

Abstract
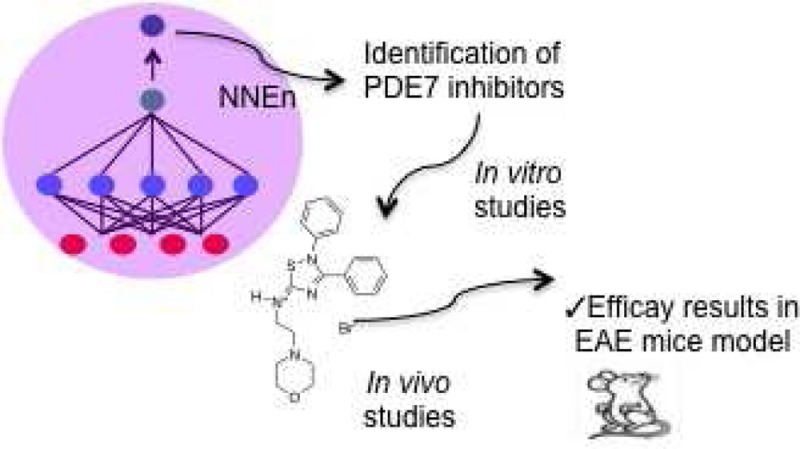
A neural network model has been developed to predict the inhibitory capacity of any chemical structure to be a phosphodiesterase 7 (PDE7) inhibitor, a new promising kind of drugs for the treatment of neurological disorders. The numerical definition of the structures was achieved using CODES program. Through the validation of this neural network model, a novel family of 5-imino-1,2,4-thiadiazoles (ITDZs) has been identified as inhibitors of PDE7. Experimental extensive biological studies have demonstrated the ability of ITDZs to inhibit PDE7 and to increase intracellular levels of cAMP. Among them, the derivative 15 showed a high in vitro potency with desirable pharmacokinetic profile (safe genotoxicity and blood brain barrier penetration). Administration of ITDZ 15 in an experimental autoimmune encephalomyelitis (EAE) mouse model results in a significant attenuation of clinical symptoms, showing the potential of ITDZs, especially compound 15, for the effective treatment of multiple sclerosis.
Keywords: Artificial neural network, virtual screening, PDE7 inhibitors, experimental autoimmune encephalomyelitis
Successful drug design requires a multidisciplinary approach. Understanding the nature of a drug and its intended target requires knowledge of their chemistries, biophysical characteristics, and, of increasing importance, their subcellular biological context. The use of information technology and management has become a critical part of the drug discovery process. Related to this, chemoinformatics has emerged as a scientific discipline encompassing the design, creation, organization, management, retrieval, analysis, dissemination, visualization, and use of chemical information. Its use to solve drug design problems has been extensively reported.1,2
Following with our efforts to develop new and efficient inhibitors of phosphodiesterase 7 (PDE7) as innovative drugs for neurological disorders,3 we consider this approach a way to boost the discovery and development of new PDE7 inhibitors based on chemical structure. Our previous experience in the use of chemoinformatic tools in drug discovery prompted us to develop a neural network model for the prediction of inhibition of PDE7 of any chemical structure. The choice of descriptors occupies a special place in the development of in silico models. The program called CODES4 is an efficient and easy-to-use program to encode chemical structures by means of neural computing (CODES is available free of charge for academic institutions). The molecular descriptors obtained from this method contain the underlying information of their chemical structure.
PDE7 is involved in pro-inflammatory processes and is necessary for the induction of T-cell proliferation.5 In addition, PDE7 is widely expressed on certain brain regions6−8 while specific inhibitors of PDE7 have been recently reported as potential new drugs for the treatment of neurological disorders due to their ability to increase levels of cAMP.3 Modulation of the inflammation process is without any doubt a neuroprotective, well-established strategy. Moreover, some unmet diseases such as multiple sclerosis involved simultaneously pathologies on immune system, T-cells, and brain cells, microglia and oligodendrocytes. Although PDE4 inhibitors are also able to increase cAMP levels and have been widely studied as efficient anti-inflammatory agents,9 a major drawback of these compounds is the significant emetic effects.10 To overcome these adverse effects, an alternative approach is to target different cAMP specific PDE families, such as PDE7. Thus, PDE7 inhibitors may represent a well targeted and innovative therapy for this pathology.11
Furthermore, development of new PDE7 inhibitors with favorable ADME properties will broaden the scope of a novel class of therapeutics with an innovative mechanism of action maintaining high levels of intracellular cAMP. These inhibitors would target a major unmet medical need in a field in which new and effective therapies are an urgent social need. Therefore, the identification of selective inhibitors targeted against PDE7 enzyme has become an attractive area of research.
Several years ago, our research group was the first one in reporting PDE7 selective inhibitors.12 Since then, a lot of efforts have been done to increase potency and selectivity of this kind of compounds, conforming a great variety of diverse chemical compounds with interesting pharmacological profiles.13
Following our ongoing research on this field, we have discovered new leads by using chemoinformatics tools.14,15 We have also shown that our compounds are able to increase cAMP in lymphocyte cultures.16 Regarding advanced studies in vivo, we have proved that PDE7 inhibitors belonging to the quinazoline family enhance neuroprotection and decrease neuroinflammation in well-characterized cellular and animal models of Parkinson’s disease,17 spinal cord injury,18 stroke,19 and also Alzheimer’s disease.20 More recently, we have reported also in vivo efficacy of the furan type PDE7 inhibitors in an animal model of multiple sclerosis.11
Herein, we describe the development of a neuronal network model in order to predict the inhibitory activity of new compounds on PDE7. Using this computational approach, we present the discovery of a new chemical family of PDE7 inhibitors, the 5-imino-1,2,4-thiadiazoles (ITDZs) recently described in our group as substrate competitive GSK-3 inhibitors.21 The enzymatic assays on PDE7A confirm the in silico results. We have also shown that the ITDZs increase cAMP levels in cell cultures, and they are able to reduce the clinical symptoms in an animal model of multiple sclerosis, where GSK-3 and PDE7 inhibitors independently have been shown an important role as therapeutic agents.11,22
Results and Discussion
Neural Network Model
As a theoretical tool, the artificial neural networks (ANNs) are a modeling methodology whose application in some areas of Medicinal Chemistry such as quantitative structure–property relationship (QSPR), quantitative structure–activity relationship (QSAR), and prediction of pharmacokinetic properties has increased spectacularly in recent years.23,24
The development of nonlinear modeling approaches, such as artificial-intelligence-based algorithms, opened up the field to the concurrent analysis of a wider variety of structures with potentially varying modes of action and noncongeneric chemicals. These artificial systems emulate the function of the brain, where a very high number of information-processing neurons are interconnected and are known for their ability to model a wide set of functions, including linear and nonlinear, without knowing the analytic forms in advance. One of the most important factors involved in the resulting quality of this kind of model is the numerical representation of the chemical structure. Encoding the chemical structure into numerical descriptors is not an easy task and often it implies a high level of quantum chemistry knowledge and very time-consuming procedures. With the objective of encoding each chemical entity, CODES software4 has been used, giving molecular descriptors from the topological point of view with the whole chemical structure information. CODES software was previously used for such kind of codification with excellent results in different QSAR or QSPR models.14,25−27 The performance and the accuracy of results are strongly dependent on the way that structures are represented but in some cases, it is difficult to manually select descriptors useful for a particular property. To overcome this problem, we used an approach based on ANNs.
A representative set of 92 PDE7A inhibitor compounds of wide structural diversity extracted from the literature was employed as input data set. The data set is characterized by a high structural diversity since it is formed by iminothiadiazoles,28,29 benzene sulfonamides,30 quinazolines,16 thiazoles,31 and spirotricyclic derivatives,32 among others (Figure 1).
Figure 1.

PDE7A inhibitor families.
Regarding the modeled variable, we used quantitative values activity against PDE7A expressed as the logarithm of the inhibitory concentration 50 (log IC50) at the micromolar range and also qualitatives where the value (1) was assigned to the range of activity that includes values lower than 0.1 μM, (0) includes activity values between 0.1 and 1 μM, and (−1) includes values greater than 1.1 μM. The structure database was divided into groups randomly in order to obtain a training set for the learning ANNs process and an external validation set to confirm the predictive power of the model obtained.
