

Abstract
Correlated presynaptic and postsynaptic activity is the key factor in inducing Hebbian plasticity and memory. However, little is known about the physiological events that could mediate such coordination. Correlated cholinergic input induces spike timing-dependent plasticity-like hippocampal synaptic plasticity. Cholinergic receptors are localized to both presynaptic and postsynaptic glutamatergic sites and thus have the potential to coordinate presynaptic and postsynaptic activity to induce plasticity. By directly monitoring presynaptic and postsynaptic activities with genetically encoded calcium indicators in mouse septohippocampal cocultures, we found interactive but independent presynaptic and postsynaptic modulations in the cholinergic-dependent synaptic plasticity. Neither presynaptic nor postsynaptic modulation alone is sufficient, but instead a coordinated modulation at both sites is required to induce the plasticity. Therefore, we propose that correlated cholinergic input can coordinate presynaptic and postsynaptic activities to induce timing-dependent synaptic plasticity, providing a novel mechanism by which neuromodulators precisely modulate network activity and plasticity with high efficiency and temporal precision.
Introduction
Synaptic plasticity has been widely studied and deemed to be a cellular mechanism underlying learning and memory (Bliss and Collingridge, 1993; Martin et al., 2000; Dan and Poo, 2006; Neves et al., 2008). Recent studies have revealed that synaptic plasticity can be greatly facilitated when presynaptic and postsynaptic activities are precisely coordinated, as evidenced in spike timing-dependent plasticity (STDP) (Bell et al., 1997; Magee and Johnston, 1997; Markram et al., 1997; Zhang et al., 1998; Egger et al., 1999; Feldman, 2000), and thus provides a potential mechanism to induce plasticity in a physiological context as a part (or the result) of continuous neural activities. Understanding the mechanisms that could coordinate presynaptic and postsynaptic activities is critical not only to our understanding of synaptic plasticity, but also to the nature of information processing and integration in general underlying higher brain functions.
Acetylcholine (ACh) (Jerusalinsky et al., 1997; Power et al., 2003; Dani and Bertrand, 2007; Kenney and Gould, 2008), as well as other modulatory neurotransmitters (Bailey et al., 2000; Reis et al., 2009), has long been suggested to be heavily involved in synaptic plasticity and various higher brain functions. ACh receptors are distributed to both presynaptic and postsynaptic sites of glutamatergic and GABAergic synapses (Levey et al., 1991; Fabian-Fine et al., 2001; Volpicelli and Levey, 2004; Dani and Bertrand, 2007; Drever et al., 2011), providing the potential capability of coordinating presynaptic and postsynaptic activities. Recent studies have suggested the importance of the timing of applied ACh in modulating high-frequency stimulation (HFS)-induced hippocampal synaptic plasticity (Ji et al., 2001; Ge and Dani, 2005). Moreover, we have recently demonstrated that the activation of endogenous cholinergic inputs can also induce timing-dependent synaptic plasticity in the hippocampus, with a temporal precision of milliseconds (Gu and Yakel, 2011); this provides an ideal model to study information integration and plasticity induction that involves neuromodulators. In the meantime, newly developed genetically encoded calcium indicators (GECIs) (Tian et al., 2012) have provided the ability to directly monitor neuronal activities at either the synapse or the network level. Differently colored (Tian et al., 2009; Zhao et al., 2011) GECIs have provided excellent tools to monitor presynaptic and postsynaptic components at the same time, greatly facilitating our understanding of the coordinated activities that mediate synaptic plasticity and other neuronal functions.
In this study, we have used a septohippocampal coculture system (Gähwiler and Hefti, 1984; Gähwiler and Brown, 1985; Rimvall et al., 1985; Fischer et al., 1999) instead of acute hippocampal slices that were used previously for two reasons: first, the ease in expressing GECIs to restricted hippocampal subregions for presynaptic and postsynaptic activity observation, and second, to be able to express the α7 nicotinic ACh receptor (nAChR) subtype to either presynaptic or postsynaptic sites (or both) in α7 nAChR knock-out (KO) slices. The latter has been our main tool to dissect out the roles of presynaptic and postsynaptic α7 nAChRs in inducing the α7 nAChR-dependent long-term potentiation (LTP) and short-term depression (STD), which has helped us reveal the individual contribution of presynaptic and postsynaptic modulation in synaptic plasticity formation.
Materials and Methods
Animals and chemicals.
α7 nAChR knock-out mice and choline acetyltransferase (ChAT)-Cre transgenic mice (of either sex) were originally purchased from Jackson Laboratory and bred at the National Institute of Environmental Health Sciences (NIEHS). Mice were tattooed and genotyped at day 4 and used for slice culture from days 8 to 12. All procedures were approved and performed in compliance with NIEHS/NIH Humane Care and Use of Animals in Research protocols. Unless otherwise indicated, general chemicals were obtained from Sigma, and culture media were from Sigma or Invitrogen.
Coculture slice preparation.
Slice cultures were prepared as described by Bastrikova et al. (2008), which was adapted from Stoppini et al. (1991). Brain slices of 300 μm were cut with a vibratome (Leica, VT1000S). The detachable parts of the vibratome and surgery instruments for dissecting brains were all autoclaved. Briefly, mice (8–12 days old) were anesthetized with isoflurane and decapitated. Brains were quickly removed into ice-cold cutting medium (MEM supplemented with 25 mm HEPES, 10 mm Tris-base, 10 mm glucose, and 3 mm MgCl2, pH 7.2). Horizontal hippocampal slices and coronal septum slices were cut in cutting medium. The hippocampus and medial septum tissues were then dissected out from the slices and placed next to each other onto the Transwell membrane inserts (Corning) that were prefilled with 1.2 ml of culture medium, which was prepared as a 2:1 mixture of basal medium Eagle (Sigma) and Earle's balanced salts solution (Sigma) and supplemented with (in mm) 20 NaCl, 5 NaHCO3, 0.2 CaCl2, 1.7 MgSO4 48 glucose, 26.7 HEPES, 10 ml/L penicillin-streptomycin (Invitrogen), insulin (1.32 mg/L) (Sigma), and 5% horse serum (Invitrogen) with pH 7.2. The slices were stored in a CO2 incubator at 34°C and fed twice a week with a half change of media. For the α7 nAChR knock-out cocultures, the septal tissue that provides the cholinergic inputs was from wild-type littermates, and only the hippocampi were from α7 nAChR knock-out mice.
Virus infection.
All of the plasmids were subcloned to an adeno-associated virus (AAV) vector with a synapsin promoter for neuronal expression. All of the viruses were packaged with serotype 9 helper at the virus core facility at NIEHS. The AAV vector with synapsin promoter (Addgene plasmid no. 26972) and ChR2 (Addgene plasmid no. 20297) plasmids were from Karl Deisseroth at Stanford University (Stanford, CA). The rat α7 nAChR plasmid was obtained from James Patrick at the Baylor College of Medicine (Houston, TX) (Séguéla et al., 1993). GCaMP3 (Addgene plasmid no. 22692) was from Loren Looger at Janelia Farm Research Campus (Ashburn, VA). R-GECO1 (Addgene plasmid no. 32444) was from Robert Campbell at the University of Alberta (Edmonton, Alberta, Canada). AAV serotype 9 helper plasmid was from James Wilson at University of Pennsylvania (Philadelphia, PA). The day after culturing, viruses (5 nl) were microinjected to the desired areas with a Drummond “Nanoject” (Drummond Scientific). Experiments were done 7 days after virus infection to allow protein expression and cholinergic innervation into the hippocampus.
Pairing two input pathways by electrical stimulation and an optogenetic approach.
Cocultured slices were cut out with the attached Transwell membrane from the inserts and put into a submerged chamber continuously perfused with 95%O2/5%CO2 balanced ACSF (in mm: 122 NaCl, 2.5 KCl, 1.3 MgCl2, 2 CaCl2, 1.2 NaH2PO4, 25 NaHCO3, 25 glucose) at a rate of 2 ml/min. Whole-cell patch clamp was performed under guidance of infrared differential interference contrast optics using an Axopatch 200B patch amplifier (Molecular Devices) with a glass pipette filled with an internal solution containing (in mm) 120 potassium gluconate, 2 NaCl, 5 MgATP, 0.3 Na2GTP, 20 KCl, 10 HEPES, 1 EGTA, and 11.3 d-glucose with pH ∼7.2–7.3 and osmolarity of ∼270–280 mOsm. Data were digitized with Digidata 1322A, collected with Clampex, and analyzed with Clampfit. Excitatory postsynaptic currents (EPSCs) were recorded under voltage-clamp at −60 mV.
