

Abstract
Developing effective immunotherapy for lung cancer is a daunting but hugely attractive challenge. Until recently, non-small cell lung cancer was thought of as a non-immunogenic tumor, but there is now evidence highlighting the integral role played by both inflammatory and immunological responses in lung carcinogenesis. Despite recent encouraging preclinical and phase I/II data, there is a paucity of phase III trials showing a clear clinical benefit for vaccines in lung cancer. There are many difficulties to overcome prior to the development of a successful therapy. Perhaps a measurable immune response may not translate into a clinically meaningful or radiological response. Patient selection may also be a problem for ongoing clinical studies. The majority of trials for lung cancer vaccines are focused on patients with advanced-stage disease, while the ideal candidates may be patients with a lower tumor burden stage I or II disease. Selecting the exact antigens to target is also difficult. It will likely require multiple epitopes of a diverse set of genes restricted to multiple haplotypes to generate a truly effective vaccine that is able to overcome the various immunologic escape mechanisms that tumors employ. This review discusses active immunotherapy employing protein/peptide vaccines, whole cell vaccines, and dendritic cell vaccines and examines the current data on some novel immunomodulating agents.
Keywords: vaccines, cancer, T lymphocytes, dendritic cells, immunomodulation
Introduction
Lung cancer accounts for approximately one third of cancer-related deaths in the United States [1]. Surgical resection is the only truly curative therapy, but with relapse rates greater than 40% after definitive resection, improved therapeutics in the adjuvant setting are required [2]. In advanced stages of disease, systemic chemotherapy and/or localized irradiation can produce objective responses and palliation of symptoms; however, these therapies offer only modest improvements in survival. Two-year survival rates for stages IIIB and IV non-small cell lung cancer (NSCLC) are 10.8% and 5.4% respectively; 5-year survival rates are 3.9% and 1.3% respectively [3]. Despite the availability of platinum-based chemotherapy doublets and agents such as docetaxel, pemetrexed, and erlotinib in refractory disease, overall outcomes are still poor.
Immunotherapy provides a unique approach to potentially consolidating surgery and/or standard chemotherapy. Stimulating the immune system by inducing a cellular immune response that harnesses CD4+ and CD8+ cytotoxic T lymphocytes (CTLs) capable of selectively destroying cancer cells by targeting tumor-associated antigens (TAAs) could result in tumor eradication. The use of lymphocytes as a therapeutic intervention takes advantage of the immune system’s specificity, surveillance properties, and memory induction. The ability of the immune system to circulate throughout the body could in theory allow for the removal of micro-metastases that remain silent until they grow to clinical significance. Furthermore, the immune system’s ability to generate long-lasting memory and an enduring immune response may protect against new tumor development.
Vaccine therapy in non-small cell lung cancer
Cellular immunity is initiated by the uptake of antigens by antigen-presenting cells (APCs) such as dendritic cells (DCs). Once internalized, short peptide sequences of the antigen are presented on the extracellular surface of the APC in conjunction with the major histocompatibility complex (MHC) molecules. DCs then move from the periphery to the draining lymph nodes where they come in contact with naïve T lymphocytes [4]. Complex reactions involving the specific T-cell receptor and the APC MHC-peptide molecule, as well as activation of costimulatory molecules such as B7.1 and B7.2, are required to activate CD8+ CTLs (Fig. 1) [5]. Once activated, these CTLs circulate and recognize cells that display the complementary peptide-MHC class I molecule on the cell surface. Cell death occurs via a combination of either granule exocytosis or expression of the FAS ligand, which leads to apoptosis (Fig. 2) [6]. Memory lymphocytes are produced, allowing a rapid T-cell response in the event of a future challenge with the same antigen [7]. Cytokines play an important role in regulating the immune system and can either direct the response toward activation or down-regulation [8].
Fig 1.

Antigen presenting cells such as dendritic cells can process antigens such as peptides or whole tumor cells, can become infected by viral vectors which then express tumor associated antigens for presentation, or can be ex vivo pulsed with peptides then injected. These antigen presenting cells present to the T-cell causing activation of the T-cell. Upon activation, CTLA4 is upregulated and inhibits activation of the T-cell. Antibodies to CTLA4 can block the inhibitory signals and cause prolonged activation. (Figure courtesy of NIH Medical Arts.)
Fig 2.

T-cell recognition requires an MHC molecule with a T-cell-specific tumor-associated antigen (TAA) peptide in its cleft. This triggers immune-mediated killing either through Fas or the release of granzymes. TCR T-cell receptor, LFA-1 lymphocyte function-associated antigen-1, Fas-L Fas ligand. (Figure courtesy of NIH Medical Arts.)
Most cancer patients develop some degree of tumor tolerance [9], which may result from a lack of proper co-stimulatory molecules, such as B7.1 or B7.2, upon initial antigen presentation [10]. Tumor cells also evade the immune system by down-regulating immune cytokines and developing resistance to CTLs [11, 12]. Lung cancer cells have been shown to produce a variety of immunosuppressive molecules including TGF-β [13], prostaglandin E2 [14], IL-10 [14], and cyclooxygenase-2 [15] that can affect DC processing and presentation, as well as the acquisition and expression of CTL effector cell function.
Current vaccine approaches focus on coupling immunogenic adjuvant agents to tumor antigens. The adjuvant agents are typically mixed with tumor cells or tumor antigens and the admixture is then used to vaccinate patients. Because the adjuvant agents are strongly immunogenic, APCs respond to the vaccine, digest the adjuvant agent along with admixed tumor antigens, and present the adjuvant agents and tumor antigens to T cells. Attempts have been made to strengthen the immune response by incorporating naturally secreted immune modulators into vaccines. Strategies include genetically modifying autologous tumor cells or allogeneic cell lines to secrete cytokines and/or co-stimulatory molecules. Another way to boost the immune system is by expressing the antigen in a viral vector, which can also be designed to encode co-stimulatory molecules or cytokines. The discovery of Toll-like receptors has increased interest in the use of DNA-based vaccines for cancer. DNA plasmid vaccines that contain unmethylated CpG sites stimulate Toll-like receptor 9 (TLR9) on APCs [16]. Other approaches include priming the immune system by using vaccines containing autologous DCs loaded with tumor antigen to elicit a tumor-specific CTL response.
Immunotherapeutic approaches in lung cancer include active immunotherapy (vaccines), passive immunotherapy (monoclonal antibodies), adoptive (T)-cell transfer, cytokine therapy, and nonspecific immunotherapy. The focus of this review is (1) protein antigen-based vaccines, (2) peptide antigen-based vaccines, (3) whole tumor cell vaccines, (4) DC vaccines, and (5) other promising nonspecific immunomodulatory treatments.
1. Protein antigen-based vaccines
1.1 Melanoma-associated antigen (MAGE)-A3
Melanoma-associated antigen (MAGE) is a TAA expressed in cancer cells. MAGE is not expressed in normal tissue, except in male germ cell lines, which are devoid of MHC molecules and are therefore unable to present MAGE-A antigens [17, 18]. MAGE-A3 is the most silent gene in germ line cells within the MAGE-A family and is considered a promising immunotherapy target [19]. MAGE-A3 is expressed in 35% of NSCLC, and the rate of expression increases as the disease spreads (30% of stage I patients and 50% of stage II patients express MAGE-A3 in their primary tumors). MAGE-A3 expression is thus associated with poor prognosis [20, 21].
A randomized phase II trial in 182 patients with completely resected stage IB or II MAGE-A3+ NSCLC comparing postoperative injections of MAGE-A3 recombinant protein combined with an adjuvant system (q3w x 5 followed by q3m x 8) vs. placebo has been reported (Table 1) [22]. After a median follow-up of 28 months, disease-free survival and overall survival hazard ratios (HR) were 0.73 (95% CI 0.45 to 1.16) and 0.66 (95% CI 0.36 to 1.20), respectively, in favor of the MAGE-A3 group. None of the outcome endpoints reached statistical significance; however, the signal with respect to survival benefit was considered strong enough to warrant a phase III evaluation. A recent paper reported an analysis of gene expression profiling of tumors prior to treatment to identify a predictive signature that correlates with clinical activity of MAGE-A3 antigen-specific immunotherapy [23]. In the overall population with resected stage IB/II NSCLC, MAGE-A3 treatment decreased the relative risk of recurrence by 25% (HR 0.75; 95% CI 0.46 to 1.23). In the population with the predictive gene signature, MAGE-A3 treatment decreased the relative risk of recurrence by 43% (HR 0.57; 95% CI 0.25 to 1.34).
Table 1.