In general, a molecule is represented by means of a 2D graphic formula. The simplest representation of this molecular structure is the linear notation converting the connection matrix of a molecule (atoms and bonds connecting them) into a sequence of alphanumeric symbols using a set of rules. The most widespread method used for linear representation is the “simplified molecular input line system” (SMILES)33,34 that is the one used as input to define the molecules (Table S1, Supporting Information).
As mentioned before, the definition of the molecules was achieved from a nonsupervised neural network using CODES program. This program codifies each molecule into a set of numerical parameters taking into account exclusively the information of its chemical structure from a topological point of view.
The dynamic matrix of each structure of the data set was obtained employing CODES. With the aim of homogenizing the data set dimensions, the dynamic matrix was reduced to four numeric parameters (a1, a2, a3, and a4) using the reduction dimension process (Table S2). This number of numeric codes was chosen based on previous studies with ANNs.27,35 These four variables were accomplished for each compound and stand for the chemical. The reduction dimension process is a strategy which decreases the complexity without chemical information data lost. This step is useful not only to encode the structures into a small number of variables, but also it makes possible the use of a set of molecules with different number and atom types.
After establishing molecular descriptors, the next step was the development of the theoretical models by means of a learning process in order to link the biological activity and the chemical structure. A supervised FFNN (feed forward neural net) network was used to perform the neural predictive models with each initial training set. Some of these initial models are gathered in Table 1 (entries 1–3). Using these models and applying in each case one of the strategies mentioned in the Methods section, new models were developed (entries 4–8). All the models present the 4 – x – 1 architecture, where the 4 value is the number of the selected descriptors, 1 is the network output, that is, the value of activity against PDE7A, and x is the number of hidden neurons.
Table 1. PDE7A Individual Model Statistical Parameters.
| model | na | architecture | r2/SD training | I.V.b | r2/SD test |
|---|---|---|---|---|---|
| 1 | 70/22 | 4:7:1 | 0.74/0.38 | 70% | 0.68/0.34 |
| 2 | 49/43 | 4:7:1 | 0.78/0.37 | – | 0.62/0.33 |
| 3 | 59/33 | 4:7:1 | 0.99/0.01 | 100% | 0.50/0.34 |
| 4 | 70/22 | 4:7:1 | 0.80/0.33 | 70% | 0.38/0.46 |
| 5 | 67/25 | 4:7:1 | 0.74/0.38 | – | 0.51/0.50 |
| 6 | 75/17 | 4:7:1 | 0.99/0.01 | 100% | 0.45/0.56 |
| 7 | 70/22 | 4:8:1 | 0.78/0.38 | 80% | 0.25/0.81 |
| 8 | 49/23 | 4:8:1 | 0.99/0.01 | 100% | 0.45/0.56 |
Number of compounds of training/test set.
% prediction in internal validation; 1– not internal validation.
The best models are presented in Table 1 along with their architecture, r2, and standard deviation (SD). The models 1–8 showed correct statistical parameters in the training process (0.99 < r2 < 0.74). However, when we moved to the results obtained for the external validation set or test, the performance declined significantly (0.25 < r2 < 0.68).
Analyzing the model 1, in qualitative terms (inactive, moderate, and active), the percentage of prediction in the external validation set does not exceed 50%; that is, the network is not predictive (Table S3 in the Supporting Information).
According to statistical parameters (Table S4 in the Supporting Information), model 1 shows lower values of index fraction correct (FC) in the test than in the training set and moderate values of false alarm rate (FAR), pointing out that there is a significant percentage of misclassified inactive compounds (classified as moderate compounds), and therefore, this model could not be used to predict real inhibitors.
Given these negative results, we proposed to use another strategy based on neural network ensemble (NNE)36 which is a learning paradigm where a collection of a finite number of neural networks is trained for the same task. It originates from Hansen and Salamon’s work, which shows that the generalization ability of neural networks, that is, training many neural networks and then combining their predictions. ANN ensemble techniques have become very popular among neural network practitioners in a variety of ANN application domains.37,38 An effective NNE should consist of a set of ANNs that not only are highly correct but make their errors on different parts of the input space as well. In general, a neural networks ensemble is constructed in two steps, that is, training a number of component neural networks and then combining the component predictions.
In order to obtain a more reliable model, we evaluated the predictive power by creating several training sets and predicting activities of unknown compounds. Instead of the selection of a sole test set, we generated multiple ones by means of NNE approach. Then we averaged external predictions. Members of NNEs were randomly generated by dividing the whole data set into 70 inhibitors for the training set (76%) and 22 inhibitors for the test set (24%), keeping the previous architecture.
All the model data is shown in Table 2 with their r2 and standard deviation. The NNE models 1–6 show correct statistical parameters in the training process (0.85 < r2 < 0.77). When we moved to the results obtained for the external validation set, the performance presented moderate predicted values (0.70 < r2 < 0.34).
Table 2. PDE7A NNE Model Statistical Parameters.
| model | strategy | no. nets | r2 /SD training | r2/SD test |
|---|---|---|---|---|
| NNE 1 | NNE A | 5 | 0.78/0.30 | 0.60/0.28 |
| NNE 2 | NNE A | 10 | 0.77/0.38 | 0.53/0.35 |
| NNE 3 | NNE A | 20 | 0.85/0.25 | 0.70/0.23 |
| NNE 4 | NNE B | 5 | 0.80/0.28 | 0.65/0.28 |
| NNE 5 | NNE B | 10 | 0.80/0.26 | 0.34/0.29 |
| NNE 6 | NNE B | 20 | 0.87/0.22 | 0.44/0.34 |
Analyzing the NNE3 model, in qualitative terms, the percentage of prediction in the training set and the external validation set, both of them were above 75%. Also, the percentage compounds well predicted in qualitative terms by the training set and the external validation of the NNE3 model is shown in the Table 3.
Table 3. Cluster Analysis of Training and Test Set of Model NNE3.
| experimental |
||||||
|---|---|---|---|---|---|---|
| training |
test |
|||||
| predicted | inactive | moderate | active | inactive | moderate | active |
| active | 10% | 3% | 71% | – | 24% | 88% |
| moderate | 54% | 93% | – | 100% | 75% | – |
| inactive | 36% | 4% | – | – | 1% | – |
According to statistical parameters, the NNE3 model shows the highest values of index fraction correct (FC) in training and test set, and lowest values of false alarm rate (FAR) than in model 1 (Table 4).
Table 4. Statistical Parameters of Training and Test Set of Model NNE3a.
| training set | test set | |
|---|---|---|
| FC | 76% | 72% |
| FAR | 2% | 18% |
| POD | 98% | 100% |
(FC) fraction correct, (FAR) false alarm rate, (POD) probability of detection.
From all models evaluated we can conclude that the NNE3 model obtained by NNEA strategy presents a good predictive capacity to be a useful tool for the discovery of new candidates, that is, new PDE7 inhibitors. This model shows an overall good classification percentage, with 82% and 74% in training (Table S5 in the Supporting Information) and test set (Table 5), respectively.
Table 5. Test Set of NNE3 Model.
| quantitative
analysis |
qualitative analysis |
|||
|---|---|---|---|---|
| compd | log IC50 | theoretical | experimental | theoretical |
| I-1 | –0.8239 | –1.1733 | 0 | 1 |
| I-4 | –0.9208 | –1.2318 | 0 | 1 |
| I-5 | –1.4949 | –1.1544 | 1 | 1 |
| I-6 | –0.7696 | –0.5978 | 0 | 0 |
| I-7 | –1.7959 | –1.2059 | 1 | 1 |
| I-11 | –1.5229 | –1.1296 | 1 | 1 |
| I-13 | –1.284 | –1.107 | 1 | 1 |
| I-14 | –1.8539 | –1.3647 | 1 | 1 |
| I-20 | –1.9586 | –1.2981 | 1 | 1 |
| I-24 | –1.8239 | –1.3433 | 1 | 1 |
| I-35 | –1.1805 | –0.7131 | 1 | 0 |
| I-37 | –0.0088 | –0.5628 | 0 | 0 |
| I-41 | –0.7959 | –0.3203 | 0 | 0 |
| I-42 | –0.7959 | –0.7512 | 0 | 0 |
| I-43 | –0.6778 | –0.7453 | 0 | 0 |
| I-48 | –1.585 | –1.2113 | 1 | 1 |
| I-53 | –0.9208 | –1.2209 | 0 | 1 |
| I-55 | –0.3768 | –0.8946 | 0 | 0 |
| I-76 | 0.1139 | –0.2832 | –1 | 0 |
| I-77 | –0.5376 | –0.393 | 0 | 0 |
| I-82 | –0.6198 | –0.9589 | 0 | 0 |
| I-84 | 0.1761 | –0.1496 | –1 | 0 |
External Validation of the Neural Model NN3
As our goal is to develop a robust neural model, we proposed to perform an external validation using our in-house chemical library composed by diverse chemical structures synthesized in our laboratory during the last years.