Evoked EPSCs were recorded from hippocampal CA1 pyramidal neurons by electrically stimulating the Schaffer collateral (SC) pathway. The stimulation intensity was adjusted to evoke a postsynaptic current of about 50–100 pA in amplitude, and the intensity was usually around 20–100 μA for 0.1 ms. Light-sensitive ChR2 was selectively expressed in cholinergic neurons by microinjecting a Cre-inducible AAV containing a double-floxed inverted ChR2 (fused with mCherry for visualization) to the septal tissue from ChAT-Cre transgenic mice. Extensive cholinergic innervations to the hippocampus can be observed 7 days later. Cholinergic terminals expressing ChR2 and mCherry were visualized with 543 nm light and activated in the stratum oriens (SO) layer above the CA1 pyramidal neuron under patch-clamp recording with 488 nm light (20 ms) generated by the Zeiss LSM 510 NLO META system. The exposure time, intensity, and area were controlled by the LSM 510 system. The intensity of the light used to activate ChR2 in processes was usually 7.5 mW for 20 ms. The cholinergic response (nAChR-mediated currents in CA1 neurons) was usually induced at around 20 ms after initiating the light exposure (Gu and Yakel, 2011). To achieve a 100 ms interval for cholinergic inputs before SC inputs, the light exposure was set at 120 ms before SC input in Clampex. To achieve a 10 ms interval for cholinergic inputs after SC inputs, the light exposure was set at 10 ms before SC input to take into account the 20 ms delay of cholinergic activation by light exposure.
Calcium imaging.
Cocultured slices with the attached membrane were cut out and put into a chamber that was continuously perfused with 95%O2/5%CO2 balanced ACSF. Calcium imaging was done with a customized spinning disc confocal microscopy. The spinning disk (PerkinElmer) was installed onto an Olympus microscope with a 60× objective lens (numerical aperture, 0.9). Lasers (Melles Griot) of 488 and 568 nm were used to excite GCaMP3 and R-GECO1, respectively. Emissions were filtered through bandpass filters 500–550 and 585–630 nm, respectively. The images were collected at 5 Hz with an iXon3 EMCCD camera (Andor) controlled by Andor iQ imaging software, which was connected with a Sutter Lambda 10 (Sutter Instrument) to select between different channels, a Neos Acousto-optic tunable filter to adjust the laser intensity, and a kinetic trigger device (Andor) to control the image timing. Images were analyzed with NIH ImageJ.
Choline-induced α7 nAChR currents in cultured hippocampal slices.
α7 nAChR-mediated currents were recorded with whole-cell patch clamp and induced by pressure applications (50 ms, 10 psi) of 10 mm choline (Fayuk and Yakel, 2004) at a 2 min interval through a glass pipette located in the stratum radiatum area (about 150 μm away from the soma of the neurons under recording). Pressure was applied through a PPM-2 pneumatic pump (Harvard Apparatus). Tetrodotoxin (1 μm) was added to the perfusion ACSF and choline solution.
Statistics.
For whole-cell recordings, the amplitude of SC-EPSCs were analyzed with Clampfit. The percent changes were calculated by comparing with the average of 10 min baseline recording. For calcium imaging, the averages of 1 s of baseline sampling (i.e., 5 time points) before SC stimulation was used to calculate the percent change of the SC-induced peak calcium intensity. Values were presented as mean ± SEM. Two-tailed Student's t tests were performed to compare changes with the baseline or control.
Results
Cholinergic inputs induce timing-dependent hippocampal plasticity in septohippocampal cocultures as in acute hippocampal slices
Previous studies have shown that the septohippocampal coculture system provides an excellent model to study cholinergic modulation of hippocampal functions (Gahwiler and Brown, 1985; Fischer et al., 1999), even including complex network activities like theta oscillations (Fischer et al., 1999). In addition, a major advantage of using the coculture system (instead of acute hippocampal slices) is that this allows for the expression of proteins (e.g., nAChR subunits or GECIs) to restricted hippocampal subregions either in wild-type or transgenic mice (Fig. 1). We have previously shown that septal cholinergic inputs, activated either by electrical stimulation or via an optogenetic approach, can induce three types of hippocampal Schaffer collateral (SC) to CA1 synaptic plasticity, depending on the timing of the cholinergic input activation relative to the SC inputs in acute hippocampal slices (Gu and Yakel, 2011). In this study, we used GECIs to measure calcium activity changes at both presynaptic and postsynaptic sites to understand their respective contributions to the expression of synaptic plasticity (Fig. 1C,D). Second, we wanted to express the α7 nAChR subunit to either presynaptic or postsynaptic sites (or both) in α7 nAChR knock-out slices to dissect out the roles of presynaptic and postsynaptic α7 nAChRs in inducing the α7 nAChR-dependent LTP and STD. To do this, medial septum tissues and hippocampi were dissected out from brain slices and placed next to each other on Transwell membranes and cultured (Fig. 1A). The following day, viruses encoding either the GECIs or the α7 nAChR subunit were microinjected into the hippocampal CA1 or CA3 regions, and experiments were done 7 days after viral infection to allow protein expression and cholinergic innervation into the hippocampus (Fig. 1B–D).
Figure 1.
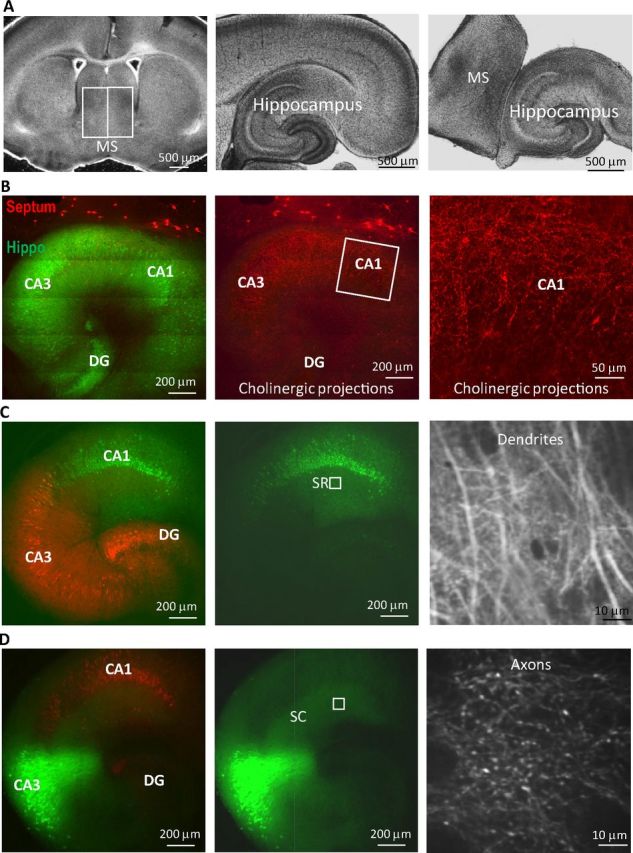
Monitoring neuronal synaptic activities with calcium imaging in septohippocampal slice cocultures. A, Septal tissue and hippocampus were dissected out from 300 μm brain slices and placed next to each other for culture. B, Abundant cholinergic innervation (red) into the hippocampus (Hippo) (green) 7 days after coculturing. C, Genetically encoded calcium indicator GCaMP3 (green, in synapsin promoter-driven AAV) was expressed in hippocampal CA1 neurons, and the dendritic spines were monitored in CA1 SR layer (box in the middle panel) as postsynaptic activities (right panel). D, GCaMP3 was expressed in CA3 neurons, but the projecting axons were monitored in CA1 SR area as presynaptic activities (right panel). The remaining areas of the hippocampus were shown with another red fluorescent protein also driven by synapsin promoter. MS, Medial septum; DG, dentate gyrus.
