1995;15:79–89. [Google Scholar]
- Crump KS. Van Landingham C. BENCH_C: a Fortran program to calculate benchmark doses from continuous data. Ruston (LA): ICF Consulting.; 1996. [Google Scholar]
- Dourson ML. Felter SP. Robinson D. Evolution of science-based uncertainty factors in noncancer risk assessment. Regulat Toxicol Pharmacol. 1996;24:108–120. doi: 10.1006/rtph.1996.0116. [DOI] [PubMed] [Google Scholar]
- European Food Safety Authority (EFSA) Opinion of the Scientific Committee on a request from EFSA related to a harmonized approach for risk assessment of substances which are both genotoxic and carcinogenic. EFSA J. 2005;282:1–31. [Google Scholar]
- European Food Safety Authority (EFSA) Opinion of the Scientific Panel on Contaminants in the Food Chain on a request from the Commission related to ochratoxin A in food. EFSA J. 2006;365:1–56. [Google Scholar]
- Faucet V. Pfohl-Leszkowicz A. Dai J. Castegnaro M. Manderville RA. Evidence for covalent DNA adduction by ochratoxin A following chronic exposure to rat and subacute exposure to pig. Chem Res Toxicol. 2004;17:1289–1296. doi: 10.1021/tx049877s. [DOI] [PubMed] [Google Scholar]
- Fink-Gremmels J. Conclusions from the Workshops on ochratoxin A in Food: Recent Developments and Significance, organized by ILSI Europe in Baden (Austria), 29 June 1 July 2005. Food Addit Contam. 22(Suppl. 1):1 5. 2005.
- Galtier P. Alvinerie M. Charpenteau JL. The pharmacokinetic profiles of ochratoxin A in pigs, rabbits and chickens. Food Cosmet Toxicol. 1981;19:735–738. doi: 10.1016/0015-6264(81)90528-9. [DOI] [PubMed] [Google Scholar]
- Gautier J. Richoz J. Welti DH. Markovic J. Gremaud E. Guengerich FP. Turesky RJ. Metabolism of ochratoxin A: absence of formation of genotoxic derivatives by human and rat enzymes. Chem Res Toxicol. 2001;14:34–45. doi: 10.1021/tx000070j. [DOI] [PubMed] [Google Scholar]
- Gay C. Estimation of population distributions of habitual nutrient intake based on a short-run weighted food diary. Br J Nutr. 2000;83:287–293. doi: 10.1017/s0007114500000362. [DOI] [PubMed] [Google Scholar]
- Gibney MJ. van der Voet H. Introduction to the Monte Carlo project and the approach to the validation of conceptual models of dietary exposure to selected food chemicals. Food Addit Contam. 20:S1 S7. 2003. [DOI] [PubMed]
- Gilbert J. Brereton P. MacDonald S. Assessment of dietary exposure to ochratoxin A in the UK using a duplicate diet approach and analysis of urine and plasma samples. Food Addit Contam. 2001;18:1088–1093. doi: 10.1080/02652030110070030. [DOI] [PubMed] [Google Scholar]
- Gross-Steinmeyer K. Weymann J. Hege HG. Metzler M. Metabolism and lack of DNA reactivity of the mycotoxin ochratoxin A in cultured rat and human primary hepatocytes. J Agricult Food Chem. 2002;50:938–945. doi: 10.1021/jf0111817. [DOI] [PubMed] [Google Scholar]
- Guenther PM. Kott PS. Carriquiry AL. Development of an approach for estimating usual nutrient intake distributions at the population level. J Nutr. 1997;127:1106–1112. doi: 10.1093/jn/127.6.1106. [DOI] [PubMed] [Google Scholar]
- Hagelberg S. Hult K. Fuchs R. Toxicokinetics of ochratoxin A in several species and its plasma-binding properties. J Appl Toxicol. 1989;9:91–96. doi: 10.1002/jat.2550090204. [DOI] [PubMed] [Google Scholar]
- Hard GC. Histopathologic evaluation of rat kidney from toxicity and carcinogenicity studies with ochratoxin. 2000. A Expert report submitted to the Joint Expert Committee on Food Additives by the International Life Sciences Institute, Washington, DC.