Virtual screening was then carried out with compound data set of our in-house chemical library composed of 715 molecules, filtered to focus on compounds with desired CNS druglike properties. Details of the workflows and tables with the compounds are detailed in the Methods section and the Supporting Information, respectively. The aim of the screen was to identify novel chemotypes that would provide starting points for optimization to compounds with good druglike properties.
As filters for our selection (Figure S1), we consider initially two different properties in these compounds thinking on a potential chronic treatment for a neurodegenerative disease: oral bioavailability and blood brain barrier (BBB) penetration. We use the Lipinski rule39 to classify the 715 molecules. Out of those, 648 structures (90.6%) meet the theoretical criteria to be bioavailable. These compounds were selected to determine the BBB permeability. Only 220 molecules (37.9%) fulfilled these criteria, and they were used in the neural model NN3 to predict the qualitative potential of PDE7 inhibition (Tables S6 and S7). The 19.5% of the in silico screened compounds (43 derivatives of our own library) were predicted as PDE7 inhibitors. From these predicted PDE7 inhibitors, only 21 of them were physical available and pure (>95% by HPLC). These compounds were evaluated experimentally on human recombinant PDE7A (Table S8), together with two reference compounds selected as positive and negative controls. Thus, MR1.51, a recently described PDE7 inhibitor,11 and Rolipram, a well-known PDE4 inhibitor,40 were well predicted, both in silico and experimentally (Table 6). The experimental evaluation of the 21 selected compounds showed a significant percent of inhibition of PDE7 at 10 μM for some derivatives, while others were inactive (Table S8). As a first step, only in the cases where percent inhibition of PDE7A is >65% the IC50 was determined. The final result of this study had provided to us two new hits (compounds 1 and 2) as PDE7A inhibitors (Table 6).
Table 6. Predicted and experimental Values of IC50 (PDE7A) of the New Hits and the Standard Reference Compounds.

Regarding the thiophene derivative hit (1), it was discarded for further development due to the fact that it was found in the literature that this kind of heterocycle was able to inhibit also PDE4,41 a cAMP isoenzyme with emetogenic properties.10 On the contrary, the 5-imino-1,2,4-thiadiazole derivative hit (2) was considered as a new lead for further development.
Recently, we have published 5-imino-1,2,4-thiadiazoles (ITDZs) as substrate competive GSK-3 inhibitors, and so we have synthetised a great number of derivatives for the kinase project. Now, we consider these number of derivatives of the family of 5-imino-1,2,4-thiadiazoles (ITDZs) structurally related with compound 2,21 as a focused chemical library, and we screen it against PDE7A both in silico with the NN3 model and experimentally using human recombinant PDE7A. Data are collected in Table 7 showing that the ITDZs family is without any doubt a new chemically diverse group of PDE7A inhibitors.
Table 7. Predicted and Experimental Values of IC50 of the New Family of PDE7A Inhibitorsa.
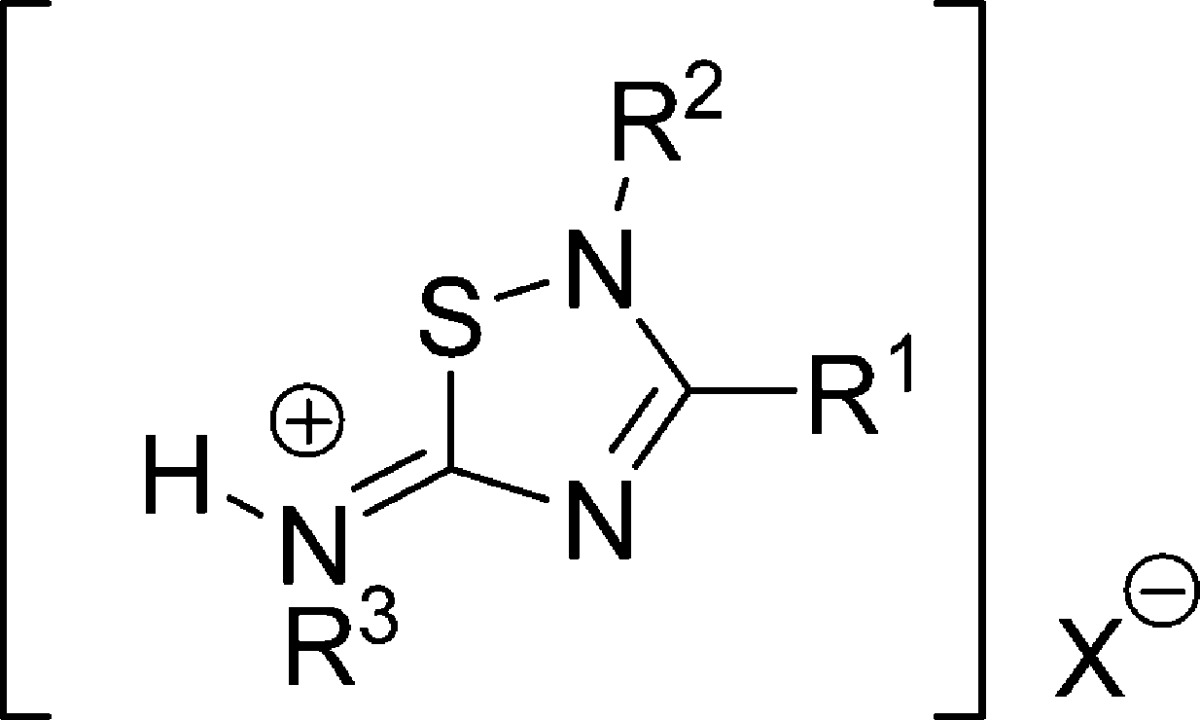
| compd | R1 | R2 | R3 | X | predicted qualitative | experimental IC50 PDE7A (μM) | experimental qualitative |
|---|---|---|---|---|---|---|---|
| 2 | Ph | Ph | CH2CO2Et | Br | 1 | 1.13 ± 0.21 | 1 |
| 3 | Ph | Ph | H | Br | 0 | 1.02 ± 0.13 | 1 |
| 4 | Ph | Ph | (CH2)4CH3 | Br | 0 | 1.44 ± 0.15 | 0 |
| 5 | Ph | Ph | cyclohex | Br | 1 | 1.64 ± 0.22 | 0 |
| 6 | Ph | Ph | CH2-3Pyr | 2Br | 0 | 0.38 ± 0.06 | 1 |
| 7 | Ph | Ph | (CH2)2OH | Br | 0 | 1.11 ± 0.17 | 1 |
| 8 | 4-OMePh | Ph | CH2-3Pyr | 2Br | 1 | 0.86 ± 0.10 | 1 |
| 9 | 4-OMePh | Ph | (CH2)2OH | Br | 1 | 1.50 ± 0.14 | 0 |
| 10 | 4-OMePh | Ph | Ph | Br | 1 | 4.36 ± 0.31 | 0 |
| 11 | 4-OMePh | Ph | CH2CO2Et | Br | 0 | 0.89 ± 0.09 | 1 |
| 12 | Ph | 4-OMePh | CH2-3Pyr | 2Br | 1 | 0.85 ± 0.07 | 1 |
| 13 | Ph | 4-OMePh | (CH2)2OH | Br | 1 | 1.18 ± 0.15 | 1 |
| 14 | Ph | 4-OMePh | CH2CO2Et | Br | 1 | 0.78 ± 0.11 | 1 |
| 15 | Ph | Ph | (CH2)2Morph | 2Br | 0 | 1.59 ± 0.16 | 0 |
| 16 | Ph | Ph | Ph | – | 0 | 3.52 ± 0.42 | 0 |
| 17 | Ph | Ph | (CH2)2OH | Cl | 0 | 1.97 ± 0.29 | 0 |
| 18 | Ph | 4-NO2Ph | CH2-3Pyr | 2Br | 1 | 0.84 ± 0.19 | 1 |
| 19 | Ph | 1-naphthyl | CH2-3Pyr | 2Br | 1 | 0.87 ± 0.09 | 1 |
| 20 | 1-naphthyl | Ph | CH2-3Pyr | 2Br | 1 | 1.08 ± 0.15 | 1 |
| 21 | 1-naphthyl | Ph | (CH2)2OH | Br | 0 | 1.24 ± 0.12 | 1 |
| 22 | pent | Ph | CH2-3Pyr | 2Br | 0 | 2.80 ± 0.17 | 0 |
(1) was assigned to the range of activity that includes values less than 0.1 μM, (0) includes activity values between 0.1 and 1 μM and (−1) includes values greater than 1.1 μM.