First we verified that the three different forms of ACh release-induced synaptic plasticity that we had previously observed in acute hippocampal slices using an optogenetic approach was well preserved with the same timing-sensitive modulation in the septohippocampal coculture system. We selectively expressed the light-activated cation channel channelrhodopsin-2 (ChR2) in cholinergic neurons by microinjecting a Cre-inducible AAV containing a double-floxed inverted ChR2 (fused with mCherry for visualization) (Tsai et al., 2009; Witten et al., 2010) to the septal tissue from ChAT-Cre transgenic mice. Extensive cholinergic innervations to the hippocampus can be observed 7 days later (Fig. 1B). Cholinergic inputs were then activated with 488 nm light exposure (20 ms) in the stratum oriens layer above the CA1 pyramidal neuron under patch-clamp recording. The SC pathway was activated via an electrode (Fig. 2A). Pairing of cholinergic fiber activation (via light exposure) with SC pathway (electrical) stimulation at three time intervals was selected to mimic the corresponding pairings of SO and SC electrical stimulation that produced the three types of synaptic plasticity previously observed in acute hippocampal slices (Gu and Yakel, 2011) (Fig. 2B).
Figure 2.
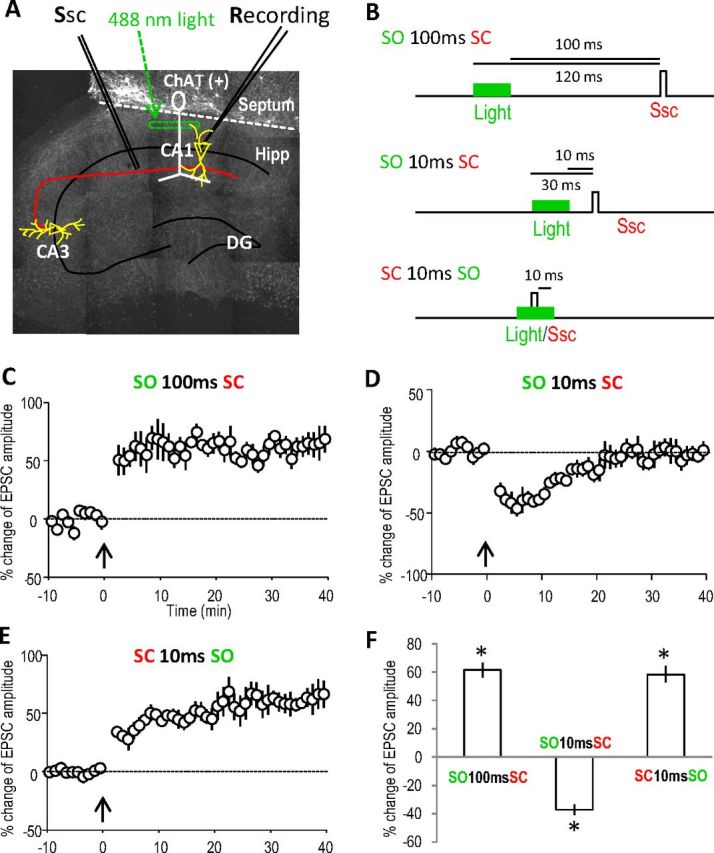
Optogenetically activated cholinergic inputs induce similar timing-dependent hippocampal plasticity in septohippocampal cocultures as in acute hippocampal slices. A, Cholinergic projections (infected with ChR2) in a small region in the hippocampal SO were exposed to 488 nm light (20 ms) for timed activation. B, Three different intervals for pairing cholinergic (SO) and SC pathways were used to induce plasticity, as used in acute hippocampal slices previously. C, LTP was induced by optically activating the cholinergic input 100 ms before stimulating the SC. The pairing protocol was introduced at the time point of 0 min as indicated by the arrow. D, STD was induced by activating the cholinergic input 10 ms before the SC. E, LTP was induced by pairing the SC 10 ms before optically activating the cholinergic input. F, Bar graph showing the amplitude changes of the three types of synaptic plasticity, analyzed at 30 min for LTP and 10 min for STD. *p < 0.001, as compared with before pairing, Student's t test, n = 5 in each group. Ssc, Stimulation of the SC; Hipp, hippocampus; DG, dentate gyrus.
Consistent with the results from acute hippocampal slices, when the cholinergic input was activated 100 ms before SC stimulation, LTP was induced (Fig. 2C). When cholinergic inputs were activated 10 ms before SC stimulation, STD was induced (Fig. 2D). Finally, when cholinergic input was activated 10 ms after SC stimulation, LTP was induced (Fig. 2E). These results demonstrated that the complex cholinergic modulation of hippocampal plasticity was well preserved in the septohippocampal coculture system as in the acute hippocampal slices (Fig. 2F). Furthermore, these results reinforced our previous observation of the robust timing-dependent cholinergic control of hippocampal function by successfully duplicating it in another system. Here, we focus on these first two forms of plasticity (i.e., the LTP and STD), both of which were previously shown to be dependent on activation of the α7 nAChR, and attempt to dissect out the individual contribution of presynaptic and postsynaptic α7 nAChRs in inducing this plasticity.
The α7 nAChR-dependent LTP involves prolonged calcium activity enhancement at both presynaptic and postsynaptic sites
Initially we examined whether the α7 nAChR-dependent LTP was due to enhanced presynaptic or postsynaptic activity. By expressing the genetically encoded calcium indicator GCaMP3 (Tian et al., 2009) in restricted hippocampal subregions, we have been able to monitor the SC stimulation-induced intracellular calcium increases in either presynaptic or postsynaptic sites during and after the induction of LTP. To measure postsynaptic activity in the CA1 region, GCaMP3 was selectively expressed in hippocampal CA1 neurons and their dendrites, and the fluorescent signal was monitored in the CA1 stratum radiatum (SR) layer (Fig. 1C). To measure presynaptic activity in the CA1 region instead, GCaMP3 was selectively expressed in CA3 neurons by viral microinjection in the CA3 region, and the fluorescent signal from the projecting axons was monitored in the CA1 SR layer (Fig. 1D). To avoid activating ChR2 while imaging GCaMP3 with 488 nm light, in these cases the cholinergic neurons were electrically stimulated with an electrode placed in the septal tissue (rather than using light to activate ChR2); this induced the same timing-dependent plasticity when paired with SC stimulation (data not shown).
Calcium imaging was carried out in the CA1 SR region 5 s before and after SC stimulation to measure the basal transient increase in intracellular calcium levels (i.e., GCaMP3 intensity increase) due to SC stimulation. When measuring the averaged fluorescence from the whole image, stimulation of the SC pathway usually induced a 5% increase over the background fluorescence, with the peak reached within 0.5 s and returning to basal levels within 2 s (Fig. 3A). This sampling was repeated every 1–2 min until stable responses were achieved (usually after about 10 min), after which the pairing protocol was introduced to induce synaptic plasticity. The sampling was then carried out for another 30 min to monitor any changes in the SC stimulation-induced calcium responses, and then the peak amplitude of the responses at 10 and 30 min after the pairing protocol were compared with the basal SC responses.
Figure 3.

The α7 nAChR-mediated LTP involves prolonged calcium activity enhancement in both presynaptic and postsynaptic sites in septohippocampal slice cocultures. LTP was induced by pairing SO pathway 100 ms before SC pathway in septohippocampal slice cocultures. A, Normalized SC pathway stimulation-induced postsynaptic GCaMP3 responses showing the prolonged enhancement of postsynaptic calcium activities after the SO pathway pairing protocol. SC pathway was stimulated at the time point of 0 s as indicated by the arrow. B, Images of SC pathway-induced postsynaptic calcium imaging from different time points before or after pairing protocol. C, Normalized presynaptic GCaMP3 responses showing the prolonged enhancement after the SO pathway pairing protocol. D, Images of presynaptic calcium imaging from different time points before or after pairing protocol. E, Bar graph showing the prolonged postsynaptic (Post) and presynaptic (Pre) responses during LTP; both were abolished in α7 nAChR knock-out slices. *p < 0.01, **p < 0.001, as compared with before pairing, Student's t test, n = 6 in each group. Ssc, Stimulation of the SC; WT, wild type.