- Hoffmann K. Boeing H. Dufour A. Volatier JL. Telman J. Virtanen M. Becker W. De Henauw S. Estimating the distribution of usual dietary intake by short-term measurements. Eur J Clin Nutr. 2002;56:S53–s62. doi: 10.1038/sj.ejcn.1601429. [DOI] [PubMed] [Google Scholar]
- Howe RB. THRESH: a computer program to compute a reference dose from quantal animal toxicity data using the benchmark dose method. Ruston (LA): ICF Kaiser Engineers; 1995. [Google Scholar]
- International Agency for Research on Cancer (IARC) Monographs on the Evaluation of Carcinogenic Risks to Humans Number 56. Lyon (France): IARC Press; 1993. Some naturally occurring substances: Food items and constituents, heterocyclic aromatic amines and mycotoxins; pp. 489–521. [Google Scholar]
- Joint Expert Committee on Food Additives (JECFA) Ochratoxin A. 1991. p. 365.p. 417. Joint FAO/WHO Expert Committee of Food Additives. 37th Meeting, 10–12 June 1990. Geneva (Switzerland): World Health Organization (WHO)
- Joint Expert Committee on Food Additives (JECFA) Ochratoxin A. 1995. Joint FAO/WHO Expert Committee of Food Additives. 44th Meeting, Technical Report Series Number 859. Geneva (Switzerland): World Health Organization (WHO).
- Joint Expert Committee on Food Additives (JECFA) Ochratoxin A. In: Safety evaluation of certain mycotoxins in food. 2001. Joint FAO/WHO Expert Committee of Food Additives, 56th Meeting, February 6 15, 2001. Geneva (Switzerland): WHO Food Additives Series No. 47. IPCS-WHO, Geneva. pp 281 415.
- Joint Expert Committee on Food Additives (JECFA) Summary and conclusions. 2007. Joint FAO/WHO Expert Committee of Food Additives. 68th Meeting, 19 28 June 2007. Geneva (Switzerland): World Health Organization (WHO).
- Kamp HG. Eisenbrand G. Janzowski C. Kiossev J. Latendresse JR. Schlatter J. Turesky RJ. ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Mol Nutr Food Res. 2005;49:1160–1167. doi: 10.1002/mnfr.200500124. [DOI] [PubMed] [Google Scholar]
- Kanisawa M. Suzuki S. Induction of renal and hepatic tumors in mice by ochratoxin A, a mycotoxin. Gann. 1978;69:599–600. [PubMed] [Google Scholar]
- Karpinski K. Nargundkar M. Technical Report No. 1992. E451311-001 (unpublished). Bureau of Biostatistics and Computer Applications, Food Directorate, Health Canada, Ottawa (Canada).
- Kempthorne O. The design and analysis of experiments. New York (UK): Wiley; 1952. pp. 271–275. [Google Scholar]
- Krogh P. Axelsen NH. Elling F. Gyrd-Hansen N. Hald B. Hyldgaard-Jensen J. Larsen AE. Madsen A. Mortensen HP. Moller T. Petersen OK. Ravnskov U. Rostgaard M. Aalund O. Experimental porcine nephropathy. Changes of renal function and structure induced by ochratoxin A contaminated feed. Acta Pathologica Microbiologica Immunologica Scandinavica Suppl. 1974;246:1–21. [PubMed] [Google Scholar]
- Krogh P. Elling F. Friis C. Hald B. Larsen AE. Lillehoj EB. Madsen A. Mortensen HP. Rasmussen F. Ravnskov U. Porcine nephropathy induced by long-term ingestion of ochratoxin A. Vet Pathol. 1979;16:466–475. doi: 10.1177/030098587901600410. [DOI] [PubMed] [Google Scholar]
- Krogh P. Gyrd-Hansen N. Hald B. Larsen S. Nielsen JP. Smith M. Ivanoff C. Meisner H. Renal enzyme activities in experimental ochratoxin A-induced porcine nephropathy: Diagnostic potential of phosphoenolpyruvate carboxykinase and gamma-glutamyl transpeptidase activity. J Toxicol Environ Hlth. 1988;23:1–14. doi: 10.1080/15287398809531092. [DOI] [PubMed] [Google Scholar]
- Kuiper-Goodman T. Risk assessment of ochratoxin A: an update. Food Addit Contam. 1996;13(Suppl. 1):53–57. [PubMed] [Google Scholar]
- Kuiper-Goodman T. Risk assessment and risk management of mycotoxins in food. In: Magan N, Olsen M, editors. Mycotoxins in food. Detection and control. Boca Raton (FL): CRC Press LLC; 2004. p. 3.p. 31. [Google Scholar]