Vaccination studies in non-small cell lung cancer
| Immunotherapy | Study | Outcome | |
|---|---|---|---|
| Protein or peptide based vaccines | MAGE-A3 | Vansteenkiste et al. [22]. Phase II; 182 patients with resected stage IB-II. | Immune response. Clinical response. Trend toward survival benefit. |
| EGF | Gonzalez et al. [24]. 83 patients with stage IIIB/IV. | Immune response. Clinical response. Survival benefit for vaccinated patients (8 months) vs. nonrandomized controls (4.5 months). | |
| MUC-1 | Butts et al. [35, 36]. Phase IIB; 171 patients with stage IIIB/IV. | Immune response. Clinical response. Trend toward survival benefit. | |
| IDM-2101 (multiple antigens) | Barve et al. [37]. Phase II; 63 patients with stage IV. | Immune response. Clinical response. Trend toward survival benefit. | |
| Tumor cell vaccines expressing | GM-CSF | Nemunaitis et al. [40]. Phase I/II; 43 patients with stage IB-IV. Nemunaitis et al. (GVAX) [41]. Phase I/II; 49 patients with stage IIIA/IV. |
Immune response. Clinical response. 3/33 advanced-stage patients (2 BACs) durable CRs lasting 6, 18, and ≥ 22 months. OS correlated with level of GM-CSF secretion by vaccine. Immune response. No clinical response. |
| TGF-β antisense | Nemunaitis et al. [51]. Phase II; 75 patients with stage II-IV. | Immune response. Clinical response. |
The MAGRIT (MAGE-A3 as Adjuvant non-small cell lunG canceR ImmunoTherapy) study is currently recruiting in 33 countries, with 2,270 patients with resected stage IB to IIIA MAGE-A3+ NSCLC expected to enroll. Following adjuvant chemotherapy, patients will be randomly assigned to receive 13 injections of vaccine or placebo over a 27-month period, with disease-free survival as the primary endpoint. The trial registry does not define the type of adjuvant chemotherapy.
1.2 Epidermal growth factor (EGF)-based vaccines
Epidermal growth factor and its cell membrane receptor (EGFR) are known to be over-expressed in many epithelial tumors, including lung cancer. In the last decade several small molecule inhibitors of EGFR-associated tyrosine kinases and monoclonal antibodies targeting EGFR have been developed. A vaccine consisting of human recombinant EGF conjugated to the carrier protein P64K, derived from Neisseria meningitides, was evaluated in 3 pilot clinical trials and a pooled analysis accruing a total of 83 patients with stage IIIB/IV NSCLC was performed (Table 1) [24]. Alum and Montanide™ ISA51 have been used as vaccine adjuvants. In one study, a single dose of cyclophosphamide (200 mg/m2) was administered 3 days prior to treatment. After EGF vaccination, 83% of vaccinated patients at least doubled their baseline anti-EGF antibody titers (seroconversion) and 49% increased their baseline levels up to 4-fold. Patients with titers > 1:4000 were considered good antibody responders (GARs). Survival was better in patients who seroconverted (mean 11.06 months, median 8.4 months) compared to patients who did not (mean 5.67 months, median 3.5 months). There was an additional survival advantage for GARs (mean 12.2 months, median 8.37 months) compared to poor antibody responders (mean 8.07 months, median 8.07 months). No significant toxicities were noted.
Neninger Vinageras et al. conducted a randomized phase II controlled trial of an EGF vaccine in patients with stage IIIB/IV NSCLC who had completed first-line chemotherapy [25]. Eighty patients were randomly assigned to receive best supportive care or EGF vaccine. When comparing vaccinated patients with controls, the median overall survival showed a trend toward a survival advantage (6.5 months vs. 5.3 months; p = 0.098). In the subset of vaccinated patients ≤ 60 years of age, survival was significantly longer (11.6 months vs. 5.3 months; p = 0.0124). The vaccination was safe and well tolerated. There was a direct correlation between GAR and survival and between decreased serum EGF and survival. Both of these parameters have been proposed as markers to optimize vaccine dosage and schedules in future clinical trials. Clinical trials to reevaluate this approach in a larger patient series are ongoing.
2. Peptide antigen-based vaccines
2.1 MUC1 and L-BLP25
Mucin 1 (MUC1) is a mucinous transmembrane glycoprotein that is over-expressed and under- or aberrantly glycosylated in many human malignancies. It is normally restricted to the apical surface of polarized epithelial cells, including those of the respiratory tract [26]. In many epithelial malignancies, MUC1 is over-expressed and loses its polarity of expression [27]. MUC1 has a large NH2-terminal ectodomain that becomes under- or aberrantly glycosylated, with shortened carbohydrate side chains, unmasking epitopes on its peptide core which can act as tumor-associated neo-epitopes. The precise role of MUC1 in promoting tumor cell growth and survival is unclear, but it is thought to be involved in tumorigenicity, tumor cell migration, and increased resistance to stress-induced apoptosis of chemotherapeutic agents [28–31].
High serum levels of MUC1 are associated with immune suppression and poor prognosis in patients with advanced adenocarcinoma [32–34]. It is thought that cells that over-express tumor-associated MUC1 may escape a strong host immune response, making MUC1 an attractive target for cancer immunotherapy.
BLP25 liposome vaccine (L-BLP25) targets the exposed core peptide of the MUC1 TAA. L-BLP25 is a lyophilized preparation consisting of BLP25 lipopeptide, immunoadjuvant monophosphoryl lipid A, and 3 lipids (cholesterol, dimyristoyl phosphatidylglycerol, and dipalmitoyl phosphatidylcholine), forming a liposomal product [35]. The vaccine is designed to induce a cellular immune response that may lead to immune rejection of tumor tissues that express MUC1 antigen.
A randomized phase IIB trial of L-BLP25 in patients with stage IIIB/IV NSCLC after stable disease or response to primary chemotherapy has been completed (Table 1; Fig. 3) [35]. L-BLP25 was given weekly for 8 weeks (administered at 4 different sites to improve vaccine uptake in draining lymph nodes), with the option to proceed to maintenance therapy consisting of vaccination every 6 weeks starting in week 13. All patients received a single infusion of cyclophosphamide 300 mg/m2 3 days before vaccine administration, which has been shown to reduce the activity of suppressor T cells [35]. The study was powered to detect a 5-month difference in survival (HR = 0.583), with a power of 80% and one-sided p < 0.25. There were 88 patients in the vaccination arm and 83 in the best supportive care (BSC) arm. Treatment was tolerable, with 96.6% of patients in the vaccine arm completing the planned 8 injections and 69.3% proceeding to the maintenance phase. The most common adverse events were grade 1 flu-like symptoms, events related to cyclophosphamide administration, and mild injection-site reactions. T-cell proliferation assays were performed at baseline and during immunization. Of 78 samples evaluated, 16 demonstrated an antigen-specific T-cell response. The median overall survival was 17.4 months for the vaccination arm vs. 13.0 months with BSC, a difference that did not reach statistical significance (p = 0.066, unadjusted Cox). The 2-year survival rate was 43.2% for the L-BLP25 arm vs. 28.9% for the BSC arm. The greatest difference in survival was observed in patients with stage IIIB loco-regional disease (adjusted HR = 0.524; 95% CI, 0.261 to 1.052; p = 0.069). In a recently reported study with a median follow-up of 53 months, the updated observed 2-year survival rate for patients with stage IIIB loco-regional disease was 60% (median survival 30.6 months) for the L-BLP25 arm vs. 36.7% for the BSC arm (median survival 13.3 months) [36].
Fig 3.

Survival analysis in a randomized phase IIB trial of BLP25 liposome vaccine in the subset of non-small cell lung cancer patients with stage IIIB locoregional disease. Median OS 13.3 months for best supportive care compared with not reached for BLP25 vaccine.
Reprinted with permission. © 2008 American Society of Clinical Oncology. All rights reserved. Butts C, et al.. J Clin Oncol 2005;23:6674–81.
A multicenter, phase III, randomized, double-blind, placebo-controlled study of the cancer vaccine Stimuvax® (L-BLP25 or BLP25 liposome vaccine) in NSCLC subjects with unresectable stage III disease is currently recruiting. The START (Stimulating Targeted Antigenic Responses To NSCLC) trial is expected to recruit more than 1,300 patients who have had a response or have stable disease after at least 2 cycles of definitive platinum-based chemoradiation.
2.2 IDM-2101: 10-epitope CTL vaccine
Tumors express a wide variety of TAAs and have a potential for TAA loss. In addition, patients have differing T-cell repertoires, suggesting that a successful vaccine may need to incorporate a wide selection of CTL specificities. The IDM-2101 vaccine was designed to overcome these potential difficulties by inducing CTL responses against 5 TAAs frequently over-expressed in NSCLC: carcinoembryonic antigen (CEA), p53, HER2/neu, and MAGE-2 and -3 [37]. IDM-2101 is composed of 10 synthetic peptides from these TAAs, 9 of which represent CTL epitopes. The tenth is a pan-DR epitope designed to augment the CTL response [38].
A phase II study investigated the efficacy of IDM-2101 in 63 HLA-A2+ patients with metastatic NSCLC (Table 1). One-year survival for the treated group was 60% and the median survival was 17.3 months. One patient had a complete response and one had a partial response [37]. A phase III validation trial in patients with advanced NSCLC is anticipated.
3. Whole tumor cell vaccines
A development in cancer therapy with potentially major significance is the use of non-antigen-specific vaccines targeting immunomodulatory molecules to therapeutically reeducate T cells to induce an antitumor immune response. Tumor cell vaccines can be divided into 2 broad categories: (1) autologous tumor cell lines with or without gene modification, such as cell suspension of resected tumor transfected with granulocyte-macrophage colony-stimulating factor (GM-CSF), and (2) allogeneic tumor cell lines.