From the analysis of predicted and experimental qualitative values obtained (Tables 7 and 8), results showed a good overall classification percentage (approximately of 62%). As concluded, thanks to this validation process, we have been able to validate our model as plausible computational tool to make a successful theoretical screening and to find two interesting hits.
Table 8. Analysis of Results from Table 7.
| experimental |
||
|---|---|---|
| predicted | active | moderate |
| active | 8/13(62%) | 1/8(12%) |
| moderate | 5/13(35%) | 5/8(62%) |
Candidate Selection for in Vivo Studies
Due to the novelty of ITDZs as PDE7 inhibitors, we decided to select one or two candidates to test the activity in vivo. Recently, a potential cardiotoxic effect of long-term PDE3A inhibition was reported.42 Thus, to avoid adverse effects in further development steps of this kind of compound, we measured the inhibition on PDE3A of our new PDE7A1 inhibitors (Table 9). When the percentage of PDE3A inhibition was greater than 40%, the corresponding compounds were discarded in order to avoid future cardiotoxic effects. According to these results, the 5-imino-1,2,4-thiadiazoles 3, 5, 7, 9, 10, 13, 14, 15, 16, 17, and 22 could be suitable drug candidates to be further explored.
Table 9. PDE3A Inhibition for Thiadiazoles 2–22.
| compd | % inhib PDE3A @10 μM |
|---|---|
| 2 | 55.20 ± 5.20 |
| 3 | 31.10 ± 1.40 |
| 4 | 72.67 ± 2.50 |
| 5 | 33.75 ± 0.40 |
| 6 | 58.84 ± 0.30 |
| 7 | 18.81 ± 3.40 |
| 8 | 55.20 ± 2.60 |
| 9 | 24.48 ± 8.50 |
| 10 | 3.81 ± 2.80 |
| 11 | 55.47 ± 4.10 |
| 12 | 40.59 ± 0.10 |
| 13 | 12.69 ± 4.30 |
| 14 | 36.86 ± 4.80 |
| 15 | 20.60 ± 10.20 |
| 16 | 8.76 ± 7.00 |
| 17 | 23.78 ± 1.30 |
| 18 | 81.05 ± 0.60 |
| 19 | 89.90 ± 2.90 |
| 20 | 67.86 ± 6.50 |
| 21 | 44.66 ± 0.10 |
| 22 | 34.10 ± 6.20 |
To know if these new PDE7 inhibitors are able to modulate cAMP cellular signaling pathways, we checked the ability of a couple of compounds, derivatives 3 and 15, to regulate intracellular cAMP levels in cell cultures (Figure 2). Two different compound concentrations (10 and 30 μM) were used, and in both cases we observed an increase of cAMP even more effective than the standard PDE7 inhibitor BRL50481.30 We have recently reported this class of compounds, the ITDZs, as substrate competitive inhibitors of GSK3 with potential for the treatment of neurodegenerative diseases.21 As synergistic interactions between PDE4B and GSK3 inhibitors have been suggested due to the influence of increased cAMP levels induced by PDE4B inhibitors on GSK3 function,43 our ITDZ compounds offer a unique potential to be explored as innovative multifunctional neurological drugs without the emetic effects present in PDE4B inhibitors. In fact, one of the member of this family (compound 7) has expressed antipsychotic capacities and ameliorated certain cognitive domains relevant to schizophrenia assessed in vivo.44
Figure 2.

Intracellular cAMP level in Raw cells treated with compound 3 and 15 at 30 and 10 μM.
Considering the great potential therapeutic use of this class of compounds, it is indispensable for pharmaceutical development to assess their safety. Consequently, we evaluate the in vitro mutagenic and genotoxic potential of 3, 7, 9, 14 and 15 following the well-known Ames test.45 The negative results of these mutagenicity studies showed the safety of this class of compounds to be further developed (Table 10). Moreover, we have previously determined some pharmacokinetic properties of ITDZs showing their ability to cross the blood-brain barrier (BBB).21 However, we found differences in solubility which might jeopardize further development. As aqueous solubility of lipophilic scaffolds is often improved by the attachment of a morpholine unit,46 we selected derivative 15 containing this unit, as candidate for further progression. Its capacity to cross the BBB21 and its safety in the Ames test, prompted us to evaluate it in chronic experimental autoimmune encephalomyelitis (EAE) mice, a well established murine model for multiple sclerosis where GSK3β22 and PDE7 inhibitors11 have shown separately efficacy.
Table 10. Ames Test Results.
| compd | dose (μg/plate) | no. of revertants without S9 | no. of revertants in the presence of S9 |
|---|---|---|---|
| 3 | 15 | 5.5 ± 2.5 | 8.0 ± 0.5 |
| 5 | 9.5 ± 0.5 | 6.0 ± 1.0 | |
| 1.6 | 8.5 ± 0.5 | 9.0 ± 0.5 | |
| 0.5 | 9.5 ± 0.5 | 9.0 ± 2.0 | |
| 0.18 | 9.0 ± 1.0 | 9.0 ± 0.5 | |
| 7 | 20 | 8.5 ± 0.5 | 10.0 ± 0.5 |
| 6 | 8.0 ± 0.5 | 9.0 ± 1.0 | |
| 2 | 11.5 ± 1.5 | 13.0 ± 2.0 | |
| 0.7 | 12.0 ± 1.0 | 12.5 ± 3.5 | |
| 0.2 | 12.0 ± 3.0 | 10.5 ± 0.5 | |
| 9 | 20 | 4.0 ± 0.5 | 9.5 ± 3.5 |
| 6.7 | 10.5 ± 2.0 | 14.5 ± 0.5 | |
| 2.2 | 7.5 ± 2.0 | 6.5 ± 0.5 | |
| 0.7 | 13.0 ± 9.0 | 8.5 ± 0.5 | |
| 0.27 | 8.5 ± 1.5 | 7.5 ± 1.5 | |
| 14 | 20 | (a) | 5.0 ± 1.0 |
| 6.7 | 9.5 ± 0.5 | 7.0 ± 0.5 | |
| 2.2 | 9.5 ± 3.0 | 7.0 ± 0.5 | |
| 0.7 | 2.5 ± 0.5 | 7.0 ± 3.0 | |
| 0.27 | 8.0 ± 1.0 | 7.5 ± 0.5 | |
| 15 | 20 | 6.0 ± 3.0 | 11.5 ± 2.0 |
| 6.7 | 25.0 ± 20.0 | 6.0 ± 0.5 | |
| 2.2 | 8.0 ± 1.5 | 9.0 ± 2.0 | |
| 0.7 | 10.5 ± 2.0 | 10.5 ± 0.5 | |
| 0.27 | 8.0 ± 4.0 | 9.5 ± 2.5 | |
| control (−) PBS | – | 11.5 ± 2.5 | |
| control (+) NPDb | – | 852 | – |
| control (−) S9 | – | – | 9.5 ± 1.5 |
| control (+) 2AFc | – | – | 412 |
Some toxicity was observed that did not allow us to evaluate the revertants.
NPD (2-nitro-o-phenylendiamine).
2AF (2-aminofluorene).