When monitoring the postsynaptic calcium activity in the CA1 SR region (i.e., when GCaMP3 was expressed in CA1 neurons and the dendrites were monitored), the SC stimulation-induced response amplitudes were significantly increased after pairing the cholinergic input 100 ms before SC pathway stimulation (43 ± 8 and 58 ± 6% increase at 10 and 30 min, respectively, n = 6; Fig. 3A,B,E). This increase lasted for at least 30 min, demonstrating that a protocol that induces LTP at CA1 synapses induces prolonged increases in postsynaptic calcium activity. When monitoring presynaptic calcium activity in the same region (i.e., when the GCaMP3 was expressed in the CA3 neurons and the projecting SC axons were monitored in the CA1 SR region), SC stimulation-induced response amplitudes were also increased after pairing cholinergic input 100 ms before SC pathway stimulation (54 ± 3 and 41 ± 7% increase at 10 and 30 min, respectively, n = 6; Fig. 3C–E), also demonstrating that the LTP-inducing protocol significantly increases presynaptic calcium activity for a prolonged period of time. These presynaptic data are significant, as our previous work had suggested that there was only a transient increase in presynaptic activity when using the paired-pulse ratio (PPR) as an indicator (Gu and Yakel, 2011).
Both presynaptic and postsynaptic α7 nAChRs are required to induce the α7 nAChR-dependent LTP
We previously showed that the α7 nAChR-dependent LTP was likely due to a postsynaptic effect, since it appeared to require the activation of the NMDAR and prolongation of the NMDAR-mediated calcium transients in the spines and GluR2-containing AMPAR synaptic insertion. To examine the contributions of presynaptic and/or postsynaptic α7 receptors to plasticity in more detail, we used a strategy of restoring the expression of the α7 nAChR subunit to slices obtained from α7 nAChR knock-out mice; in these mice, this form of LTP was absent. Initially, we verified that in septohippocampal cocultures from α7 nAChR knock-out mice (the septal tissue providing cholinergic inputs was from wild-type mice), the pairing protocol that normally would induce the α7 nAChR-dependent LTP, failed to induce any significant prolonged increase in calcium signals at either postsynaptic or presynaptic sites (−6 ± 4 and −3 ± 5% change at 30 min, respectively, n = 5; Fig. 3E).
Next, we then restored the α7 nAChR (Séguéla et al., 1993) to either postsynaptic (by expressing the α7 nAChR along with GCaMP3 into CA1 neurons) or presynaptic sites (by expressing the α7 receptor into CA3 neurons in α7 nAChR knock-out slices). The α7 nAChR was also subcloned to the AAV vector under the synapsin promoter. The restricted localization of α7 nAChRs and GCaMP3 to either CA1 or CA3 was verified with the GCaMP3 expression pattern before imaging experiments. When the α7 receptor was restored in the postsynaptic CA1 neurons only, the SC stimulation-induced GCaMP3 responses were transiently increased in the postsynaptic sites after the pairing (44 ± 5% and −1 ± 2% change at 10 and 30 min, respectively, n = 5; Fig. 4A,B,E). Similarly, when the α7 nAChR was restored in the presynaptic CA3 neurons only, the SC stimulation-induced GCaMP3 responses were also transiently increased in the presynaptic sites after the pairing (50 ± 7% and 4 ± 4% increase at 10 and 30 min, respectively, n = 5; Fig. 4C–E). When we expressed the α7 nAChR in both CA1 and CA3 neurons in the same slice (i.e., both postsynaptically and presynaptically), this restored the LTP that was seen in wild-type slices (i.e., a prolonged enhancement in the SC stimulation-induced GCaMP3 responses both presynaptically and postsynaptically; 31 ± 6 and 70 ± 7% increase at 30 min, respectively, n = 5; Fig. 4E) These data demonstrate that both presynaptic and postsynaptic α7 nAChRs play a role in regulating synaptic activities and that both presynaptic and postsynaptic α7 receptors must be present to induce the α7 nAChR-dependent LTP. Furthermore these findings suggest that coordination between the presynaptic and postsynaptic modulation is required to induce this form of LTP.
Figure 4.
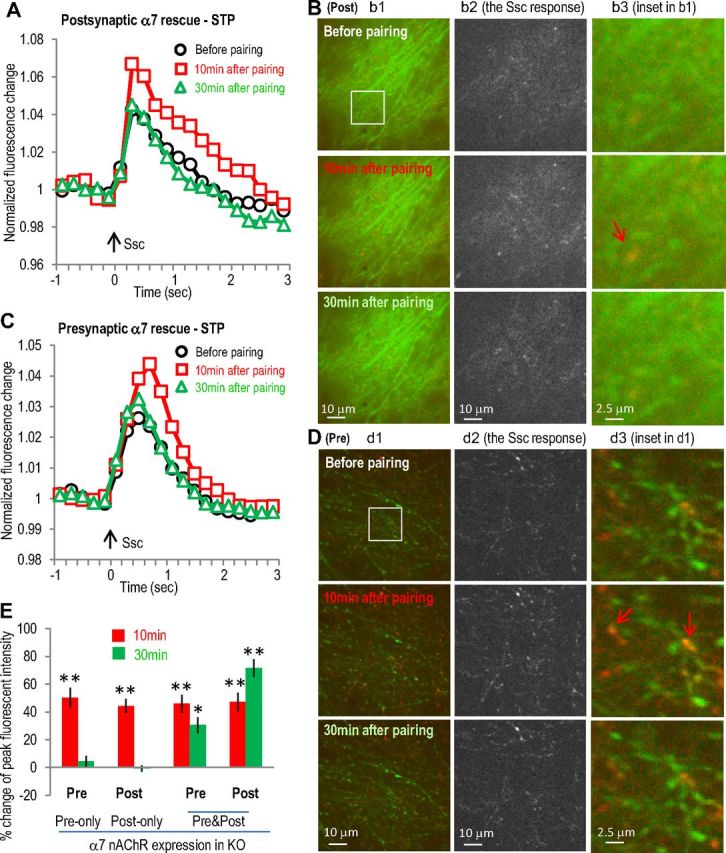
Expression of α7 nAChR in postsynaptic or presynaptic sites in α7 nAChR KO slice only induced transient activity enhancement in postsynaptic or presynaptic sites, respectively. A, α7 nAChR was coexpressed with GCaMP3 in hippocampal CA1 area of α7 nAChR knock-out mice (where the α7 nAChR-dependent LTP was abolished) as postsynaptic rescue of α7 nAChR. Normalized postsynaptic GCaMP3 responses show transient enhancement after the SO pathway pairing protocol. B, Images of postsynaptic calcium imaging from different time points before or after pairing protocol, with left panels showing the original images, middle panel showing the active calcium responses to SC stimulation, and right panels showing some potential dendritic spines. The potentiation was present in most of the dendrites responsive to SC stimulation (middle panels). C, α7 nAChR was coexpressed with GCaMP3 in hippocampal CA3 area as presynaptic rescue. Normalized presynaptic GCaMP3 responses show transient enhancement of presynaptic calcium activities after the SO pathway pairing protocol. D, Images of presynaptic calcium imaging from different time points before or after pairing protocol, with individual panels similar as described in B. E, Bar graph showing the requirement of both presynaptic and postsynaptic presence of α7 nAChRs to induce LTP. Red signals (in B and D) show the SC stimulation-induced calcium increases (also shown separately in the middle panels) over the basal signals before stimulation (green). Arrows indicate some synaptic sites with significant transient calcium increase. *p < 0.01, **p < 0.001, as compared with before pairing, Student's t test, n = 5 in each group. Ssc, Stimulation of the SC.
Both presynaptic and postsynaptic α7 nAChRs are required to induce the α7 nAChR-dependent STD
We have previously shown that the STD induced by pairing cholinergic input stimulation 10 ms before SC pathway stimulation was also mediated by the α7 nAChR, although a presynaptic mechanism was previously suggested from the change in the paired-pulse ratio. Therefore, we tested whether a presynaptic mechanism alone is sufficient to explain this STD. In wild-type cocultured slices, we observed a transient decrease in the SC stimulation-induced GCaMP3 responses at both postsynaptic (−39 ± 5 and 8 ± 5% change at 10 and 30 min, respectively, n = 5; Fig. 5A,B,E) and presynaptic sites (−37 ± 6 and −3 ± 3% change at 10 and 30 min, respectively, n = 5; Fig. 5C–E). Furthermore, both responses were abolished in α7 nAChR knock-out slices (Fig. 5E). When we restored the α7 nAChR to either the presynaptic (5 ± 7% change at 10 min, n = 5; Fig. 5F) or the postsynaptic (6 ± 7% change at 10 min, n = 5; Fig. 5F) sites alone (in α7 nAChR knock-out slices), we could not restore the STD (Fig. 5F); restoring the α7 receptor to both presynaptic and postsynaptic sites in the same slice was required to restore the STD in the α7 knock-out slices (Fig. 5F). This suggests that a presynaptic mechanism alone is not sufficient to induce this form of STD, again further implicating some type of coordination between presynaptic and postsynaptic sites.