3.1 Cell suspension of whole tumor transfected with GM-CSF
GVAX® is a vaccine composed of whole tumor cells genetically modified to secreteGM-CSF. The initial vaccine used autologous irradiated tumor cells transfected with a non-replicating adenoviral vector engineered to secrete GM-CSF [39]. In the phase I trial involving patients with stage IV NSCLC, the vaccine was well tolerated, with toxicities restricted to grade 1 and 2 local skin reactions. The vaccine demonstrated activity, with 5 patients having stable disease and 2 patients with no demonstrable disease (having had surgical resection of all known metastatic sites prior to vaccination) achieving prolonged remissions of more than 40 months. Based on these promising results, a larger phase I/II trial was reported in 2004 (Table 1) [40]. This trial consisted of 2 cohorts: patients with stage IB or II NSCLC and patients with stage III or IV disease. Only 43 of 83 patients who underwent tumor harvest were treated, of whom 33 had advanced disease. Three of the 33 advanced-stage patients (2 of whom had bronchioloalveolar carcinoma) had durable complete responses lasting 6, 18, and ≥ 22 months. Interestingly, secretion of GM-CSF after vaccination was shown to correlate with outcome. A longer survival was observed in patients receiving vaccines secreting GM-CSF at more than 40 ng/24 h/106 cells (median survival = 17 months; 95% CI 6 to 23 months) than in patients receiving vaccines secreting less GM-CSF (median survival = 7 months; 95% CI 4 to 10 months) (p = 0.028). The results of this study suggested a vaccine dose-related survival advantage.
A subsequent trial evaluated unmodified tumor cells combined with an allogeneic bystander cell line (K562, human erythroleukemia cell line) genetically modified to secrete higher levels of GM-CSF. The combination is referred to as bystander GVAX®. This vaccine does not require genetic modification of individual autologous tumor cell preparations and provides substantially higher and more consistent GM-CSF secretion. Forty-nine patients with stage IIIA to IV NSCLC received vaccine, but unlike in the previous study, none achieved a partial or complete tumor response (Table 1) [41]. There is speculation that derivation from a bystander cell line may detract from GM-CSF’s ability to boost immune responses against the TAA by the induction of myeloid suppressor cells and associated impairment of antigen-specific T-cell responses. It has been concluded that GM-CSF has to be secreted by the tumor cell itself or as recombinant purified protein.
Recent data from phase III clinical trials utilizing this approach in other cancer settings have not demonstrated improved survival compared with standard chemotherapy leading to uncertainty of the future of this platform.
3.2 Allogeneic tumor cell vaccines: belagenpumatucel-L (Lucanix )
Transforming growth factor-beta (TGF-β) is part of a family of multifunctional proteins (isoforms) that regulate the growth and function of many normal and neoplastic cell processes [42, 43]. Through what has been termed the “TGF-β paradox,” TGF-β can have either oncogenic or tumor suppressor effects and can stimulate or inhibit cell growth. Many tumors, including lung cancer, produce high levels of latent and active TGF-β [13]. Elevated levels of TGF-β correlate with tumor-associated immunosuppression and suppression of natural killer (NK) cells and DC activity, and has been identified as a poor prognostic factor in NSCLC [44].
A number of T cell- and DC-associated mechanisms have been proposed to explain the activity of TGF-β, including impairment of high affinity IL-2 receptor function and expression, leading to inhibition of CTL activation [45]. TGF-β2 is also able to convert naïve T cells to T regulatory cells by inducing the transcription factor FOXP3 [46]. T regulatory cells specifically prevent immune activation [47]. DCs play a crucial role in the induction of antitumor immunity in tumor-bearing hosts by a process of antigenic cross-presentation [48]. Immature DCs are unable to stimulate potent immune responses. TGF-β2 can block maturation of DCs and inhibit their ability to present antigens [49, 50].
The vaccine belagenpumatucel-L (Lucanix ) is a mixture of 4 allogeneic NSCLC cell lines genetically modified to secrete an antisense oligonucleotide to TGF-β2. The rationale for using a cell cocktail vaccine is based on the observation that NSCLC tumor cell lines share immunogenic epitopes with primary tumors. MHC class I-restricted CTLs generated against a specific human lung adenocarcinoma cell line have exhibited demonstrable cytotoxicity against other lung cancer cell lines. To increase the array of tumor antigens presented in the vaccine, belagenpumatucel-L employs 4 cell lines that express low levels of TGF-β due to prior transfection with a TGF-β2 antisense plasmid. It is anticipated that down-regulation of TGF-β expression in the vaccine will mitigate a major source of immune suppression at the vaccine injection site. The hypothesis is that injecting allogeneic tumor cells with down-regulated TGF-β2 will enhance local immune recognition and activation of effector cells, leading to a systemic immune response capable of targeting the patient’s native tumor.
In a randomized phase II trial [51], 75 patients with stage II to IV NSCLC were randomized to intradermal injections of 1 to 3 dose levels (1.25 × 107 cells/injection, 2.5 × 107 cells/injection, or 5.0 × 107 cells/injection) on a monthly or every other month schedule to a maximum of 16 injections (Table 1; Fig. 4). No significant adverse events were reported, and a dose-related survival difference was demonstrated in patients who received ≥ 2.5 × 107 cells/injection (p = 0.0155). Analysis suggested that cohorts 2 and 3, who received doses ≥ 2.5 × 107 cells/injection, had a significant survival advantage compared to cohort 1 (p = 0.0069). Of the 61 patients with late-stage NSCLC (IIIB/IV), 15% had a partial response. The estimated probabilities of surviving 1 and 2 years were 68% and 52% respectively for the higher-dose groups combined, and 39% and 20% respectively for the low-dose group. The estimated median survival for patients receiving ≥ 2.5 × 107 cells/injection was 581 days, compared with 252 days for patients receiving 1.25 × 107 cells/injection (p = 0.0186). The difference in overall survival between dose cohorts 1, 2, and 3 in stage III/IV patients was not statistically significant (p = 0.5148). Increased cytokine production (IFN-γ, p = 0.006; IL-6, p = 0.004; and IL-4, p = 0.007) was observed among clinical responders (partial response or stable disease), who also displayed an elevated antibody-mediated response to the vaccine (p = 0.014). A phase III investigation using doses ≥ 2.5 × 107 cells/injection is ongoing as maintenance therapy in patients with unresectable stage III/IV NSCLC who have responded to or have stable disease after first-line platinum-based chemotherapy.
Fig 4.
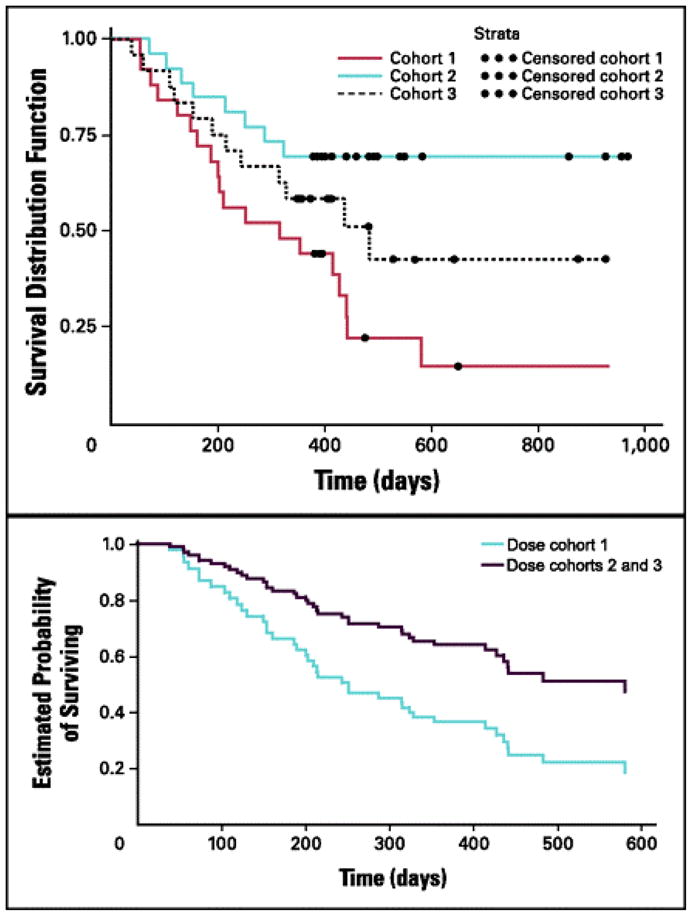
Phase II study of belagenpumatucel-L, a TGF-β2 antisense gene-modified allogeneic tumor cell vaccine in non-small cell lung cancer. Panel A: Dose-related survival between cohorts (1.25, 2.5,or 5.0 × 107 cells/injection) for all patients (N = 75; P = .0155). Panel B: Overall survival for cohort 1 versus cohorts 2 and 3 for advanced-stage patients (n = 61; P = .0186).
Reprinted with permission. © 2008 American Society of Clinical Oncology. All rights reserved. Nemunaitis J, et al. J Clin Oncol 2006;24:4721–30.
4. Dendritic cell vaccines
Some investigators have suggested that targeting tumor antigens alone may not elicit a strong enough immune response to be of therapeutic benefit. The discovery of DCs as professional APCs heralded their use in the development of cancer vaccines [52]. It has been proposed that manipulating DCs as a vaccine adjuvant may be an effective way to stimulate antitumor immunity and overcome tolerance. The principal purpose of DC-based immunotherapy is to induce an antigen-specific immune response [53]. Once stimulated by maturation factors such as inflammatory cytokines, or via CD40, DCs up-regulate adhesion and co-stimulatory molecules to become terminally differentiated stimulators of T-cell immunity [54]. The effector arms of this antitumor response are CD4+ and CD8+ cells, which can only become activated against antigen when presented by APCs [53].