- Kuiper-Goodman T. Ominski K. Marquardt RR. Malcolm S. McMullen E. Lombaert GA. Morton T. Estimating human exposure to ochratoxin A in Canada. In: Creppy EE, Castegnaro M, Dirheimer G, editors. Human ochratoxicosis and its pathologies. Vol. 231. INSERM, Montrouge (France): John Libbey Eurotext; 1993. pp. 167–174. [Google Scholar]
- Kuiper-Goodman T. Scott PM. Risk assessment of the mycotoxin ochratoxin A. Biomed Environ Sci. 1989;2:179–248. [PubMed] [Google Scholar]
- Lambe J. The use of food consumption data in assessments of exposure to food chemicals including the application of probabilistic modelling. Proc. Nutrit. Soc. 2002;61:11–18. doi: 10.1079/pns2001125. [DOI] [PubMed] [Google Scholar]
- Lock EA. Hard GC. Chemically induced renal tubule tumors in the laboratory rat and mouse: review of the NCI/NTP database and categorization of renal carcinogens based on mechanistic information. Crit Rev Toxicol. 2004;34:211–299. doi: 10.1080/10408440490265210. [DOI] [PubMed] [Google Scholar]
- Lombaert GA. Pellaers P. Chettiar M. Lavalee D. Scott PM. Lau BPY. Survey of Canadian retail coffees for ochratoxin A. Food Addit Contam. 2002;19:869–877. doi: 10.1080/02652030210145027. [DOI] [PubMed] [Google Scholar]
- Lombaert GA. Pellaers P. Neumann G. Kitchen D. Huzel V. Trelka R. Kotello S. Scott PM. Ochratoxin A in dried vine fruits on the Canadian retail market. Food Addit Contam. 2004;21:578–585. doi: 10.1080/02652030410001687681. [DOI] [PubMed] [Google Scholar]
- Lombaert GA. Pellaers P. Roscoe V. Mankotia M. Neil R. Scott PM. Mycotoxins in infant cereal foods from the Canadian retail market. Food Addit Contam. 2003;20:494–504. doi: 10.1080/0265203031000094645. [DOI] [PubMed] [Google Scholar]
- Mally A. Pepe G. Ravoori S. Fiore M. Gupta RC. Dekant W. Mosesso P. Ochratoxin A causes DNA damage and cytogenetic effects but no DNA adducts in rats. Chem Res Toxicol. 2005b;18:1253–1261. doi: 10.1021/tx049650x. [DOI] [PubMed] [Google Scholar]
- Mally A. Völkel W. Amberg A. Kurz M. Wanek P. Eder E. Hard G. Dekant W. Functional, biochemical, and pathological effects of repeated oral administration of ochratoxin A to rats. Chem Res Toxicol. 2005a;18:1242–1252. doi: 10.1021/tx049651p. [DOI] [PubMed] [Google Scholar]
- Mally A. Zepnik H. Wanek P. Eder E. Dingley K. Ihmels H. Völkel W. Dekant W. Ochratoxin A: lack of formation of covalent DNA adducts. Chem Res Toxicol. 2004;17:234–242. doi: 10.1021/tx034188m. [DOI] [PubMed] [Google Scholar]
- Manderville RA. A case for the genotoxicity of ochratoxin A by bioactivation and covalent DNA adduction. Chem Res Toxicol. 2005;18:1091–1097. doi: 10.1021/tx050070p. [DOI] [PubMed] [Google Scholar]
- Mantle P. Kulinskaya E. Nestler S. Renal tumourigenesis in male rats in response to chronic dietary ochratoxin A. Food Addit Contam. 2005;22(Suppl. 1):58–64. doi: 10.1080/02652030500358431. [DOI] [PubMed] [Google Scholar]
- Munro IC. Moodie CA. Kuiper-Goodman T. Scott PM. Grice HC. Toxicologic changes in rats fed graded dietary levels of ochratoxin A. Toxicol Appl Pharmacol. 1974;28:180–184. doi: 10.1016/0041-008x(74)90003-9. [DOI] [PubMed] [Google Scholar]
- National Research Council (NRC) Nutrient adequacy: assessment using food consumption surveys. Washington (DC): National Academies Press; 1986. National Research Council Subcommittee on Criteria for Dietary Evaluation. [PubMed] [Google Scholar]
- National Toxicology Program (NTP) Toxicology and carcinogenesis studies of ochratoxin A (CAS No. 303-47-9) in F344/N rats (gavage studies) Research Triangle Park (NC): Department of Health and Human Services; 1989. Technical Report Number 358. [PubMed] [Google Scholar]
- Ng W. Mankotia M. Pantazopoulos P. Neil RJ. Scott PM. Ochratoxin A in wine and grape juice sold in Canada. Food Addit Contam. 2004;21:971–981. doi: 10.1080/02652030400000653. [DOI] [PubMed] [Google Scholar]