Figure 5.
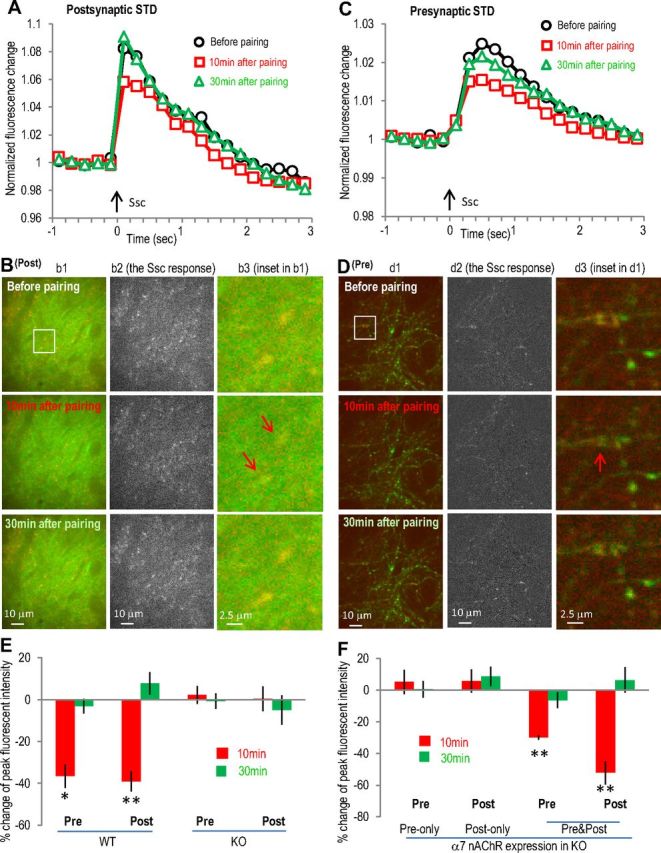
The α7 nAChR-mediated STD involves transient calcium activity depression in both postsynaptic and presynaptic sites. The α7 nAChR-dependent STD was induced by pairing SO 10 ms before SC. A, Normalized postsynaptic GCaMP3 responses showing the transient depression of postsynaptic calcium activities after the SO pathway pairing protocol. B, Images of postsynaptic (Post) calcium imaging from different time points before or after pairing protocol, with individual panels similar as described in Figure 4B. C, Normalized presynaptic GCaMP3 responses showing the transient depression after the SO pathway pairing protocol. D, Images of presynaptic (Pre) calcium imaging from different time points before or after pairing protocol. E, F, Bar graphs showing that the presence of α7 nAChRs at both presynaptic and postsynaptic sites was required to induce the STD. Red signals (in B and D) show the SC stimulation-induced calcium increases (also shown separately in the middle panels) over the basal signals before stimulation (green). Arrows indicate some synaptic sites with significant transient calcium decrease. *p < 0.01, **p < 0.001, as compared with before pairing, Student's t test, n = 5 in each group. Ssc, Stimulation of the SC.
The expression of functional α7 nAChRs in pyramidal neurons was verified by directly measuring choline (10 mm)-induced α7 nAChR currents (Fayuk and Yakel, 2004). The α7 nAChR currents were induced in all of the CA1 and CA3 pyramidal neurons we tested (8–12 neurons for either group), with peak amplitudes of ∼50–100 pA that were completely blocked by the α7-selective antagonist methyllycaconitine (MLA) (10 nm); choline-induced responses were absent in α7 nAChR knock-out slices (Fig. 6A,B). Virus-introduced α7 nAChRs (fused and visualized with yellow fluorescent protein, YFP, in AAV virus under the synapsin promoter) restored α7 nAChR currents in both CA1 and CA3 neurons from α7 nAChR knock-out slices, with peak amplitudes not significantly different than those from wild-type slices (Fig. 6A,B) in both CA1 and CA3 neurons. Expression of the reporter YFP alone (in AAV under the synapsin promoter) did not result in any α7 nAChR currents in knock-out slices and had no obvious effects in wild-type slices (data were combined with the respective knock-out and wild-type groups as shown in Fig. 6B).
Figure 6.
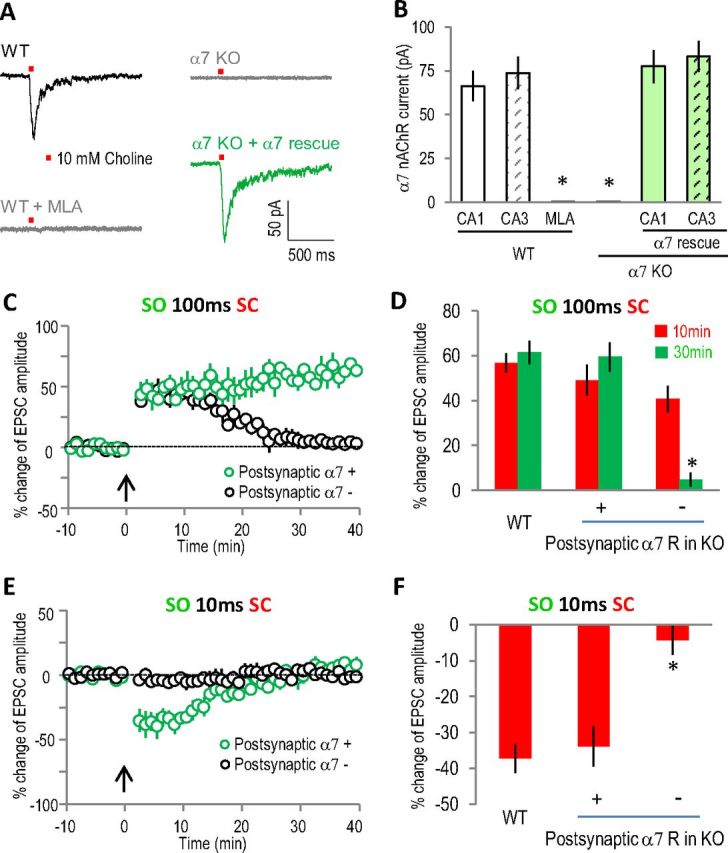
Postsynaptic functional α7 nAChRs in CA1 pyramidal neuron were required to induce either LTP or STD. A, Typical traces of α7 nAChR currents from whole-cell patch clamp recordings of CA1 pyramidal neurons. The α7 nAChR currents were induced by pressure application (10 psi for 50 ms) of 10 mm choline to the SR area (about 150 μm from the cell body). The α7 nAChR currents were completely blocked by MLA (10 nm) and absent in α7 nAChR knock-out slices. B, Bar graph showing the expression of functional α7 nAChRs in CA1 and CA3 pyramidal neurons in septohippocampal slice cultures and the efficient rescue by virus-introduced α7 nAChRs to the knock-out slices. *p < 0.001 as compared with wild-type, Student's t test, n = 8–12 in each group. C, E, Whole-cell recordings of EPSCs showing normal LTP and STD in α7 nAChR-positive CA1 pyramidal neurons in α7 nAChR knock-out rescue (at both CA1 and CA3 sites) slices, while both forms of plasticity were disrupted in neighboring α7 nAChR-negative CA1 pyramidal neurons. D, F, Bar graphs showing the difference of the plasticity between α7 nAChR-positive and their neighboring α7 nAChR-negative CA1 pyramidal neurons. *p < 0.001, as compared with α7 nAChR-positive CA1 pyramidal neurons, Student's t test, n = 5 in each group. WT, Wild type.