Upon administration of a DC-based vaccine, the DCs should migrate to secondary lymphoid organs and induce an antigen-specific immune response [55, 56]. In the small phase I/II trials of DC vaccines in patients with NSCLC that have been reported, the most common antigen used has been CEA [57–62], a glycoprotein that acts as an adhesion molecule and is over-expressed in 70% of NSCLC [63]. Human leukocyte antigen (HLA)-restricted class I CEA peptides or altered peptides were used as antigens in these studies, as the CEA peptide must be modified for effective immunogenicity.
Hirschowitz et al. reported a study of 16 patients with NSCLC ranging from stage IA to IIIB who received DC-based vaccines [64]. The objectives of the study were to evaluate tolerability and measure immunologic responses in a heterogeneous group of NSCLC patients. Autologous DCs generated ex vivo from monocyte precursors (CD14+) were loaded with apoptotic bodies from an allogeneic adenocarcinoma cell line that overexpressed the TAAs HER2/neu, CEA, Wilm’s tumor 1, MAGE-2, and survivin. Two doses of vaccine consisting of 8 to 9 × 107 cells were administered intradermally one month apart. Immune reactivity measured in vitro showed 3 distinct patterns: 5 of 16 patients showed no measurable immune reactivity, 5 showed reactivity to autologous DCs in the absence of tumor antigen (tumor-independent response), and 6 showed a tumor-specific immune response. Immunologic responses were independent of stage and prior therapy. There was no correlation between immune response and clinical outcome. In a continuation of the study, 14 additional patients were given the vaccine [65]. This study evaluated immunologic responses to immature, antigen-pulsed autologous DC vaccines in 2 distinct groups of NSCLC patients. Seven patients had undergone surgical resection (stage I/II) with or without adjuvant therapy, and 7 had unresectable stage III disease and were treated with chemoradiation alone. Four of the 7 stage III unresectable and 6 of the 7 stage I/II surgically resected patients, including 3 of 3 resected patients who had also received adjuvant chemoradiation, demonstrated immune responses to the vaccine. No clear indication of therapeutic efficacy was noted.
In another study, patients with NSCLC were vaccinated with DCs loaded with MUC1 peptides or tumor lysate [66]. A survival advantage was observed in vaccinated patients whose tumors were found to express MUC1 compared with patients with MUC1-negative tumors (16.75 months vs. 3.8 months; p = 0.0101). Seven of 9 patients with MUC1+ tumors had clinical responses, including one complete response. All of the patients who exhibited partial responses were vaccinated with peptides rather than tumor lysate.
To achieve a greater immune response through better antigen presentation, researchers have used different mechanisms of delivering vaccine to APCs. Morse et al. added antigen directly to ex vivo-generated DCs via an engineered fowlpox virus that expressed full-length CEA and TRICOM, an adjuvant agent consisting of 3 co-stimulatory molecules: B7.1, intracellular adhesion molecule (ICAM)-1, and lymphocyte function-associated antigen (LFA)-3 [60]. Only 3 of 14 patients enrolled had NSCLC. T-cell responses were induced in 10 of 14 patients (71%), with 5 having stable disease for 3 months. The authors did not report the responders’ underlying disease. Another study used a modified vaccinia virus engineered to express the entire MUC1 gene to vaccinate patients with advanced cancer expressing MUC1 [67]. Three of 14 patients enrolled had NSCLC. Of the 14, 4 had stable disease for 6 to 9 months and 5 had an induced T-cell response.
5. Nonspecific immunomodulatory therapies
5.1 Talactoferrin
Talactoferrin alfa (also known as recombinant human lactoferrin) is a recombinant version of the glycoprotein expressed in and purified from Aspergillus niger var. awamor [68]. Talactoferrin is structurally and functionally similar to native human lactoferrin purified from human milk and is known to differ only in the nature of glycosylation [69]. Lactoferrin is a member of the transferrin family of non-heme iron-binding proteins and is found in mammalian serum and exocrine secretions such as milk, seminal fluid, intestinal secretions, tears, sweat, saliva, nasal secretions, and in secretory granules of neutrophils [70]. Lactoferrins have demonstrated anti-inflammatory [71], anti-infective, [72] and antitumor properties [73]. Lactoferrins have a wide array of immunomodulatory functions, including activation of NK and lymphokine-activated killer cells [74], and enhancement of PMN and macrophage cytotoxicity [75]. Lactoferrins have demonstrated anticancer activity in a broad range of tumor types [74].
Talactoferrin is an orally active immunomodulatory protein that specifically binds receptors on cells lining the upper gastrointestinal tract, initiating an immunostimulatory cascade in the gut-associated lymphoid tissue (GALT) [76]. Talactoferrin is thought to act as a chemokine, binding chemokine receptors and inducing the chemotaxis of immune cells, including effector cells such as lymphocytes and APCs. Once administered, talactoferrin is transported by M cells into Peyer’s patches in the small intestine. Here, DCs bearing tumor antigens to GALT are recruited and matured. Talactoferrin also induces the production of key cytokines in the GALT, including IL-18 and IFN-γ [76]. These cytokines play an important role in stimulating both innate and adaptive immunity by promoting the maturation and proliferation of immune cells such as anticancer NK cells, CD8+ lymphocytes, and NK-T cells. In preclinical experiments, oral administration of talactoferrin has produced an increase in the total cellularity of small intestinal Peyer’s patches, including an increase in the number of NK-T cells and CD8+ T lymphocytes [76]. It is hoped that administration of talactoferrin can activate tumor-draining lymph nodes, leading to cellular infiltration of tumors and ultimately to tumor-cell death.
In a phase I study in refractory solid tumors, oral talactoferrin alfa was well tolerated and showed promising disease activity [76]. Three of the 10 patients in the study had NSCLC, and 2 of these 3 showed the most significant slowing in rate of growth and development of new metastatic lesions. Both patients remained on study for over 6 months. In the aggregate, the average rate of tumor growth dropped from 33.3%/month to 9.9%/month, a relative decrease of > 70%. It was hoped that this sustained decrease in the rate of tumor growth could be translated into improved progression-free and overall survival in phase II and III studies of talactoferrin alfa [76].
Two company-sponsored trials have been conducted in patients with NSCLC. The first was a randomized phase II study in patients with locally advanced or metastatic (stage IIIB/IV) NSCLC who had failed first- or second-line chemotherapy. The study enrolled 100 patients who received one vial b.i.d. of either 1.5 g talactoferrin (n = 47) or placebo (n = 53) for up to 3 cycles. Each 14-week cycle consisted of 12 consecutive weeks on study drug, followed by 2 weeks off. Patients in the talactoferrin arm had a 62% increase in median overall survival compared to the placebo arm (6.0 months vs. 3.7 months; p < 0.05; HR 0.69) and also showed a trend toward an improvement in progression-free survival. Talactoferrin was well tolerated. The second phase II study used talactoferrin or placebo in combination with carboplatin and paclitaxel in 110 patients with locally advanced and/or metastatic NSCLC (stage IIIB/IV). The response rate increased from 27% in the placebo arm to 42% in the talactoferrin arm (15% absolute improvement, 56% relative improvement; p = 0.08). Median overall survival increased 22% in the intent-to-treat population (10.4 months for talactoferrin vs. 8.5 months for placebo). Safety data indicated that talactoferrin with chemotherapy was well tolerated in this patient population.
FORTIS-M is an ongoing phase III randomized, double-blind, placebo-controlled study of oral talactoferrin plus BSC in patients with NSCLC who have failed ≥ 2 prior treatment regimens.
5.2 Toll-like receptor 9 agonists
Oligonucleotides containing certain CpG sequence motifs can stimulate an innate and adaptive immune response. TLRs are a family of highly conserved receptors that regulate innate antigen-specific immunity by recognizing pathogen-associated molecular patterns [77]. TLR9 is expressed on B and T lymphocytes, plasmacytoid cells, and DCs. Activation of TLR9 may reduce immune tolerance and improve tumor antigen recognition and cell death via both innate and specific immune systems [78]. Through the TLR9 signaling pathway, immunostimulatory oligonucleotides activate a complex cascade that leads to stimulation of an immune response and increased production of proinflammatory cytokines and chemokines [79, 80]. TLR9 agonists enhance the maturation of plasmacytoid dendritic cells (pDCs). Mature pDCs exhibit increased cell-surface expression of MHC class II molecules and the costimulatory molecule B7. They secrete cytokines such as IFN-α that are associated with a TH1 response and secondarily activate NK cells, monocytes, and neutrophils [81, 82]. Induction of this rapid innate immune response can be followed by an adaptive (antigen-specific) immune response through presentation of tumor antigens to CTLs. TLR9 agonists can reduce the threshold for antigenic stimulation of B cells, enhancing differentiation into antibody-secreting plasma cells and potentially promoting antibody-dependent cellular cytotoxicity [83]. It has been shown that TLR9 is expressed in many lung cancer cell lines and lung cancer specimens, whereas nonmalignant lung tissue expresses low levels of TLR9 [84]. It is thought that this stimulation of the immune system leads to the antitumor effects of CpG oligonucleotides [79, 80, 85].