- Ng W. Mankotia M. Pantazopoulos P. Neil RJ. Scott PM. Survey of dry pasta for ochratoxin A. J Food Protect. 2009;72:890–893. doi: 10.4315/0362-028x-72.4.890. [DOI] [PubMed] [Google Scholar]
- Nusser SM. Carriquiry AL. Dodd KW. Fuller WA. A semi-parametric transformation approach to estimating usual daily intake distributions. J Am Stat Assoc. 1996;91:1440–1449. [Google Scholar]
- O'Brien E. Dietrich DR. Ochratoxin A: The continuing enigma. Crit Rev Toxicol. 2005;35:33–60. doi: 10.1080/10408440590905948. [DOI] [PubMed] [Google Scholar]
- O'Brien J. Renwick AG. Constable A. Dybing E. Müller DJG. Schlatter J. Slob W. Tueting W. van Benthem J. Williams GM. Wolfreys A. Approaches to the risk assessment of genotoxic carcinogens in food: a critical appraisal. Food Chem Toxicol. 2006;44:1613–1635. doi: 10.1016/j.fct.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Obrecht-Pflumio S. Chassat T. Dirheimer G. Marzin D. Genotoxicity of ochratoxin A by Salmonella mutagenicity test after bioactivation by mouse kidney microsomes. Mutat Res. 1999;446:95–102. doi: 10.1016/s1383-5718(99)00152-7. [DOI] [PubMed] [Google Scholar]
- Olsen M. Thorup I. Knudsen I. Larsen JJ. Hald B. Olsen J. Health evaluation of ochratoxin A in food products Number 545. Copenhagen (Denmark): Nordic Council of Ministers; 1991. [Google Scholar]
- Petersen BJ. Probabilistic modelling: theory and practice. Food Addit Contam. 2000;17:591–599. doi: 10.1080/026520300412500. [DOI] [PubMed] [Google Scholar]
- Petzinger E. Ziegler K. Ochratoxin A from a toxicological perspective. J Vet Pharmacol Ther. 2000;23:91–98. doi: 10.1046/j.1365-2885.2000.00244.x. [DOI] [PubMed] [Google Scholar]
- Pfohl-Leszkowicz A. Castegnaro M. Further arguments in favour of direct covalent binding of ochratoxin A (OTA) after metabolic biotransformation. Food Addit Contam. 2005;22(Suppl. 1):75–87. doi: 10.1080/02652030500309400. [DOI] [PubMed] [Google Scholar]
- Pfohl-Leszkowicz A. Manderville RA. Ochratoxin A: an overview on toxicity and carcinogenicity in animals and humans. Mol Nutr Food Res. 2007;51:61–99. doi: 10.1002/mnfr.200600137. [DOI] [PubMed] [Google Scholar]
- Pfohl-Leszkowicz A. Tozlovanu M. Manderville R. Peraica M. Castegnaro M. Stefanovic V. New molecular and field evidences for the implication of mycotoxins but not aristolochic acid in human nephropathy and urinary tract tumors. Mol Nutr Food Res. 2007;51:1131–1146. doi: 10.1002/mnfr.200700045. [DOI] [PubMed] [Google Scholar]
- Rached E. Hard GC. Blumbach K. Weber K. Draheim R. Lutz WK. Özden S. Steger U. Dekant W. Mally A. Ochratoxin A: 13-week oral toxicity and cell proliferation in male F344/N rats. Toxicol Sci. 2007;97:288–298. doi: 10.1093/toxsci/kfm042. [DOI] [PubMed] [Google Scholar]
- Roscoe V. Lombaert GA. Huzel V. Neumann G. Melietio J. Kitchen D. Kotello S. Krakalovich T. Trelka R. Scott PM. Mycotoxins in breakfast cereals from the Canadian retail market: a 3-year survey. Food Addit Contam. 2008;25:347–355. doi: 10.1080/02652030701551826. [DOI] [PubMed] [Google Scholar]
- Rydén E. Ekström CHL. Hellmér L. Bolcsfoldi G. Comparison of the sensitivities of Salmonella typhimurium strains TA102 and TA2638A to 16 mutagens. Mutagenesis. 2000;15:495–502. doi: 10.1093/mutage/15.6.495. [DOI] [PubMed] [Google Scholar]
- Schlatter C. Studer-Rohr J. Rásonyi T. Carcinogenicity and kinetic aspects of ochratoxin A. Food Addit Contam. 1996;13(Suppl.):43–44. [PubMed] [Google Scholar]
- SCOOP. Assessment of dietary intake of ochratoxin A by the population of EU Member States. 2002. Report of Tasks for Scientific Cooperation Number 3.2.7. (Miraglia M and Brera C, Coordinators), Rome (Italy)/ISS, National Institute of Health.