To further verify the direct involvement of postsynaptic α7 nAChRs in CA1 pyramidal neurons in this plasticity, we compared the plasticity in individual α7 nAChR-positive (fused with YFP) and neighboring α7 nAChR-negative CA1 pyramidal neurons from the knock-out rescue (both CA1 and CA3 rescue) slices with whole-cell patch clamp. Consistent with the GCaMP3 imaging data, both LTP and STD were induced in α7 nAChR-positive CA1 pyramidal neurons (Fig. 6C–F). However, only STP was induced in α7 nAChR-negative CA1 pyramidal neurons with the LTP pairing protocol, and no plasticity was induced with the STD protocol (Fig. 6C–F), which is consistent with the presynaptic-only rescue imaging data. These results strongly suggest the direct involvement of α7 nAChRs in postsynaptic CA1 pyramidal neurons in inducing either LTP or STD.
Differential presynaptic and postsynaptic mechanisms in the expression of both α7 nAChR-dependent LTP and STD revealed with dual-color GECI imaging
Although the α7 nAChR rescue experiments suggest that the induction of both the α7 nAChR-dependent LTP and STD requires coordination between presynaptic and postsynaptic activities, the similar time courses between the presynaptic and postsynaptic changes could suggest that both of these two forms of plasticity were expressed primarily via presynaptic mechanisms; the observed postsynaptic change during the plasticity expression may simply be the result of presynaptic modulation. However, we did see some differences in the time course of presynaptic and postsynaptic changes during either LTP or STD (especially during LTP). For example, the increase at 30 min (58%) was larger than that at 10 min (43%) at postsynaptic sites, but smaller (41% at 30 min versus 54% at 10 min) at presynaptic sites during the LTP (Fig. 3E). Another important issue is that the presynaptic and postsynaptic activities observed above were not from the same slices, since the recordings were done separately. To clarify this question, we expressed two differently colored GECIs (green GCaMP3 and red calcium indicator R-GECO1) (Tian et al., 2009; Zhao et al., 2011) in the same slices, one at presynaptic sites and the other at postsynaptic sites. In this way we could observe the presynaptic and postsynaptic activities in the same slice simultaneously during the expression of either LTP or STD. A high degree of correlation between the changes in the presynaptic and postsynaptic activities would suggest a major role for the presynaptic sites in the expression of either form of plasticity.
We expressed green GCaMP3 in CA3 neurons and red R-GECO1 in CA1 neurons in the same slices to observe green presynaptic and red postsynaptic activities simultaneously while monitoring in the CA1 SR region. With this dual-color imaging, we found that in many cases the activity changes in the presynaptic and postsynaptic sites were not correlated for either LTP or STD. For the example shown in Figure 7A–D during the α7 nAChR-dependent LTP, both presynaptic and postsynaptic activities were increased at 10 and 30 min after the pairing protocol. However, while the postsynaptic increase was similar to presynaptic increase at 10 min, it was significantly larger at 30 min (Fig. 7C). The ratio of postsynaptic to presynaptic response was significantly larger at 30 min than that at 10 min (1.46 ± 0.09 at 30 min versus 0.82 ± 0.06 at 10 min, n = 5, p < 0.001, t test), suggesting that at least the later stage postsynaptic increase was not mainly driven by an increased presynaptic release. Instead, the data suggest that an independent postsynaptic mechanism is driving the expression of later stage LTP.
Figure 7.
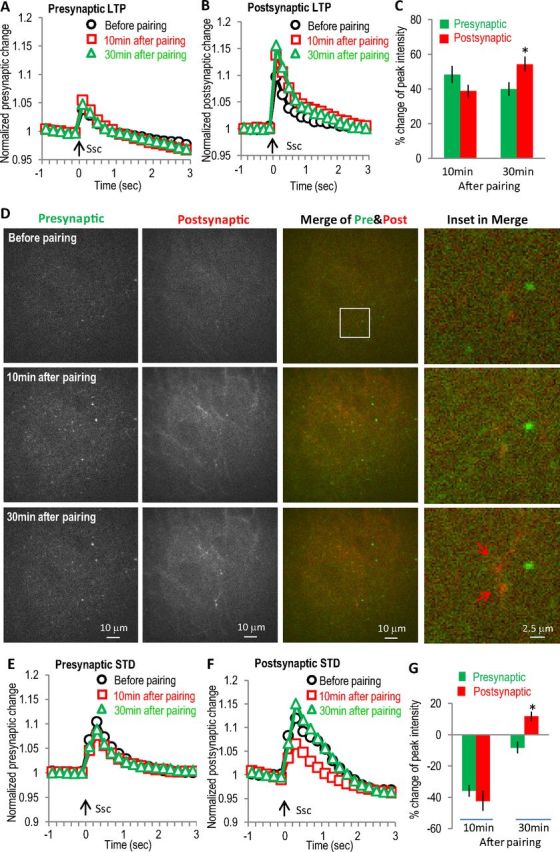
Differential presynaptic and postsynaptic changes in α7 nAChR-dependent LTP and STD revealed with dual-color GECI imaging. GECIs (green GCaMP3 and red R-GECO1) were expressed separately in hippocampal CA3 and CA1 to monitor presynaptic and postsynaptic activities simultaneously for LTP (A–D) and STD (E–G). A, Normalized presynaptic GCaMP3 responses showing smaller enhancement at 30 min than that at 10 min. B, Normalized postsynaptic R-GECO1 responses showing stronger enhancement at 30 min than that at 10 min. C, Bar graph showing the differential changes of the later stage (at 30 min) between presynaptic and postsynaptic activities during the LTP. D, Images of presynaptic and postsynaptic SC stimulation-induced calcium increases at different time points during the LTP. Left two panels show the active presynaptic and postsynaptic calcium responses to SC stimulation, respectively. The third panels are merged images of presynaptic and postsynaptic responses, with the right panels showing some potential spines. E, Normalized presynaptic GCaMP3 responses showing the transient depression after the SO pathway pairing protocol. Arrow indicates stimulation of the SC pathway. F, Normalized postsynaptic R-GECO1 responses showing the transient depression after pairing the SO pathway. G, Bar graph showing the differential recovery between presynaptic and postsynaptic activities during the STD. Ssc, Stimulation of the SC.
Similarly, differential modulation of presynaptic and postsynaptic activities was seen in the expression of STD. As shown in Figure 7E–G during the α7 nAChR-dependent STD, both presynaptic and postsynaptic activities were similarly decreased at 10 min; however, they were significantly different at 30 min. The postsynaptic to presynaptic ratio were significantly changed at 30 min as compared with that at 10 min (−1.52 ± 0.32 at 30 min versus 1.2 ± 0.04 at 10 min, n = 5, p < 0.001, t test). These results again strongly suggest the existence of an independent postsynaptic mechanism during the expression of STD.
In summary, our data show that both presynaptic and postsynaptic mechanisms contributed to the expression of the α7 nAChR-dependent LTP and STD. More importantly, we have provided direct evidence that the α7 nAChR-dependent modulation of both presynaptic and postsynaptic sites is required to induce both of these forms of plasticity. Therefore, we conclude that cholinergic inputs, through activation of α7 nAChRs located at both synaptic sites, coordinate presynaptic and postsynaptic activities to induce synaptic plasticity.
Discussion
Previously, we have shown that activating the septal cholinergic terminals in acute hippocampal slices induced three different types of hippocampal SC to CA1 synaptic plasticity, depending on the precise timing of cholinergic stimulation relative to the SC input (Gu and Yakel, 2011). Two of these forms of plasticity, including long-term potentiation and short-term depression, were dependent on the activation of the α7 nAChR. Here, we have directly monitored the presynaptic and postsynaptic activity changes with GECIs to investigate their individual contributions to the induction and expression of the α7 nAChR-dependent LTP and STD. To do this, we used a coculture system whereby a slice from the septum was cultured next to a slice with hippocampus; in these cocultures, we confirmed that all three forms of plasticity that we had observed in acute slices was preserved. Then we expressed the GECIs into either the postsynaptic CA1 neurons or presynaptic CA3 neurons and monitored in the CA1 stratum radiatum region for postsynaptic (dendrites of CA1 neurons) and presynaptic (axons of CA3 neurons) activities, respectively. We observed a prolonged enhancement (>30 min) of the SC-induced calcium responses both postsynaptically and presynaptically in the α7 nAChR-dependent LTP. During the α7 nAChR-dependent STD there was a short-term depression of the calcium responses both presynaptically and postsynaptically. Dual-color calcium imaging revealed a differential time course and pattern of postsynaptic versus presynaptic modulation during both LTP and STD (especially during the later stages), suggesting the existence of independent postsynaptic modulatory mechanisms. A similar time course between the presynaptic and postsynaptic changes would otherwise suggest that the postsynaptic change was merely the result of the presynaptic change.