PF-3512676 (formerly known as CpG 7909) is a synthetic, nuclease-resistant, TLR9-activating oligodeoxynucleotide that mimics the natural ligand of TLR9 (unmethylated CpG motifs), thereby inducing a cascade of immune reactions and potentially promoting an antitumor immune response [82, 86]. In a multicenter randomized phase II trial of PF-3512676 in 112 chemotherapy-naive patients with stage IIIB/IV NSCLC [16], patients were randomized 2:1 to receive 4 to 6 cycles of a standard regimen of carboplatin and paclitaxel alone or with 0.2 mg/kg of s.c. PF-3512676 on days 8 and 15 of each 3-week cycle. Median survival was 12.3 months in the PF-3512676 arm vs. 6.8 months in the chemotherapy-alone arm; 1-year survival was 50% and 33%, respectively. HR for overall survival was 0.747 (95% CI, 0.484 to 1.153; p = 0.188). On the basis of this phase II trial, 2 randomized, international, multicenter phase III trials commenced comparing PF-3512676 plus platinum-based chemotherapy (paclitaxel plus carboplatin or gemcitabine plus cisplatin) with chemotherapy alone in chemotherapy-naïve patients with locally advanced or metastatic NSCLC. Both trials were discontinued after interim analysis by an independent data monitoring safety committee concluded that PF-3512676 provided no additional benefit in terms of overall survival or progression-free survival over standard platinum-based doublet chemotherapy.
5.3 Neu-glycosylated gangliosides and 1E10 anti-idiotype vaccine
An attractive focus for immunotherapy using anti-idiotype monoclonal antibodies (mAb) are Neu-glycosylated (NeuGc)-containing gangliosides. Carbohydrate determinants are known to undergo extensive changes during the course of malignant transformation [87]. GM2 is a monosialoganglioside that contains one N-acetyl sialic acid residue. Many human cancers express the nonhuman sialic acid N-glycolyl sialic acid (NeuGc), which is known to be immunogenic in humans. It is designated as the Hanganutziu-Deicher (H-D) antigen. These glycolipids have a differential pattern of expression in normal human tissue and malignant cells, making them a potentially relevant therapeutic target [88, 89]. 1E10 is a vaccine preparation containing a murine anti-idiotype mAb that reacts with NeuGc-containing ganglioside antigen [90]. Phase I clinical trials have shown a favorable toxicity profile in various cancer types, including small cell lung cancer, using aluminum hydroxide-precipitated 1E10 mAb [91]. 1E10 was evaluated in 71 patients with stage IIIB/IV NSCLC in a compassionate-use study [92]. All patients (15 adenocarcinomas, 45 squamous cell carcinomas, and 11 large cell carcinomas) had completed standard chemotherapy and radiotherapy for their stage of disease. Patients received 5 biweekly injections of 1 mg 1E10/alum, and those who maintained a good performance status received 10 more doses at 28-day intervals. Overall survival from the time of initial vaccination was 9.93 months (95% CI, 8.61 to 11.25); one-year survival rate was 34%. Median survival of the 56 patients who entered the study with either partial response or stable disease and with a performance status of 1 after first-line chemo/radiotherapy was 11.5 months (95% CI, 7.97 to 15.03 months); one-year survival rate was 39%. Patients who commenced treatment with progressive disease or performance status 2 had a median overall survival of 6.5 months (95% CI, 4.31 to 8.69 months) and a one-year survival rate of 13% (p = 0.002). Ongoing trials are investigating the clinical effect of 1E10 mAb vaccine in NSCLC patients with partial response or stable disease and a performance status of 1, and evaluating the correlation between the immune responses induced by this anti-idiotype vaccine and patient survival.
4.4 Anti-CTLA-4 therapy
Cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) is a critical element in the homeostatic mechanism of immunosuppression and T-cell regulation. CTLA-4 is a transmembrane protein expressed on the surface of activated T lymphocytes and monocytes. Weak antigens such as TAAs cause an antigen-specific T-cell response via an interaction with the T-cell receptor. This interaction relies on nonantigen-specific signals generated when CD28 on the T cell binds to B7 on the APC. Once this occurs, CTLA-4 becomes expressed on the T-cell surface [93]. CTLA-4 has a higher binding affinity to B7 than to CD28, which leads to a diminished immune response by decreasing IL-2 and IL-2 receptor expression [94–96].
Two potential immunological strategies targeting such molecules on activated T cells include (1) administering antibodies with agonistic effects on co-stimulatory molecules such as CD137, which is rapidly induced on CD4+ and CD8+ T cells after activation and enhances T-cell activation/proliferation on cross-linking [97]; or (2) administering antibodies with antagonistic effects for co-inhibitory molecules such as CTLA-4. Preliminary phase I data in melanoma have confirmed the safety of CTLA-4 antibodies, and there are ongoing phase II studies in multiple tumor types, including NSCLC. A phase II study evaluating the safety and efficacy of ipilimumab (BMS-734016) plus paclitaxel/carboplatin vs. paclitaxel/carboplatin alone in previously untreated patients with both NSCLC and SCLC is currently recruiting [98].
Conclusion
Developing an effective immunotherapy for lung cancer is a daunting challenge. Until recently, NSCLC was considered non-immunogenic or poorly immunogenic. Although it is unlikely that the immune system will be able to adequately and consistently deal with bulky disease, effective immunotherapy would be an ideal adjuvant following initial clinical responses to definitive surgical multimodality or medical therapy.
At present, there are limited data from phase III trials showing a clear clinical benefit for vaccines in lung cancer. One explanation may be that a measurable immune response does not translate into a clinically meaningful response. Patient selection may also be a problem for ongoing clinical studies. The majority of these trials are focused on patients with advanced-stage disease, while the ideal candidates for lung cancer vaccines may be patients with stage I or II disease who are considered at high risk of recurrence post-resection. Selecting the optimal tumor antigens to target is also problematic. A truly effective therapeutic lung cancer vaccine may require multiple epitopes of a diverse set of genes restricted to multiple haplotypes in order to combat the various tumor escape mechanisms.
It is hoped that ongoing studies will provide additional information on optimal patient selection and vaccine administration needed to bridge the gap between promising concept and therapeutic reality, thereby expanding the thoracic oncologist’s armamentarium in the fight against lung cancer.
References
- 1.Cancer Facts & Figures. American Cancer Society; Atlanta, GA: 2006. [Google Scholar]
- 2.Sugimura H, Nichols FC, Yang P, Allen MS, Cassivi SD, Deschamps C, Williams BA, Pairolero PC. Survival after recurrent nonsmall-cell lung cancer after complete pulmonary resection. Ann Thorac Surg. 2007;83:409–417. doi: 10.1016/j.athoracsur.2006.08.046. discussioin 417–408. [DOI] [PubMed] [Google Scholar]
- 3.Shepherd FA, Dancey J, Ramlau R, Mattson K, Gralla R, O'Rourke M, Levitan N, Gressot L, Vincent M, Burkes R, Coughlin S, Kim Y, Berille J. Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J Clin Oncol. 2000;18:2095–2103. doi: 10.1200/JCO.2000.18.10.2095. [DOI] [PubMed] [Google Scholar]
- 4.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 5.Lechler R, Chai JG, Marelli-Berg F, Lombardi G. The contributions of T-cell anergy to peripheral T-cell tolerance. Immunology. 2001;103:262–269. doi: 10.1046/j.1365-2567.2001.01250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Russell JH, Ley TJ. Lymphocyte-mediated cytotoxicity. Annu Rev Immunol. 2002;20:323–370. doi: 10.1146/annurev.immunol.20.100201.131730. [DOI] [PubMed] [Google Scholar]
- 7.Farber DL. T cell memory: heterogeneity and mechanisms. Clin Immunol. 2000;95:173–181. doi: 10.1006/clim.2000.4858. [DOI] [PubMed] [Google Scholar]