In Vivo Studies. Experimental Autoimmune Encephalomyelitis Model
EAE was induced in C57BL/6J mice by subcutaneous (s.c.) immunization with 100 μg of MOG35–55 peptide in complete Freund’s adjuvant on day 0. Clinical signs and score were monitored up to day 41. Mice began to show neurological deficits on day 11, reaching a maximum score around day 16. A therapeutic regimen of administration was chosen to test the inhibitor in the EAE model, which means that the compound is administered when the animals present the disease with the worst neurological score. Thus, a daily intraperitoneal (i.p.) administration of ITDZ 15 for 29 days started at day 5 after disease onset. The dose was selected considering the IC50 value on the two targets (PDE7 and GSK-3) and the dose used to observe cellular anti-inflammatory activity.21 As we can see in Figure 3, a clear and significant attenuation of clinical symptoms during the days of treatment was observed. The cumulative clinical scores were calculated by adding up the daily score for individual animals over the time period when clinical signs were evident. Statistically different cumulative clinical scores is indicated.
Figure 3.

Therapeutic effect of compound 15 in MOG35–55-induced EAE in C57BL6J mice. (A) Mean ± SEM clinical score as a function of days after immunization of mice with MOG35–55 peptide. Mice developed clinical signs on day 11, and from the 5th day after the onset of the disease they were treated daily either with compound 15 (triangles) or with vehicle (squares) until day 45. (B) Cumulative clinical scores (±SEM) during EAE with ITDZ 15 (gray filled bar) or with vehicle (black filled bar). *p < 0.05 compared to vehicle-treated animals, unpaired t test. ●, Vehicle (n = 7); ▲, 10 mg/kg compound 15 (n = 4) i.p. daily.
This experiment showed the efficacy of PDE7-GSK3 dual inhibitors on a well established model of multiple sclerosis. Specifically, compound 15 administrated to the EAE mice when the disease is established and the worst neurological score was measured showed a pronounced recovery of the clinical symptoms.
Concluding Remarks
In summary, we have developed a neural network model to determine the inhibition of PDE7A of any new chemical structure with a percentage of prediction of 74%. The model has been established using CODES program that defines each molecule from the 2D graphical representation. It is interesting to emphasize that CODES is an efficient and easy way to encode structures and it does not need 3D information, thus avoiding the risky choice of the appropriate physicochemical descriptors and problems associated with the conformation.
Thanks to this neural network model, we found a new family of PDE7A inhibitors that contains a 5-imino-1,2,4-thiadiazole ring in their structure, the ITDZ compounds. Biological in vitro studies on PDE7A of a great number of compounds showed without any doubt that ITDZs are PDE7A inhibitors in the low micromolar range able to increase levels of intracellular cAMP. Among the tested derivatives, compound 15 was chosen for in vivo studies based on its good pharmacokinetic, pharmakodynamic, and safety profile properties. This compound is able to reverse clinical symptoms in an EAE mice model when it is administered following a therapeutic regimen. As ITDZs were previously known as substrate competitive GSK-3 inhibitors, these results emphasize the idea of the great potential of multitarget drugs, especially PDE7A-GSK3 dual inhibitors as therapeutic agents for CNS diseases.
Methods
Computational Methods
Development of Neural Models
The following strategy was pursued to develop a neuronal network model to predict PDE7A inhibition.
Database
A series of 92 inhibitors was collected from literature whose biological activity is expressed as IC50 (μM). This database is formed by different families which show IC50 values in PDE7A. The original database was divided randomly into training sets and test sets of different size. Regarding the output (values of activity), we used for the training of the neural net: quantitative values that measure the inhibition as log IC50, and qualitative values that refer to whether the compounds are active (1), moderate (0), or inactive (−1). Active compounds (1) represent the compounds whose activity comprises the values until 0.1 μM, moderate compounds (0) represents the compounds with activity between 0.1 and 1 μM, and inactive compounds (−1) represents the compounds with activity higher than 1.1 μM.
Input Data
The first step is to draw the selected compounds using CHEMDRAW software (v. 8.0) and encode them using SMILES system that denotes a molecular structure as a graph.33 Subsequently, the molecular descriptors are obtained using CODES software (v1.0, revision 3). CODES encodes each molecule into a dynamic matrix. CODES consists of two levels, topological and neural, and its philosophy relies on a Gestalt isomorphism47 between both levels. While the topological space is the chemical structure by itself, the neural one consists of an interactive and competitive network. Each point of atom of the topological space corresponds with each unit or neuron of the neural space, and each type of atom takes a different initial value based on the atom nature, the number of atoms, bonds, the connectivity with the rest of the molecule, and chirality (if applicable). Attending to connectivity, CODES considers both bonding and nonbonding interactions between atoms. If atoms are not bonded in the topological space, it means an inhibitory connection in the neural level (value −1), otherwise the neural space considers an excitatory connection and the value depends on bond type (values: 1 for simple bond, 2 for double bond, 3 for triple bond, and 1 + 1/2 for aromatic bonds). The stereochemistry is also taken into account during the codification process and R o S configuration is expressed by a corrective nonlinear function (Table S1 in the Supporting Information).
The neural network employs a sigmoidal function in the codification process, and the network is characterized by a nonsupervised learning. In the learning process, CODES records all the activities reached in every cycle (or iteration) of the network. This process finishes when the equilibrium state is reached. So, all activities values of each atom of the structure during each cycle or iteration are gathered in a matrix from the initial to final step forming the dynamic matrix, which contains the whole codification process. It is interesting to emphasize that CODES does not need three-dimensional information because the topological space and its conversion to a neural space only needs details about atoms and the relationship between them (bonds); this is the chemical structure by itself. Thus, CODES avoids the risky choice of appropriate physicochemical descriptors and problems associated with the conformation.
Based on the topological matrix, we used the whole previous matrix of each compound. The next step is reduction of dimension of matrices of each chemical molecule in order to have the same number of descriptors.
Reduction of dimension (RD) philosophy resides in reducing the complexity of any system without loss of any information about the molecule. By training supervised multilayer neural network, namely, ReNDeR (Reversible Nonlinear Dimension Reduction), high-dimensional data can be converted to low-dimensional codes. This network consists of an entry layer, three hidden layers (coding, two or three neurons, and decoding), and an output layer, with its simetric architecture as shown: (AxR)-c-h-c-(AxR). The input and the output layers (AxR) contain the same information and the same number of neurons. On the other hand, c means the hidden layers which represent the chosen number of variables the matrix will be reduced in.
In the developed model, the process of dimension reduction is carried out in order to compress the dynamic matrix data to a set of four numeric codes for each molecule (hidden neurons: A, B, C, and D). Reduction of dimension is carried out using TSAR48 software which applies the Monte Carlo algorithm (Table S2 in the Supporting Information).
Convergence parameters are 0.005 rms (root mean square) 500 cycles past best, 6000 iterations/cycle, and a data excluded of 1%. The process is finished when Best rms and Test rms are constant and their values are not higher than 0.02. The neural network is considered trained when the line diagrams of the convergence plot are unchanging.
Development of Neural Network Model
This procedure is carried out by a standard feed-forward network with back-propagation using TSAR software (v3.0)48 with an architecture 4-n-1, where four is the number of parameters above-described, n is the hidden neurons, and one is the output value (IC50).
In each training set established, we performed a systematic study of the neural network learning process. In a first approach, we evaluated the suitable number of hidden neurons. Thus, several initial trainings were carried out with the appropriate architecture. Convergence parameters are 0.01 rms 200–300 cycles past best, 1000–2000 iterations/cycle, and a data excluded of 30–50%. In order to have a model to predict PDE7A inhibition, we have developed individual networks and neural network ensembles. Individual networks: models were evaluated using statistical parameters and internal validation method, and all were retrained in the second learning stage. We used two different approaches for the second learning. Type 1 consists in retraining the selected model several times, while type 2 consists also in several retrainings using the previous model in the following one. NNE: each ensemble was evaluated using statistical parameters and internal validation method.
Validation of the Models
All models were evaluated using statistical parameters: fraction correct (FC), false alarm rate (FAR), and probability of detection (POD). We should have counted such parameters by each final model:
Fraction correct (FC) is the fraction of compounds that were classified correctly
where TA is true active, TM is true moderate, and TI is true inactive.
False alarm rate (FAR) represents the fraction of inactive compounds that were wrongly classified.
where FA is false active.