To determine whether presynaptic or postsynaptic α7 receptors (or both) might be required for either LTP or STD, we expressed α7 receptors to either presynaptic or postsynaptic sites in slices from α7 nAChR knock-out (KO) mice. We found that the α7 receptors were required both presynaptically and postsynaptically to restore either LTP or STD in the α7 nAChR KO slices. This is consistent with the idea that there are independent postsynaptic and presynaptic modulations during the plasticity. Taken together, these results demonstrate that modulation at either presynaptic or postsynaptic sites alone is insufficient. Instead, coordinated modulation at both sites is required to induce either the LTP or the STD. Therefore, it appears that cholinergic inputs, through α7 nAChRs at both presynaptic and postsynaptic sites, can thus coordinate presynaptic and postsynaptic activities to induce timing-dependent synaptic plasticity.
Neuromodulator systems have been heavily implicated in higher brain functions and the induction of synaptic plasticity (Jerusalinsky et al., 1997; Bailey et al., 2000; Power et al., 2003; Dani and Bertrand, 2007; Kenney and Gould, 2008; Reis et al., 2009). The α7 nAChRs, as well as other neuromodulator receptors, are known to be expressed at both the presynaptic and postsynaptic sites of glutamatergic and GABAergic terminals and can modulate synaptic excitability via presynaptic or postsynaptic mechanisms (Levey et al., 1991; Fabian-Fine et al., 2001; Sher et al., 2004; Volpicelli and Levey, 2004; Dani and Bertrand, 2007; Drever et al., 2011). The nAChR-dependent modulation of synaptic plasticity has been widely observed in the hippocampus and is thought to involve either presynaptic or postsynaptic mechanisms. For example, activation of nAChRs (both α7 and non-α7 receptors) in the CA1, dentate regions, and entorhinal cortex enhances synaptic plasticity (Fujii et al., 1999; Mann and Greenfield, 2003; Tu et al., 2009; Ondrejcak et al., 2012). In addition, the activation of α7 nAChRs on hippocampal interneurons can block concurrent STP and LTP in pyramidal cells, whereas presynaptic nAChRs can enhance the release of glutamate and, thus, increase the probability of inducing LTP (Ji et al., 2001). However, the precise molecular mechanisms involving the induction and expression of synaptic plasticity and the role of presynaptic and postsynaptic changes remains unresolved and still much debated (Lisman, 2009; Glanzman, 2010). It is likely that in most cases, both presynaptic and postsynaptic components will be involved in the coordination and forming of synaptic plasticity. Here, we have provided direct evidence for presynaptic and postsynaptic modulation to cooperate in the induction of synaptic plasticity and, importantly, we show here that neuromodulator systems can serve as the mediator of such coordination.
The molecular mechanisms underlying the α7 nAChR-mediated plasticity are not clear at this time, but may involve NMDA receptor activation and retrograde signaling, the two major molecular mechanisms proposed to connect the presynaptic and postsynaptic activities in inducing synaptic plasticity. The NMDA receptor, because of its depolarization-sensitive Mg2+ block, has been proposed as the coincidence detector for presynaptic and postsynaptic activities in both HFS-induced plasticity and STDP. We have previously shown that the α7 nAChR-dependent LTP also involves NMDA receptor activation and prolonged postsynaptic intracellular calcium increase, suggesting that α7 nAChR activation may facilitate NMDA receptor activation. Retrograde signals (e.g., brain-derived neurotrophic factor or endocannabinoid) are released from the postsynaptic sites in an activity-dependent manner and affect presynaptic sites, thereby inducing plasticity (under the correct conditions). Neuromodulators can also facilitate retrograde messenger release with reduced calcium requirement, or in some cases even without calcium increases or postsynaptic firing (Kim et al., 2002; Best and Regehr, 2008; Regehr et al., 2009). However, other networks of neurons (for example GABAergic interneurons) or non-neuronal cells (such as astrocytes or glia) could aid in communication between the presynaptic and postsynaptic sites that is responsible for the prolonged increase in calcium responses observed both presynaptically and postsynaptically. For example, non-neuronal cells in the brain also express a variety of neurotransmitter receptors and ion channels (including the α7 nAChRs) (Vélez-Fort et al., 2009; Shen and Yakel, 2012) and participate in synaptic signaling and plasticity in the brain (Araque et al., 2002; Agulhon et al., 2008; Fiacco et al., 2009).
Neuromodulator-dependent associative heterosynaptic plasticity has several advantages over Hebbian homosynaptic plasticity (Bailey et al., 2000). First, it incorporates extrinsic signals into local networks, and second it significantly prolongs the expression of LTP and with higher efficiency and specificity. Currently, NMDA receptor-dependent homosynaptic plasticity is primarily induced by controlled postsynaptic depolarization through HFS or current injection. Little is known about how endogenous neuronal firing patterns lead to synaptic plasticity. Our study here has provided direct evidence that the precisely timed modulatory transmitter release can induce glutamatergic synaptic plasticity through coordinating the presynaptic and postsynaptic activities. Here, we propose that precisely timed activation of cholinergic inputs through the α7 nAChRs, as well as potentially other neuromodulator systems, can coordinate the presynaptic and postsynaptic activities to induce synaptic plasticity with high efficiency and temporal precision.
Footnotes
This work was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences. We thank Drs. Charles Romeo, Negin Martin, and Bernd Gloss for virus packaging; Charles J. Tucker and Agnus Janoshazi for assistance with fluorescent microscopy; and Drs. Serena Dudek and Christian Erxleben for comments on the manuscript. We also thank Dr. Karl Deisseroth at Stanford University for ChR2 and synapsin promoter-driven AAV vectors, Dr. Loren Looger at Janelia Farm Research Campus for GCaMP3, Dr. Robert Campbell at University of Alberta for R-GECO1, and Dr. James Wilson at University of Pennsylvania for AAV serotype 9 helper plasmid.
The authors declare no competing financial interests.