- 8.Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–758. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- 9.Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–839. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- 10.Dorfman DM, Schultze JL, Shahsafaei A, Michalak S, Gribben JG, Freeman GJ, Pinkus GS, Nadler LM. In vivo expression of B7-1 and B7-2 by follicular lymphoma cells can prevent induction of T-cell anergy but is insufficient to induce significant T-cell proliferation. Blood. 1997;90:4297–4306. [PubMed] [Google Scholar]
- 11.Garcia-Hernandez ML, Hernandez-Pando R, Gariglio P, Berumen J. Interleukin-10 promotes B16-melanoma growth by inhibition of macrophage functions and induction of tumour and vascular cell proliferation. Immunology. 2002;105:231–243. doi: 10.1046/j.1365-2567.2002.01363.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Skinnider BF, Mak TW. The role of cytokines in classical Hodgkin lymphoma. Blood. 2002;99:4283–4297. doi: 10.1182/blood-2002-01-0099. [DOI] [PubMed] [Google Scholar]
- 13.Fischer JR, Darjes H, Lahm H, Schindel M, Drings P, Krammer PH. Constitutive secretion of bioactive transforming growth factor beta 1 by small cell lung cancer cell lines. Eur J Cancer. 1994;30A:2125–2129. doi: 10.1016/0959-8049(94)00364-b. [DOI] [PubMed] [Google Scholar]
- 14.Huang M, Sharma S, Mao JT, Dubinett SM. Non-small cell lung cancer-derived soluble mediators and prostaglandin E2 enhance peripheral blood lymphocyte IL-10 transcription and protein production. J Immunol. 1996;157:5512–5520. [PubMed] [Google Scholar]
- 15.Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty GJ, Sharma S, Huang M, Pold M, Batra RK, Dubinett SM. Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. J Biol Chem. 2001;276:20809–20812. doi: 10.1074/jbc.C100140200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Manegold C, Gravenor D, Woytowitz D, Mezger J, Hirsh V, Albert G, Al-Adhami M, Readett D, Krieg AM, Leichman CG. Randomized phase II trial of a toll-like receptor 9 agonist oligodeoxynucleotide, PF-3512676, in combination with first-line taxane plus platinum chemotherapy for advanced-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:3979–3986. doi: 10.1200/JCO.2007.12.5807. [DOI] [PubMed] [Google Scholar]
- 17.Takahashi K, Shichijo S, Noguchi M, Hirohata M, Itoh K. Identification of MAGE-1 and MAGE-4 proteins in spermatogonia and primary spermatocytes of testis. Cancer Res. 1995;55:3478–3482. [PubMed] [Google Scholar]
- 18.Tomita Y, Kimura M, Tanikawa T, Nishiyama T, Morishita H, Takeda M, Fujiwara M, Sato S. Immunohistochemical detection of intercellular adhesion molecule-1 (ICAM-1) and major histocompatibility complex class I antigens in seminoma. J Urol. 1993;149:659–663. doi: 10.1016/s0022-5347(17)36174-8. [DOI] [PubMed] [Google Scholar]
- 19.Gaugler B, Van den Eynde B, van der Bruggen P, Romero P, Gaforio JJ, De Plaen E, Lethe B, Brasseur F, Boon T. Human gene MAGE-3 codes for an antigen recognized on a melanoma by autologous cytolytic T lymphocytes. J Exp Med. 1994;179:921–930. doi: 10.1084/jem.179.3.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gure AO, Chua R, Williamson B, Gonen M, Ferrera CA, Gnjatic S, Ritter G, Simpson AJ, Chen YT, Old LJ, Altorki NK. Cancer-testis genes are coordinately expressed and are markers of poor outcome in non-small cell lung cancer. Clin Cancer Res. 2005;11:8055–8062. doi: 10.1158/1078-0432.CCR-05-1203. [DOI] [PubMed] [Google Scholar]
- 21.Sienel W, Varwerk C, Linder A, Kaiser D, Teschner M, Delire M, Stamatis G, Passlick B. Melanoma associated antigen (MAGE)-A3 expression in Stages I and II non-small cell lung cancer: results of a multi-center study. Eur J Cardiothorac Surg. 2004;25:131–134. doi: 10.1016/j.ejcts.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 22.Vansteenkiste J, Zielinski M, Linder A, Dahabre J, Esteban E, Malinowski W, Jassem J, Passlick B, Lehmann F, Brichard V. Final results of a multi-center, double-blind, randomized, placebo-controlled phase II study to assess the efficacy of MAGE-A3 immunotherapeutic as adjuvant therapy in stage IB/II non-small cell lung cancer (NSCLC) [abstract] J Clin Oncol. 2007;25(18S):7554. [Google Scholar]
- 23.Vansteenkiste J, Zielinski M, Dahabreh I, Linder A, Lehmann F, Gruselle O, Therasse P, Louahed J, Brichard V. Association of gene expression signature and clinical efficacy of MAGE-A3 antigen-specific cancer immunotherapeutic (ASCI) as adjuvant therapy in resected stage IB/II non-small cell lung cancer (NSCLC) [abstract] J Clin Oncol. 2008;26(15S):7501. [Google Scholar]
- 24.Gonzalez G, Crombet T, Neninger E, Viada C, Lage A. Therapeutic vaccination with epidermal growth factor (EGF) in advanced lung cancer: analysis of pooled data from three clinical trials. Hum Vaccin. 2007;3:8–13. doi: 10.4161/hv.3.1.3537. [DOI] [PubMed] [Google Scholar]
- 25.Neninger Vinageras E, de la Torre A, Osorio Rodriguez M, Catala Ferrer M, Bravo I, Mendoza del Pino M, Abreu Abreu D, Acosta Brooks S, Rives R, del Castillo Carrillo C, Gonzalez Duenas M, Viada C, Garcia Verdecia B, Crombet Ramos T, Gonzalez Marinello G, Lage Davila A. Phase II randomized controlled trial of an epidermal growth factor vaccine in advanced non-small-cell lung cancer. J Clin Oncol. 2008;26:1452–1458. doi: 10.1200/JCO.2007.11.5980. [DOI] [PubMed] [Google Scholar]
- 26.Vlad AM, Kettel JC, Alajez NM, Carlos CA, Finn OJ. MUC1 immunobiology: from discovery to clinical applications. Adv Immunol. 2004;82:249–293. doi: 10.1016/S0065-2776(04)82006-6. [DOI] [PubMed] [Google Scholar]
- 27.Ho SB, Niehans GA, Lyftogt C, Yan PS, Cherwitz DL, Gum ET, Dahiya R, Kim YS. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res. 1993;53:641–651. [PubMed] [Google Scholar]
- 28.Rahn JJ, Chow JW, Horne GJ, Mah BK, Emerman JT, Hoffman P, Hugh JC. MUC1 mediates transendothelial migration in vitro by ligating endothelial cell ICAM-1. Clin Exp Metastasis. 2005;22:475–483. doi: 10.1007/s10585-005-3098-x. [DOI] [PubMed] [Google Scholar]
- 29.Yin L, Li Y, Ren J, Kuwahara H, Kufe D. Human MUC1 carcinoma antigen regulates intracellular oxidant levels and the apoptotic response to oxidative stress. J Biol Chem. 2003;278:35458–35464. doi: 10.1074/jbc.M301987200. [DOI] [PubMed] [Google Scholar]
- 30.Yin L, Huang L, Kufe D. MUC1 oncoprotein activates the FOXO3a transcription factor in a survival response to oxidative stress. J Biol Chem. 2004;279:45721–45727. doi: 10.1074/jbc.M408027200. [DOI] [PubMed] [Google Scholar]
- 31.Ren J, Agata N, Chen D, Li Y, Yu WH, Huang L, Raina D, Chen W, Kharbanda S, Kufe D. Human MUC1 carcinoma-associated protein confers resistance to genotoxic anticancer agents. Cancer Cell. 2004;5:163–175. doi: 10.1016/s1535-6108(04)00020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reddish MA, MacLean GD, Poppema S, Berg A, Longenecker BM. Pre-immunotherapy serum CA27.29 (MUC-1) mucin level and CD69+ lymphocytes correlate with effects of Theratope sialyl-Tn-KLH cancer vaccine in active specific immunotherapy. Cancer Immunol Immunother. 1996;42:303–309. doi: 10.1007/s002620050287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agrawal B, Krantz MJ, Reddish MA, Longenecker BM. Cancer-associated MUC1 mucin inhibits human T-cell proliferation, which is reversible by IL-2. Nat Med. 1998;4:43–49. doi: 10.1038/nm0198-043. [DOI] [PubMed] [Google Scholar]
- 34.Hiltbold EM, Vlad AM, Ciborowski P, Watkins SC, Finn OJ. The mechanism of unresponsiveness to circulating tumor antigen MUC1 is a block in intracellular sorting and processing by dendritic cells. J Immunol. 2000;165:3730–3741. doi: 10.4049/jimmunol.165.7.3730. [DOI] [PubMed] [Google Scholar]
- 35.Butts C, Murray N, Maksymiuk A, Goss G, Marshall E, Soulieres D, Cormier Y, Ellis P, Price A, Sawhney R, Davis M, Mansi J, Smith C, Vergidis D, Ellis P, MacNeil M, Palmer M. Randomized phase IIB trial of BLP25 liposome vaccine in stage IIIB and IV non-small-cell lung cancer. J Clin Oncol. 2005;23:6674–6681. doi: 10.1200/JCO.2005.13.011. [DOI] [PubMed] [Google Scholar]
- 36.Butts C, Maksymiuk A, Goss G. A multicentre phase IIB randomized controlled study of BLP25 liposome vaccine (L-BLP25 or Stimuvax) for active specific immunotherapy of non-small cell lung cancer (NSCLC): updated survival analysis [abstract] J Thorac Oncol. 2007;S2:B1–01. [Google Scholar]