Probability of detection (POD) represents the fraction of active cases that were truly classified.
where FI is false inactive.
Also, we carried out an internal validation by cross-validation expressed (r2) by leave-group-out method. On the other hand, we carried out an external validation by a test set.
Virtual Screening
We used the workflow shown in Figure S1 (Supporting Information). According to this workflow, we had employed physicochemical property filters to avoid low levels of a potential drug absorption or distribution. One of the common employed filters is the empirical Lipinski rule39 (log P < 5, a key property for an oral drug; molecular weight < 500 given the fact larger molecules showed reduced passive penetration across membranes; H-bond donors < 5; H-bond acceptor < 10). All these properties were calculated with Molinspiration cheminformatics software (http://www.molinspiration.com/) using the SMILE code for each molecule.
The second filter was the ability of a compound to penetrate the blood-brain barrier (BBB), a compulsory property of any drug to act on the CNS. The majority of compounds, that are able to cross the BBB, use passive diffusion. It was shown49,50 that physicochemical properties are involved in brain penetration. Therefore, a Volsurf model51 was used for the classification of compounds according to logBB predicted given by two rules.
If log P – (N + O) > 0 or if N + O < 5 (the number of nitrogen and oxygen atoms), then log BB is positive considering as high chance to cross this barrier. Both rules could be useful to estimate blood-brain partitioning.
According to this data, it was calculated log BB for each compound with suitable druglike properties using the above two rules. Afterward, 220 compounds were selected to perform the prediction of their potential activity against PDE7.
As standard references, we considered Rolipram,40 a PDE4 inhibitor as negative inhibitor of PDE7, and the furan derivative MR1.51,11 a recently described PDE7 inhibitor.
Experimental Methods
Radiometric Phosphodiesterase Inhibition Assay
The methodology used for measuring human recombinant PDE7A1 and PDE3A activity was based in a Scintillation Proximity Assay (SPA) from Perkin-Elmer (TRKQ7090). The activity of the phosphodiesterase is measured by coincubating the enzyme with [3H]cAMP and the hydrolysis of the nucleotide is quantified by radioactivity measurement after binding of [3H]AMP to scintillation binding bead.
Either 0.02 units of PDE7A1 (Calbiochem # 524751) or 0.02 units of PDE3A (Calbiochem # 524742) were incubated in a 96-well flexiplate with 5 nCi of [3H]cAMP and inhibitors in 100 μL of assay buffer (contained in the kit) for 20 min at 30 °C. After the incubation time, 50 μL of a solution of SPA-beads (approximately 1 mg/well) was added to each well and the plate was shaken for 1 h at room temperature. Finally, beads were settled for 30 min and radioactivity was detected in a Microbeta Trilux reader.
IC50 values were calculated by nonlinear regression fitting using GraphPad Prism. Data (radioactivity vs log concentration) was fitted to a sigmoidal dose–response equation: Y = bottom + (top – bottom)/(1 + 10(logIC50–X)n), where bottom and top were the minimum and maximal inhibition for PDE, respectively, IC50 was the concentration of compound that inhibited the PDE activity by 50%, and n was the slope of the concentration–response curve.
cAMP Measurements in Raw Cells
Quantification of cAMP was carried out using the EIA (enzyme immunoassay) kit from GE Healthcare. Briefly, Raw cells were seeded at 3 × 104/well in 96-well dishes and incubated overnight before the assay. After 60 min incubation with compounds 3 and 15, cAMP intracellular levels were determined following the manufacturer’s instructions.
Mutagenicity Assay
The method of direct incubation in plate45 using culture of Salmonella typhimurium TA98 strain was performed on derivatives 3, 7, 9, 14, and 15. The influence of metabolic activation was tested by adding S9 fraction of mouse liver. Positive controls of NDP and 2AF were run in parallel. The revertant number was manually counted and compared to the natural revertant. The compound is considered mutagenic when the number of revertant colonies is at least 2-fold of the spontaneous revertant frequencies for at least two consecutive dose levels.52−54 The maximum assayed doses were determined according to toxic effect on S. typhimurium previously determined for each compound.
EAE Induction and Treatment
Six-week-old female C57BL6 mice (15–20 g) were purchased from Harlan (Spain). All experimental procedures followed the European Communities Council Directive of November 24, 1986 (86/609/EEC). The protocol was approved by the ethic committee of the University of Barcelona and of the Generalitat de Catalunya. The mice were maintained on a 12 h light/dark cycle at a constant environmental temperature with free access to food and water for 1 week prior to experimentation.
EAE was induced by subcutaneous immunization with 100 μg MOG35–55 peptide (EspiKem S.r.l., Italy) emulsified in 100 μL of complete Freund’s adjuvant (CFA) (Sigma-Aldrich) enriched with Mycobacterium tuberculosis (H37Ra strain, Difco, Detroit, MI). Mice were immediately intraperitoneally injected with 200 ng of Bordetella pertussis toxin (Sigma-Aldrich) and again 48 h after the immunization.
Animals were scored daily for EAE. Disease severity of EAE was graded according to a five-point scale: grade 0 = no disability; 1 = a flaccid tail; 2 = a mild but definite weakness of one or both hind legs; 3 = moderate paraparesis of one hind leg; 4 = no hind leg movement; 5 = a moribund state with little or no spontaneous movement and impaired respiration.55 Mice with a score of 4.0 were euthanized.
Stock solution of the compound (100 mg/mL in DMSO) was diluted 1:50 in a solution of 5% Tocrisolve (Tocris, U.K.) in distilled water. Mice were treated through daily intraperitoneal (i.p.) injection starting on day 5 after the onset of the disease at a dose of 10 mg/kg of animal (n = 7) or with only vehicle (n = 7) until day 41.
GraphPad Prism 4.01 (GraphPad Software Inc., San Diego, CA) was used to analyze data reported in the Figure 3 legend.
Acknowledgments
We are grateful to Profs. Hugo Cerecetto and Mercedes González (Universidad de la República, Uruguay) for their helpful assistant in the genotoxicity assays.
Supporting Information Available
Codification of PDE7 inhibitors used in the development of the network model and tables relating the different steps of this process. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Conceived and designed the experiments: A.M., C.G., and N.E.C. Supervised the experiments: C.G. and N.E.C. Performed the computational experiments: M.R. Synthesis of ITDZs: V.P. Performed and supervised in vitro studies: J.B., D.I.P., C.P., M.I.C., and M.I.L. Performed and supervised in vivo studies: R.M.-A., N.P.-F., and G.M. Chemical library maintenance: S.C.
The authors gratefully acknowledge the financial support of Ministry of Science and Innovation (MICINN) Project Nos. SAF2009-13015-C02-01, SAF2009-13015-C02-02, PI10-01874, and CTQ2009-07664; Instituto de Salud Carlos III (ISCiii) Projects RD07/0060/0015 (RETICS program) and CIBERNED; Fundación Española para la Ciencia y la Tecnología (FECYT) Project No. FCT-09-INC-0367; Programa de cooperación bilateral CSIC-Universidad de la República (2009UY0006). M.R., V.P. and D.I.P. acknowledge pre- and postdoctoral fellowship from the CSIC (JAE program) respectively.
The authors declare no competing financial interest.
Supplementary Material
References
- Livingstone D. J., and Davis A. M. (2012) Drug design strategies: Quantitative approaches, The Royal Society of Chemistry, Cambridge. [Google Scholar]
- Clark T., and Banting L. (2012) Drug design strategies: Computational techniques and applications, The Royal Society of Chemistry, Cambridge. [Google Scholar]
- Gil C.; Campillo N. E.; Perez D. I.; Martinez A. (2008) Phosphodiesterase 7 (PDE7) inhibitors as new drugs for neurological and inflammatory disorders. Expert Opin. Ther. Pat. 18, 1127–1139. [Google Scholar]
- CODES v1.0. Original idea Prof. Manfred Stud.