References
- Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD. What is the role of astrocyte calcium in neurophysiology? Neuron. 2008;59:932–946. doi: 10.1016/j.neuron.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araque A, Martín ED, Perea G, Arellano JI, Buño W. Synaptically released acetylcholine evokes Ca2+ elevations in astrocytes in hippocampal slices. J Neurosci. 2002;22:2443–2450. doi: 10.1523/JNEUROSCI.22-07-02443.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Giustetto M, Huang YY, Hawkins RD, Kandel ER. Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory? Nat Rev Neurosci. 2000;1:11–20. doi: 10.1038/35036191. [DOI] [PubMed] [Google Scholar]
- Bastrikova N, Gardner GA, Reece JM, Jeromin A, Dudek SM. Synapse elimination accompanies functional plasticity in hippocampal neurons. Proc Natl Acad Sci U S A. 2008;105:3123–3127. doi: 10.1073/pnas.0800027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CC, Han VZ, Sugawara Y, Grant K. Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature. 1997;387:278–281. doi: 10.1038/387278a0. [DOI] [PubMed] [Google Scholar]
- Best AR, Regehr WG. Serotonin evokes endocannabinoid release and retrogradely suppresses excitatory synapses. J Neurosci. 2008;28:6508–6515. doi: 10.1523/JNEUROSCI.0678-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GLA. Synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Drever BD, Riedel G, Platt B. The cholinergic system and hippocampal plasticity. Behav Brain Res. 2011;221:505–514. doi: 10.1016/j.bbr.2010.11.037. [DOI] [PubMed] [Google Scholar]
- Egger V, Feldmeyer D, Sakmann B. Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nat Neurosci. 1999;2:1098–1105. doi: 10.1038/16026. [DOI] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A. Ultrastructural distribution of the α7 nicotinic acetylcholine receptor subunit in rat hippocampus. J Neurosci. 2001;21:7993–8003. doi: 10.1523/JNEUROSCI.21-20-07993.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayuk D, Yakel JL. Regulation of nicotinic acetylcholine receptor channel function by acetylcholinesterase inhibitors in rat hippocampal CA1 interneurons. Mol Pharmacol. 2004;66:658–666. doi: 10.1124/mol.104.000042. [DOI] [PubMed] [Google Scholar]
- Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, Agulhon C, McCarthy KD. Sorting out astrocyte physiology from pharmacology. Annu Rev Pharmacol Toxicol. 2009;49:151–174. doi: 10.1146/annurev.pharmtox.011008.145602. [DOI] [PubMed] [Google Scholar]
- Fischer Y, Gähwiler BH, Thompson SM. Activation of intrinsic hippocampal theta oscillations by acetylcholine in rat septohippocampal cocultures. J Physiol. 1999;519:405–413. doi: 10.1111/j.1469-7793.1999.0405m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Ji Z, Morita N, Sumikawa K. Acute and chronic nicotine exposure differentially facilitate the induction of LTP. Brain Res. 1999;846:137–143. doi: 10.1016/s0006-8993(99)01982-4. [DOI] [PubMed] [Google Scholar]
- Gähwiler BH, Brown DA. Functional innervation of cultured hippocampal neurones by cholinergic afferents from co-cultured septal explants. Nature. 1985;313:577–579. doi: 10.1038/313577a0. [DOI] [PubMed] [Google Scholar]
- Gähwiler BH, Hefti F. Guidance of acetylcholinesterase-containing fibres by target tissue in co-cultured brain slices. Neuroscience. 1984;13:681–689. doi: 10.1016/0306-4522(84)90088-5. [DOI] [PubMed] [Google Scholar]
- Ge S, Dani JA. Nicotinic acetylcholine receptors at glutamate synapses facilitate long-term depression or potentiation. J Neurosci. 2005;25:6084–6091. doi: 10.1523/JNEUROSCI.0542-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glanzman DL. Common mechanisms of synaptic plasticity in vertebrates and invertebrates. Curr Biol. 2010;20:R31–R36. doi: 10.1016/j.cub.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Z, Yakel JL. Timing-dependent septal cholinergic induction of dynamic hippocampal synaptic plasticity. Neuron. 2011;71:155–165. doi: 10.1016/j.neuron.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerusalinsky D, Kornisiuk E, Izquierdo I. Cholinergic neurotransmission and synaptic plasticity concerning memory processing. Neurochem Res. 1997;22:507–515. doi: 10.1023/a:1027376230898. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Kenney JW, Gould TJ. Modulation of hippocampus-dependent learning and synaptic plasticity by nicotine. Mol Neurobiol. 2008;38:101–121. doi: 10.1007/s12035-008-8037-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Isokawa M, Ledent C, Alger BE. Activation of muscarinic acetylcholine receptors enhances the release of endogenous cannabinoids in the hippocampus. J Neurosci. 2002;22:10182–10191. doi: 10.1523/JNEUROSCI.22-23-10182.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey AI, Kitt CA, Simonds WF, Price DL, Brann MR. Identification and localization of muscarinic acetylcholine receptor proteins in brain with subtype-specific antibodies. J Neurosci. 1991;11:3218–3226. doi: 10.1523/JNEUROSCI.11-10-03218.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE. The pre/post LTP debate. Neuron. 2009;63:281–284. doi: 10.1016/j.neuron.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Mann EO, Greenfield SA. Novel modulatory mechanisms revealed by the sustained application of nicotine in the guinea-pig hippocampus in vitro. J Physiol. 2003;551:539–550. doi: 10.1113/jphysiol.2003.045492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Sakmann B. Regulation of synaptic efficacy by coincidence of postsynaptic APs and EPSPs. Science. 1997;275:213–215. doi: 10.1126/science.275.5297.213. [DOI] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG. Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci. 2000;23:649–711. doi: 10.1146/annurev.neuro.23.1.649. [DOI] [PubMed] [Google Scholar]
- Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- Ondrejcak T, Wang Q, Kew JN, Virley DJ, Upton N, Anwyl R, Rowan MJ. Activation of α7 nicotinic acetylcholine receptors persistently enhances hippocampal synaptic transmission and prevents Aβ-mediated inhibition of LTP in the rat hippocampus. Eur J Pharmacol. 2012;677:63–70. doi: 10.1016/j.ejphar.2011.12.008. [DOI] [PubMed] [Google Scholar]
- Power AE, Vazdarjanova A, McGaugh JL. Muscarinic cholinergic influences in memory consolidation. Neurobiol Learn Mem. 2003;80:178–193. doi: 10.1016/s1074-7427(03)00086-8. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Carey MR, Best AR. Activity-dependent regulation of synapses by retrograde messengers. Neuron. 2009;63:154–170. doi: 10.1016/j.neuron.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reis HJ, Guatimosim C, Paquet M, Santos M, Ribeiro FM, Kummer A, Schenatto G, Salgado JV, Vieira LB, Teixeira AL, Palotás A. Neurotransmitters in the central nervous system and their implication in learning and memory processes. Curr Med Chem. 2009;16:796–840. doi: 10.2174/092986709787549271. [DOI] [PubMed] [Google Scholar]
- Rimvall K, Keller F, Waser PG. Development of cholinergic projections in organotypic cultures of rat septum, hippocampus and cerebellum. Brain Res. 1985;351:267–278. doi: 10.1016/0165-3806(85)90198-1. [DOI] [PubMed] [Google Scholar]
- Séguéla P, Wadiche J, Dineley-Miller K, Dani JA, Patrick JW. Molecular cloning, functional properties, and distribution of rat brain α7: a nicotinic cation channel highly permeable to calcium. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JX, Yakel JL. Functional α7 nicotinic ACh receptors on astrocytes in rat hippocampal CA1 slices. J Mol Neurosci. 2012 doi: 10.1007/s12031-012-9719-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher E, Chen Y, Sharples TJ, Broad LM, Benedetti G, Zwart R, McPhie GI, Pearson KH, Baldwinson T, De Filippi G. Physiological roles of neuronal nicotinic receptor subtypes: new insights on the nicotinic modulation of neurotransmitter release, synaptic transmission and plasticity. Curr Top Med Chem. 2004;4:283–297. doi: 10.2174/1568026043451393. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Tian L, Hires SA, Mao T, Huber D, Chiappe ME, Chalasani SH, Petreanu L, Akerboom J, McKinney SA, Schreiter ER, Bargmann CI, Jayaraman V, Svoboda K, Looger LL. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat Methods. 2009;6:875–881. doi: 10.1038/nmeth.1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Akerboom J, Schreiter ER, Looger LL. Neural activity imaging with genetically encoded calcium indicators. Prog Brain Res. 2012;196:79–94. doi: 10.1016/B978-0-444-59426-6.00005-7. [DOI] [PubMed] [Google Scholar]
- Tsai HC, Zhang F, Adamantidis A, Stuber GD, Bonci A, de Lecea L, Deisseroth K. Phasic firing in dopaminergic neurons is sufficient for behavioral conditioning. Science. 2009;324:1080–1084. doi: 10.1126/science.1168878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B, Gu Z, Shen JX, Lamb PW, Yakel JL. Characterization of a nicotine-sensitive neuronal population in rat entorhinal cortex. J Neurosci. 2009;29:10436–10448. doi: 10.1523/JNEUROSCI.2580-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vélez-Fort M, Audinat E, Angulo MC. Functional alpha 7-containing nicotinic receptors of NG2-expressing cells in the hippocampus. Glia. 2009;57:1104–1114. doi: 10.1002/glia.20834. [DOI] [PubMed] [Google Scholar]
- Volpicelli LA, Levey AI. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog Brain Res. 2004;145:59–66. doi: 10.1016/S0079-6123(03)45003-6. [DOI] [PubMed] [Google Scholar]
- Witten IB, Lin SC, Brodsky M, Prakash R, Diester I, Anikeeva P, Gradinaru V, Ramakrishnan C, Deisseroth K. Cholinergic interneurons control local circuit activity and cocaine conditioning. Science. 2010;330:1677–1681. doi: 10.1126/science.1193771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LI, Tao HW, Holt CE, Harris WA, Poo M. A critical window for cooperation and competition among developing retinotectal synapses. Nature. 1998;395:37–44. doi: 10.1038/25665. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE. An expanded palette of genetically encoded Ca2+ indicators. Science. 2011;333:1888–1891. doi: 10.1126/science.1208592. [DOI] [PMC free article] [PubMed] [Google Scholar]