- 37.Barve M, Bender J, Senzer N, Cunningham C, Greco FA, McCune D, Steis R, Khong H, Richards D, Stephenson J, Ganesa P, Nemunaitis J, Ishioka G, Pappen B, Nemunaitis M, Morse M, Mills B, Maples PB, Sherman J, Nemunaitis JJ. Induction of immune responses and clinical efficacy in a phase II trial of IDM-2101, a 10-epitope cytotoxic T-lymphocyte vaccine, in metastatic non-small-cell lung cancer. J Clin Oncol. 2008;26:4418–4425. doi: 10.1200/JCO.2008.16.6462. [DOI] [PubMed] [Google Scholar]
- 38.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, Snoke K, Serra HM, Kubo RT, Sette A, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994;1:751–761. doi: 10.1016/s1074-7613(94)80017-0. [DOI] [PubMed] [Google Scholar]
- 39.Salgia R, Lynch T, Skarin A, Lucca J, Lynch C, Jung K, Hodi FS, Jaklitsch M, Mentzer S, Swanson S, Lukanich J, Bueno R, Wain J, Mathisen D, Wright C, Fidias P, Donahue D, Clift S, Hardy S, Neuberg D, Mulligan R, Webb I, Sugarbaker D, Mihm M, Dranoff G. Vaccination with irradiated autologous tumor cells engineered to secrete granulocyte-macrophage colony-stimulating factor augments antitumor immunity in some patients with metastatic non-small-cell lung carcinoma. J Clin Oncol. 2003;21:624–630. doi: 10.1200/JCO.2003.03.091. [DOI] [PubMed] [Google Scholar]
- 40.Nemunaitis J, Sterman D, Jablons D, Smith JW, 2nd, Fox B, Maples P, Hamilton S, Borellini F, Lin A, Morali S, Hege K. Granulocyte-macrophage colony-stimulating factor gene-modified autologous tumor vaccines in non-small-cell lung cancer. J Natl Cancer Inst. 2004;96:326–331. doi: 10.1093/jnci/djh028. [DOI] [PubMed] [Google Scholar]
- 41.Nemunaitis J, Jahan T, Ross H, Sterman D, Richards D, Fox B, Jablons D, Aimi J, Lin A, Hege K. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther. 2006;13:555–562. doi: 10.1038/sj.cgt.7700922. [DOI] [PubMed] [Google Scholar]
- 42.Sporn MB, Roberts AB, Wakefield LM, Assoian RK. Transforming growth factor-beta: biological function and chemical structure. Science. 1986;233:532–534. doi: 10.1126/science.3487831. [DOI] [PubMed] [Google Scholar]
- 43.Massague J. The TGF-beta family of growth and differentiation factors. Cell. 1987;49:437–438. doi: 10.1016/0092-8674(87)90443-0. [DOI] [PubMed] [Google Scholar]
- 44.Kong F, Jirtle RL, Huang DH, Clough RW, Anscher MS. Plasma transforming growth factor-beta1 level before radiotherapy correlates with long term outcome of patients with lung carcinoma. Cancer. 1999;86:1712–1719. [PubMed] [Google Scholar]
- 45.Ruffini PA, Rivoltini L, Silvani A, Boiardi A, Parmiani G. Factors, including transforming growth factor beta, released in the glioblastoma residual cavity, impair activity of adherent lymphokine-activated killer cells. Cancer Immunol Immunother. 1993;36:409–416. doi: 10.1007/BF01742258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen W, Wahl SM. TGF-beta: the missing link in CD4+CD25+ regulatory T cell-mediated immunosuppression. Cytokine Growth Factor Rev. 2003;14:85–89. doi: 10.1016/s1359-6101(03)00003-0. [DOI] [PubMed] [Google Scholar]
- 47.Woo EY, Chu CS, Goletz TJ, Schlienger K, Yeh H, Coukos G, Rubin SC, Kaiser LR, June CH. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early-stage non-small cell lung cancer and late-stage ovarian cancer. Cancer Res. 2001;61:4766–4772. [PubMed] [Google Scholar]
- 48.Enk AH, Jonuleit H, Saloga J, Knop J. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int J Cancer. 1997;73:309–316. doi: 10.1002/(sici)1097-0215(19971104)73:3<309::aid-ijc1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 49.Kao JY, Gong Y, Chen CM, Zheng QD, Chen JJ. Tumor-derived TGF-beta reduces the efficacy of dendritic cell/tumor fusion vaccine. J Immunol. 2003;170:3806–3811. doi: 10.4049/jimmunol.170.7.3806. [DOI] [PubMed] [Google Scholar]
- 50.Kobie JJ, Wu RS, Kurt RA, Lou S, Adelman MK, Whitesell LJ, Ramanathapuram LV, Arteaga CL, Akporiaye ET. Transforming growth factor beta inhibits the antigen-presenting functions and antitumor activity of dendritic cell vaccines. Cancer Res. 2003;63:1860–1864. [PubMed] [Google Scholar]
- 51.Nemunaitis J, Dillman RO, Schwarzenberger PO, Senzer N, Cunningham C, Cutler J, Tong A, Kumar P, Pappen B, Hamilton C, DeVol E, Maples PB, Liu L, Chamberlin T, Shawler DL, Fakhrai H. Phase II study of belagenpumatucel-L, a transforming growth factor beta-2 antisense gene-modified allogeneic tumor cell vaccine in non-small-cell lung cancer. J Clin Oncol. 2006;24:4721–4730. doi: 10.1200/JCO.2005.05.5335. [DOI] [PubMed] [Google Scholar]
- 52.Cranmer LD, Trevor KT, Hersh EM. Clinical applications of dendritic cell vaccination in the treatment of cancer. Cancer Immunol Immunother. 2004;53:275–306. doi: 10.1007/s00262-003-0432-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15:138–147. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- 54.van Kooten C, Banchereau J. Functions of CD40 on B cells, dendritic cells and other cells. Curr Opin Immunol. 1997;9:330–337. doi: 10.1016/s0952-7915(97)80078-7. [DOI] [PubMed] [Google Scholar]
- 55.Gatti E, Pierre P. Understanding the cell biology of antigen presentation: the dendritic cell contribution. Curr Opin Cell Biol. 2003;15:468–473. doi: 10.1016/s0955-0674(03)00069-3. [DOI] [PubMed] [Google Scholar]
- 56.Offringa R, van der Burg SH, Ossendorp F, Toes RE, Melief CJ. Design and evaluation of antigen-specific vaccination strategies against cancer. Curr Opin Immunol. 2000;12:576–582. doi: 10.1016/s0952-7915(00)00145-x. [DOI] [PubMed] [Google Scholar]
- 57.Babatz J, Rollig C, Lobel B, Folprecht G, Haack M, Gunther H, Kohne CH, Ehninger G, Schmitz M, Bornhauser M. Induction of cellular immune responses against carcinoembryonic antigen in patients with metastatic tumors after vaccination with altered peptide ligand-loaded dendritic cells. Cancer Immunol Immunother. 2006;55:268–276. doi: 10.1007/s00262-005-0021-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, Davis MM, Engleman EG. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci U S A. 2001;98:8809–8814. doi: 10.1073/pnas.141226398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Itoh T, Ueda Y, Kawashima I, Nukaya I, Fujiwara H, Fuji N, Yamashita T, Yoshimura T, Okugawa K, Iwasaki T, Ideno M, Takesako K, Mitsuhashi M, Orita K, Yamagishi H. Immunotherapy of solid cancer using dendritic cells pulsed with the HLA-A24-restricted peptide of carcinoembryonic antigen. Cancer Immunol Immunother. 2002;51:99–106. doi: 10.1007/s00262-001-0257-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morse MA, Clay TM, Hobeika AC, Osada T, Khan S, Chui S, Niedzwiecki D, Panicali D, Schlom J, Lyerly HK. Phase I study of immunization with dendritic cells modified with fowlpox encoding carcinoembryonic antigen and costimulatory molecules. Clin Cancer Res. 2005;11:3017–3024. doi: 10.1158/1078-0432.CCR-04-2172. [DOI] [PubMed] [Google Scholar]
- 61.Morse MA, Deng Y, Coleman D, Hull S, Kitrell-Fisher E, Nair S, Schlom J, Ryback ME, Lyerly HK. A Phase I study of active immunotherapy with carcinoembryonic antigen peptide (CAP-1)-pulsed, autologous human cultured dendritic cells in patients with metastatic malignancies expressing carcinoembryonic antigen. Clin Cancer Res. 1999;5:1331–1338. [PubMed] [Google Scholar]
- 62.Ueda Y, Itoh T, Nukaya I, Kawashima I, Okugawa K, Yano Y, Yamamoto Y, Naitoh K, Shimizu K, Imura K, Fuji N, Fujiwara H, Ochiai T, Itoi H, Sonoyama T, Hagiwara A, Takesako K, Yamagishi H. Dendritic cell-based immunotherapy of cancer with carcinoembryonic antigen-derived, HLA-A24-restricted CTL epitope: Clinical outcomes of 18 patients with metastatic gastrointestinal or lung adenocarcinomas. Int J Oncol. 2004;24:909–917. [PubMed] [Google Scholar]
- 63.Hammarstrom S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999;9:67–81. doi: 10.1006/scbi.1998.0119. [DOI] [PubMed] [Google Scholar]
- 64.Hirschowitz EA, Foody T, Kryscio R, Dickson L, Sturgill J, Yannelli J. Autologous dendritic cell vaccines for non-small-cell lung cancer. J Clin Oncol. 2004;22:2808–2815. doi: 10.1200/JCO.2004.01.074. [DOI] [PubMed] [Google Scholar]