- Nakata A.; Ogawa K.; Sasaki T.; Koyama N.; Wada K.; Kotera J.; Kikkawa H.; Omori K.; Kaminuma O. (2002) Potential role of phosphodiesterase 7 in human T cell function: comparative effects of two phosphodiesterase inhibitors. Clin. Exp. Immunol. 128, 460–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miro X.; Perez-Torres S.; Palacios J. M.; Puigdomenech P.; Mengod G. (2001) Differential distribution of cAMP-specific phosphodiesterase 7A mRNA in rat brain and peripheral organs. Synapse 40, 201–214. [DOI] [PubMed] [Google Scholar]
- Sasaki T.; Kotera J.; Omori K. (2002) Novel alternative splice variants of rat phosphodiesterase 7B showing unique tissue-specific expression and phosphorylation. Biochem. J. 361, 211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes-Irisarri E.; Perez-Torres S.; Mengod G. (2005) Neuronal expression of cAMP-specific phosphodiesterase 7B mRNA in the rat brain. Neuroscience 132, 1173–1185. [DOI] [PubMed] [Google Scholar]
- Houslay M. D.; Schafer P.; Zhang K. Y. (2005) Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discovery Today 10, 1503–1519. [DOI] [PubMed] [Google Scholar]
- Robichaud A.; Stamatiou P. B.; Jin S. L.; Lachance N.; MacDonald D.; Laliberte F.; Liu S.; Huang Z.; Conti M.; Chan C. C. (2002) Deletion of phosphodiesterase 4D in mice shortens alpha(2)-adrenoceptor-mediated anesthesia, a behavioral correlate of emesis. J. Clin. Invest. 110, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo M.; Brea J. M.; Perez D. I.; Soteras I.; Val C.; Perez C.; Morales-Garcia J. A.; Alonso-Gil S.; Paul-Fernandez N.; Martin-Alvarez R.; Cadavid I.; Loza I.; Perez-Castillo A.; Mengod G.; Campillo N. E.; Martinez A.; Gil C. (2012) Effect of phosphodiesterase 7 (PDE7) inhibitors in experimental autoimmune encephalomyelitis mice. Discovery of a new chemically diverse family of compounds. J. Med. Chem. 55, 3274–3284. [DOI] [PubMed] [Google Scholar]
- Martinez A.; Castro A.; Gil C.; Miralpeix M.; Segarra V.; Domenech T.; Beleta J.; Palacios J. M.; Ryder H.; Miro X.; Bonet C.; Casacuberta J. M.; Azorin F.; Piña B.; Puigdomenech P. (2000) Benzyl derivatives of 2,1,3-benzo- and benzothieno[3,2-a]thiadiazine 2,2-dioxides: first phosphodiesterase 7 inhibitors. J. Med. Chem. 43, 683–689. [DOI] [PubMed] [Google Scholar]
- Castro A.; Jerez M. J.; Gil C.; Martinez A. (2005) Cyclic nucleotide phosphodiesterases and their role in immunomodulatory responses: advances in the development of specific phosphodiesterase inhibitors. Med. Res. Rev. 25, 229–244. [DOI] [PubMed] [Google Scholar]
- Castro A.; Jerez M. J.; Gil C.; Calderon F.; Domenech T.; Nueda A.; Martinez A. (2008) CODES, a novel procedure for ligand-based virtual screening: PDE7 inhibitors as an application example. Eur. J. Med. Chem. 43, 1349–1359. [DOI] [PubMed] [Google Scholar]
- Gil C.; Castro A.; Jerez M. J.; Ke H.; Wang H.; Ballester S.; González-García C.; Martínez A. (2008) New PDE7 inhibitors leads for neurodegenerative diseases discovered by using a pharmacophoric model. Drugs Future 33(Supp. A), 228. [Google Scholar]
- Castaño T.; Wang H.; Campillo N. E.; Ballester S.; Gonzalez-Garcia C.; Hernandez J.; Perez C.; Cuenca J.; Perez-Castillo A.; Martinez A.; Huertas O.; Gelpi J. L.; Luque F. J.; Ke H.; Gil C. (2009) Synthesis, structural analysis, and biological evaluation of thioxoquinazoline derivatives as phosphodiesterase 7 inhibitors. ChemMedChem 4, 866–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales-Garcia J.; Redondo M.; Gil C.; Alonso-Gil S.; Martinez A.; Santos A.; Perez-Castillo A. (2011) Phosphodiesterase 7 inhibition preserves dopaminergic neurons in cellular and rodent models of Parkinson disease. PLoS One 6, e17240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterniti I.; Mazzon E.; Gil C.; Impllizzari D.; Palomo V.; Redondo M.; Perez D. I.; Esposito E.; Martinez A.; Cuzzocrea S. (2011) PDE 7 inhibitors: new potential drugs for the therapy of spinal cord injury. PLoS One 6, e15937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo M.; Zarruk J. G.; Ceballos P.; Perez D. I.; Perez C.; Perez-Castillo A.; Moro M. A.; Brea J.; Val C.; Cadavid M. I.; Loza M. I.; Campillo N. E.; Martinez A.; Gil C. (2012) Neuroprotective efficacy of quinazoline type phosphodiesterase 7 inhibitors in cellular cultures and experimental stroke model. Eur. J. Med. Chem. 47, 175–185. [DOI] [PubMed] [Google Scholar]
- Perez R.; Antequera D.; Redondo M.; Gil C.; Martínez A.; Carro E. (2011) Phosphodiesterase 7 inhibitor S14 regulates astrocytes-induced degradation of brain amyloid-b. Alzheimer's Dementia 7(Supp. 1), S452. [Google Scholar]
- Palomo V.; Perez D. I.; Perez C.; Morales-Garcia J. A.; Soteras I.; Alonso-Gil S.; Encinas A.; Castro A.; Campillo N. E.; Perez-Castillo A.; Gil C.; Martinez A. (2012) 5-Imino-1,2,4-thiadiazoles: first small molecules as substrate competitive inhibitors of glycogen synthase kinase 3. J. Med. Chem. 55, 1645–1661. [DOI] [PubMed] [Google Scholar]
- De Sarno P.; Axtell R. C.; Raman C.; Roth K. A.; Alessi D. R.; Jope R. S. (2008) Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J. Immunol. 181, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duch W.; Swaminathan K.; Meller J. (2007) Artificial intelligence approaches for rational drug design and discovery. Curr. Pharm. Des. 13, 1497–1508. [DOI] [PubMed] [Google Scholar]
- Yap C. W.; Xue Y.; Li H.; Li Z. R.; Ung C. Y.; Han L. Y.; Zheng C. J.; Cao Z. W.; Chen Y. Z. (2006) Prediction of compounds with specific pharmacodynamic, pharmacokinetic or toxicological property by statistical learning methods. Mini Rev. Med. Chem. 6, 449–459. [DOI] [PubMed] [Google Scholar]
- Guerra A.; Páez J. A.; Campillo N. E. (2008) Artificial neural networks in ADMET modeling: Prediction of blood–brain barrier permeation. QSAR Comb. Sci. 27, 586–594. [Google Scholar]
- Guerra A.; Campillo N. E.; Paez J. A. (2010) Neural computational prediction of oral drug absorption based on CODES 2D descriptors. Eur. J. Med. Chem. 45, 930–940. [DOI] [PubMed] [Google Scholar]
- Castaño T.; Encinas A.; Perez C.; Castro A.; Campillo N. E.; Gil C. (2008) Design, synthesis, and evaluation of potential inhibitors of nitric oxide synthase. Bioorg. Med. Chem. 16, 6193–6206. [DOI] [PubMed] [Google Scholar]
- Vergne F.; Bernardelli P.; Lorthiois E.; Pham N.; Proust E.; Oliveira C.; Mafroud A. K.; Royer F.; Wrigglesworth R.; Schellhaas J.; Barvian M.; Moreau F.; Idrissi M.; Tertre A.; Bertin B.; Coupe M.; Berna P.; Soulard P. (2004) Discovery of thiadiazoles as a novel structural class of potent and selective PDE7 inhibitors. Part 1: design, synthesis and structure-activity relationship studies. Bioorg. Med. Chem. Lett. 14, 4607–4613. [DOI] [PubMed] [Google Scholar]