- Scott PM. Biomarkers of human exposure to ochratoxin A. Food Addit Contam. 2005;22(Suppl. 1):99–107. doi: 10.1080/02652030500410315. [DOI] [PubMed] [Google Scholar]
- Scott PM. Kanhere SR. Determination of ochratoxin A in beer. Food Addit Contam. 1995;12:591–598. doi: 10.1080/02652039509374347. [DOI] [PubMed] [Google Scholar]
- Scott PM. Kanhere SR. Lau BP. Lewis DA. Hayward S. Ryan JJ. Kuiper-Goodman T. Survey of Canadian human blood plasma for ochratoxin A. Food Addit Contam. 1998;15:555–562. doi: 10.1080/02652039809374681. [DOI] [PubMed] [Google Scholar]
- Scudamore KA. Banks J. MacDonald SJ. Fate of ochratoxin A in the processing of whole wheat grains during milling and bread production. Food Addit Contam. 2003;20:1153–63. doi: 10.1080/02652030310001605979. [DOI] [PubMed] [Google Scholar]
- Slob W. Modeling long-term exposure of the whole population to chemicals in food. Risk Analysis. 1993;16:195–200. doi: 10.1111/j.1539-6924.1993.tb00011.x. [DOI] [PubMed] [Google Scholar]
- Stefanovic V. Toncheva D. Atanasova S. Polenakovic M. Etiology of Balkan Endemic Nephropathy and associated urothelial cancer. Am J Nephrol. 2006;26:1–11. doi: 10.1159/000090705. [DOI] [PubMed] [Google Scholar]
- Tennant R. Non-genotoxic chemical carcinogens: Evidence for multiple mechanisms. In: Cockburn A, Smith L, editors. Non-genotoxic carcinogenesis. Ernst Schering Research Foundation Workshop Number 10. Berlin (Germany): Springer; 1994. pp. 1–16. [Google Scholar]
- Thuvander A. Möller T. Barbieri HE. Jansson A. Salomonsson AC. Olsen M. Dietary intake of some important mycotoxins by the Swedish population. Food Addit Contam. 2001;18:696–706. doi: 10.1080/02652030121353. [DOI] [PubMed] [Google Scholar]
- Tozlovanu M. Faucet-Marquis V. Pfohl-Leszkowicz A. Manderville RA. Genotoxicity of the hydroquinone metabolite of ochratoxin A: structure activity relationships for covalent DNA adduction. Chem Res Toxicol. 2006;19:1241–1247. doi: 10.1021/tx060138g. [DOI] [PubMed] [Google Scholar]
- Tressou J. Crépet A. Bertail P. Feinberg MH. Leblanc JCh. Probabilistic exposure assessment to food chemicals based on extreme value theory. Application to heavy metals from fish and sea products. Food Chem Toxicol. 2004a;42:1349–1358. doi: 10.1016/j.fct.2004.03.016. [DOI] [PubMed] [Google Scholar]
- Tressou J. Leblanc JCh. Feinberg M. Bertail P. Statistical methodology to evaluate food exposure to a contaminant and influence of sanitary limits: application to ochratoxin A. Regulat Toxicol Pharmacol. 2004b;40:252–263. doi: 10.1016/j.yrtph.2004.07.005. [DOI] [PubMed] [Google Scholar]
- Turesky RJ. Perspective: ochratoxin A is not a genotoxic carcinogen. Chem Res Toxicol. 2005;18:1082–1090. doi: 10.1021/tx050076e. [DOI] [PubMed] [Google Scholar]
- US Department of Agriculture (USDA) CSFII data set and documentation: the 1994 1996, 1998 continuing surveys of food intakes by individuals. Beltsville (MD): Food Surveys Research Group, Beltsville Human Nutrition Research Center, Agricultural Research Service; 2000. [Google Scholar]
- US Environmental Protection Agency (USEPA) Guidelines for carcinogen risk assessment. Washington (DC): USEPA; 2005. EPA/630/P-03/001F. [Google Scholar]
- World Health Organization/International Programme on Chemical Safety (WHO-IPCS) Environmental Health Criteria Number 210. Geneva (Switzerland): WHO; 1999. Principles for the assessment of risks to human health from exposure to chemicals. [Google Scholar]
- Zepnik H. Pähler A. Schauer U. Dekant W. Ochratoxin A-induced tumor formation: is there a role of reactive ochratoxin A metabolites? Toxicol Sci. 2001;59:59–67. doi: 10.1093/toxsci/59.1.59. [DOI] [PubMed] [Google Scholar]