- 65.Hirschowitz EA, Foody T, Hidalgo GE, Yannelli JR. Immunization of NSCLC patients with antigen-pulsed immature autologous dendritic cells. Lung Cancer. 2007;57:365–372. doi: 10.1016/j.lungcan.2007.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kontani K, Taguchi O, Ozaki Y, Hanaoka J, Sawai S, Inoue S, Abe H, Hanasawa K, Fujino S. Dendritic cell vaccine immunotherapy of cancer targeting MUC1 mucin. Int J Mol Med. 2003;12:493–502. [PubMed] [Google Scholar]
- 67.Rochlitz C, Figlin R, Squiban P, Salzberg M, Pless M, Herrmann R, Tartour E, Zhao Y, Bizouarne N, Baudin M, Acres B. Phase I immunotherapy with a modified vaccinia virus (MVA) expressing human MUC1 as antigen-specific immunotherapy in patients with MUC1-positive advanced cancer. J Gene Med. 2003;5:690–699. doi: 10.1002/jgm.397. [DOI] [PubMed] [Google Scholar]
- 68.Anderson BF, Baker HM, Norris GE, Rice DW, Baker EN. Structure of human lactoferrin: crystallographic structure analysis and refinement at 2.8 A resolution. J Mol Biol. 1989;209:711–734. doi: 10.1016/0022-2836(89)90602-5. [DOI] [PubMed] [Google Scholar]
- 69.Sun XL, Baker HM, Shewry SC, Jameson GB, Baker EN. Structure of recombinant human lactoferrin expressed in Aspergillus awamori. Acta Crystallogr D Biol Crystallogr. 1999;55:403–407. doi: 10.1107/s0907444998011226. [DOI] [PubMed] [Google Scholar]
- 70.Levay PF, Viljoen M. Lactoferrin: a general review. Haematologica. 1995;80:252–267. [PubMed] [Google Scholar]
- 71.Bellamy W, Takase M, Wakabayashi H, Kawase K, Tomita M. Antibacterial spectrum of lactoferricin B, a potent bactericidal peptide derived from the N-terminal region of bovine lactoferrin. J Appl Bacteriol. 1992;73:472–479. doi: 10.1111/j.1365-2672.1992.tb05007.x. [DOI] [PubMed] [Google Scholar]
- 72.Legrand D, Elass E, Carpentier M, Mazurier J. Interactions of lactoferrin with cells involved in immune function. Biochem Cell Biol. 2006;84:282–290. doi: 10.1139/o06-045. [DOI] [PubMed] [Google Scholar]
- 73.Bezault J, Bhimani R, Wiprovnick J, Furmanski P. Human lactoferrin inhibits growth of solid tumors and development of experimental metastases in mice. Cancer Res. 1994;54:2310–2312. [PubMed] [Google Scholar]
- 74.Tsuda H, Sekine K, Fujita K, Ligo M. Cancer prevention by bovine lactoferrin and underlying mechanisms--a review of experimental and clinical studies. Biochem Cell Biol. 2002;80:131–136. doi: 10.1139/o01-239. [DOI] [PubMed] [Google Scholar]
- 75.Gahr M, Speer CP, Damerau B, Sawatzki G. Influence of lactoferrin on the function of human polymorphonuclear leukocytes and monocytes. J Leukoc Biol. 1991;49:427–433. doi: 10.1002/jlb.49.5.427. [DOI] [PubMed] [Google Scholar]
- 76.Hayes TG, Falchook GF, Varadhachary GR, Smith DP, Davis LD, Dhingra HM, Hayes BP, Varadhachary A. Phase I trial of oral talactoferrin alfa in refractory solid tumors. Invest New Drugs. 2006;24:233–240. doi: 10.1007/s10637-005-3690-6. [DOI] [PubMed] [Google Scholar]
- 77.Chen K, Huang J, Gong W, Iribarren P, Dunlop NM, Wang JM. Toll-like receptors in inflammation, infection and cancer. Int Immunopharmacol. 2007;7:1271–1285. doi: 10.1016/j.intimp.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 78.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 79.Klinman DM, Yi AK, Beaucage SL, Conover J, Krieg AM. CpG motifs present in bacteria DNA rapidly induce lymphocytes to secrete interleukin 6, interleukin 12, and interferon gamma. Proc Natl Acad Sci U S A. 1996;93:2879–2883. doi: 10.1073/pnas.93.7.2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 81.Krieg AM. Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Curr Oncol Rep. 2004;6:88–95. doi: 10.1007/s11912-004-0019-0. [DOI] [PubMed] [Google Scholar]
- 82.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 83.Traggiai E, Becker S, Subbarao K, Kolesnikova L, Uematsu Y, Gismondo MR, Murphy BR, Rappuoli R, Lanzavecchia A. An efficient method to make human monoclonal antibodies from memory B cells: potent neutralization of SARS coronavirus. Nat Med. 2004;10:871–875. doi: 10.1038/nm1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Droemann D, Albrecht D, Gerdes J, Ulmer AJ, Branscheid D, Vollmer E, Dalhoff K, Zabel P, Goldmann T. Human lung cancer cells express functionally active Toll-like receptor 9. Respir Res. 2005;6:1. doi: 10.1186/1465-9921-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang H, Rayburn E, Zhang R. Synthetic oligodeoxynucleotides containing deoxycytidyl-deoxyguanosine dinucleotides (CpG ODNs) and modified analogs as novel anticancer therapeutics. Curr Pharm Des. 2005;11:2889–2907. doi: 10.2174/1381612054546707. [DOI] [PubMed] [Google Scholar]
- 86.Leichman G, Gravenor D, Woytowitz D, Mezger J, Albert G, Schmalbach T, Al-Adhami M, Manegold C. CPG 7909, a TLR9 agonist, added to first line taxane/platinum for advanced non-small cell lung cancer, a randomized, controlled phase II study [abstract] J Clin Oncol. 2005;23(16S):7039. [Google Scholar]
- 87.Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc Natl Acad Sci U S A. 2002;99:10231–10233. doi: 10.1073/pnas.172380699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yin J, Hashimoto A, Izawa M, Miyazaki K, Chen GY, Takematsu H, Kozutsumi Y, Suzuki A, Furuhata K, Cheng FL, Lin CH, Sato C, Kitajima K, Kannagi R. Hypoxic culture induces expression of sialin, a sialic acid transporter, and cancer-associated gangliosides containing non-human sialic acid on human cancer cells. Cancer Res. 2006;66:2937–2945. doi: 10.1158/0008-5472.CAN-05-2615. [DOI] [PubMed] [Google Scholar]
- 89.Carr A, Rodriguez E, Arango Mdel C, Camacho R, Osorio M, Gabri M, Carrillo G, Valdes Z, Bebelagua Y, Perez R, Fernandez LE. Immunotherapy of advanced breast cancer with a heterophilic ganglioside (NeuGcGM3) cancer vaccine. J Clin Oncol. 2003;21:1015–1021. doi: 10.1200/JCO.2003.02.124. [DOI] [PubMed] [Google Scholar]
- 90.Vazquez AM, Perez A, Hernandez AM, Macias A, Alfonso M, Bombino G, Perez R. Syngeneic anti-idiotypic monoclonal antibodies to an anti-NeuGc-containing ganglioside monoclonal antibody. Hybridoma. 1998;17:527–534. doi: 10.1089/hyb.1998.17.527. [DOI] [PubMed] [Google Scholar]
- 91.Neninger E, Diaz RM, de la Torre A, Rives R, Diaz A, Saurez G, Gabri MR, Alonso DF, Wilkinson B, Alfonso AM, Combet T, Perez R, Vazquez AM. Active immunotherapy with 1E10 anti-idiotype vaccine in patients with small cell lung cancer: report of a phase I trial. Cancer Biol Ther. 2007;6:145–150. doi: 10.4161/cbt.6.2.3574. [DOI] [PubMed] [Google Scholar]
- 92.Alfonso S, Diaz RM, de la Torre A, Santiesteban E, Aguirre F, Perez K, Rodriguez JL, del Barroso MC, Hernandez AM, Toledo D, Gabri MR, Alonso DF, Viada C, Gomez RE, Suarez E, Vazquez AM, Perez R, Macias AE. 1E10 anti-idiotype vaccine in non-small cell lung cancer: experience in stage IIIb/IV patients. Cancer Biol Ther. 2007;6:1847–1852. doi: 10.4161/cbt.6.12.5000. [DOI] [PubMed] [Google Scholar]
- 93.Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, Thompson CB, Bluestone JA. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1:405–413. doi: 10.1016/1074-7613(94)90071-x. [DOI] [PubMed] [Google Scholar]
- 94.Manzotti CN, Tipping H, Perry LC, Mead KI, Blair PJ, Zheng Y, Sansom DM. Inhibition of human T cell proliferation by CTLA-4 utilizes CD80 and requires CD25+ regulatory T cells. Eur J Immunol. 2002;32:2888–2896. doi: 10.1002/1521-4141(2002010)32:10<2888::AID-IMMU2888>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 95.Krummel MF, Allison JP. CTLA-4 engagement inhibits IL-2 accumulation and cell cycle progression upon activation of resting T cells. J Exp Med. 1996;183:2533–2540. doi: 10.1084/jem.183.6.2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–465. doi: 10.1084/jem.182.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sznol M, Hodi F, Margolin K, McDermott D, Ernstoff M, Kirkwood J, Wojtaszek C, Feltquate D, Logan T. Phase I study of BMS-663513, a fully human anti-CD137 agonist monoclonal antibody, in patients (pts) with advanced cancer (CA) [abstract] J Clin Oncol. 2008;26(15S):3007. [Google Scholar]
- 98.Bhowmick NA, Moses HL. Tumor-stroma interactions. Curr Opin Genet Dev. 2005;15:97–101. doi: 10.1016/j.gde.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]