- Vergne F.; Bernardelli P.; Lorthiois E.; Pham N.; Proust E.; Oliveira C.; Mafroud A. K.; Ducrot P.; Wrigglesworth R.; Berlioz-Seux F.; Coleon F.; Chevalier E.; Moreau F.; Idrissi M.; Tertre A.; Descours A.; Berna P.; Li M. (2004) Discovery of thiadiazoles as a novel structural class of potent and selective PDE7 inhibitors. Part 2: metabolism-directed optimization studies towards orally bioavailable derivatives. Bioorg. Med. Chem. Lett. 14, 4615–4621. [DOI] [PubMed] [Google Scholar]
- Smith S. J.; Cieslinski L. B.; Newton R.; Donnelly L. E.; Fenwick P. S.; Nicholson A. G.; Barnes P. J.; Barnette M. S.; Giembycz M. A. (2004) Discovery of BRL 50481 [3-(N,N-dimethylsulfonamido)-4-methyl-nitrobenzene], a selective inhibitor of phosphodiesterase 7: in vitro studies in human monocytes, lung macrophages, and CD8+ T-lymphocytes. Mol. Pharmacol. 66, 1679–1689. [DOI] [PubMed] [Google Scholar]
- Pitts W. J.; Vaccaro W.; Huynh T.; Leftheris K.; Roberge J. Y.; Barbosa J.; Guo J.; Brown B.; Watson A.; Donaldson K.; Starling G. C.; Kiener P. A.; Poss M. A.; Dodd J. H.; Barrish J. C. (2004) Identification of purine inhibitors of phosphodiesterase 7 (PDE7). Bioorg. Med. Chem. Lett. 14, 2955–2958. [DOI] [PubMed] [Google Scholar]
- Lorthiois E.; Bernardelli P.; Vergne F.; Oliveira C.; Mafroud A. K.; Proust E.; Heuze L.; Moreau F.; Idrissi M.; Tertre A.; Bertin B.; Coupe M.; Wrigglesworth R.; Descours A.; Soulard P.; Berna P. (2004) Spiroquinazolinones as novel, potent, and selective PDE7 inhibitors. Part 1. Bioorg. Med. Chem. Lett. 14, 4623–4626. [DOI] [PubMed] [Google Scholar]
- Weinenger D. (1988) SMILES: A chemical language for information systems.1. Introduction to methodology and encoding rules. J. Chem. Inf. Comp. Sci. 28, 31–36. [Google Scholar]
- Weinenger D. (1989) SMILES: 2. Algorithm ofr generation of unique SMILES notation. J. Chem. Inf. Comp. Sci. 29, 97–101. [Google Scholar]
- Dorronsoro I.; Chana A.; Abasolo I.; Castro A.; Gil C.; Stud M.; Martinez A. (2004) CODES/neural network model: a usedful tool for in silico prediction of oral absortion and blood-brain barrier permeability of structurally diverse drugs. QSAR Comb. Sci. 23, 89–98. [Google Scholar]
- Akhand M. A.; Islam M. M.; Murase K. (2009) A comparative study of data sampling techniques for constructing neural network ensembles. Int. J. Neural Syst. 19, 67–89. [DOI] [PubMed] [Google Scholar]
- Pugalenthi G.; Tang K.; Suganthan P. N.; Chakrabarti S. (2009) Identification of structurally conserved residues of proteins in absence of structural homologs using neural network ensemble. Bioinformatics 25, 204–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M.; Carreiras M. C.; Marco J. L.; Caballero J. (2006) Modeling of acetylcholinesterase inhibition by tacrine analogues using Bayesian-regularized Genetic Neural Networks and ensemble averaging. J. Enzyme Inhib. Med. Chem. 21, 647–661. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Wang H. C.; Liu Y. D.; Chen Y. X.; Robinson H.; Ke H. M. (2005) Multiple elements jointly determine inhibitor selectivity of cyclic nucleotide phosphodiesterases 4 and 7. J. Biol. Chem. 280, 30949–30955. [DOI] [PubMed] [Google Scholar]
- Nankervis J. L.; Feil S. C.; Hancock N. C.; Zheng Z. H.; Ng H. L.; Morton C. J.; Holien J. K.; Ho P. W. M.; Frazzetto M. M.; Jennings I. G.; Manallack D. T.; Martin T. J.; Thompson P. E.; Parker M. W. (2011) Thiophene inhibitors of PDE4: Crystal structures show a second binding mode at the catalytic domain of PDE4D2. Bioorg. Med. Chem. Lett. 21, 7089–7093. [DOI] [PubMed] [Google Scholar]
- Movsesian M. A.; Kukreja R. C. (2011) Phosphodiesterase inhibition in heart failure. Handb. Exp. Pharmacol. 237–249. [DOI] [PubMed] [Google Scholar]
- Lipina T. V.; Wang M.; Liu F.; Roder J. C. (2012) Synergistic interactions between PDE4B and GSK-3: DISC1 mutant mice. Neuropharmacology 62, 1252–1262. [DOI] [PubMed] [Google Scholar]
- Lipina T. V., Palomo V., Gil C., Martinez A., Roder J. C.. Dual inhibitor of PDE7 and GSK-3 - VP1.15 acts as antipsychotic and cognitive enhancer in C57BL/6J mice. Neuropharmacology, published on line Jun 28, 2012. DOI: 2010.1016/j.neuropharm.2012.2006.2032. [DOI] [PubMed] [Google Scholar]
- Maron D. M.; Ames B. N. (1983) Revised methods for the Salmonella mutagenicity test. Mutat. Res. 113, 173–215. [DOI] [PubMed] [Google Scholar]
- Wuitschik G.; Carreira E. M.; Wagner B.; Fischer H.; Parrilla I.; Schuler F.; Rogers-Evans M.; Müller K. (2010) Oxetanes in drug discovery: structural and synthetic insights. J. Med. Chem. 53, 3227–3246. [DOI] [PubMed] [Google Scholar]
- Kohler W. (1969) The Hervert & Sidney Longfeld memorial lectures. The task of Gestalt psychology, Princeton, NJ, pp 66–92. [Google Scholar]
- TSAR v3.3. Oxford Molecular Ltd., http:www.oxomol.com.
- Cruciani G.; Pastor M.; Guba W. (2000) VolSurf: a new tool for the pharmacokinetic optimization of lead compounds. Eur. J. Pharm. Sci. 11(Suppl 2), S29–S39. [DOI] [PubMed] [Google Scholar]
- Norinder U.; Haeberlein M. (2002) Computational approaches to the prediction of the blood-brain distribution. Adv. Drug Delivery Rev. 54, 291–313. [DOI] [PubMed] [Google Scholar]
- Crivori P.; Cruciani G.; Carrupt P. A.; Testa B. (2000) Predicting blood-brain barrier permeation from three-dimensional molecular structure. J. Med. Chem. 43, 2204–2216. [DOI] [PubMed] [Google Scholar]
- Chu K. C.; Patel K. M.; Lin A. H.; Tarone R. E.; Linhart M. S.; Dunkel V. C. (1981) Evaluating statistical analyses and reproducibility of microbial mutagenicity assays. Mutat. Res. 85, 119–132. [DOI] [PubMed] [Google Scholar]
- Benitez D.; Cabrera M.; Hernandez P.; Boiani L.; Lavaggi M. L.; Di Maio R.; Yaluff G.; Serna E.; Torres S.; Ferreira M. E.; Vera de Bilbao N.; Torres E.; Perez-Silanes S.; Solano B.; Moreno E.; Aldana I.; Lopez de Cerain A.; Cerecetto H.; Gonzalez M.; Monge A. (2011) 3-Trifluoromethylquinoxaline N,N′-dioxides as anti-trypanosomatid agents. Identification of optimal anti-T. cruzi agents and mechanism of action studies. J. Med. Chem. 54, 3624–3636. [DOI] [PubMed] [Google Scholar]
- Cabrera M.; Lopez G. V.; Gomez L. E.; Breijo M.; Pintos C.; Botti H.; Raymondo S.; Vettorazzi A.; Cerain A. L.; Monge A.; Rubbo H.; Gonzalez M.; Cerecetto H. (2011) Genetic toxicology and preliminary in vivo studies of nitric oxide donor tocopherol analogs as potential new class of antiatherogenic agents. Drug Chem. Toxicol. 34, 285–293. [DOI] [PubMed] [Google Scholar]
- McFarlin D. E.; Blank S. E.; Kibler R. F. (1974) Recurrent experimental allergic encephalomyelitis in the Lewis rat. J. Immunol. 113, 712–715. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.