

Abstract
We identified the far upstream element binding protein 1 (FBP1), an activator of transcription of the proto-oncogene c-myc, in a functional yeast survival screen for tumor-related antiapoptotic proteins and demonstrated strong overexpression of FBP1 in human hepato-cellular carcinoma (HCC). Knockdown of the protein in HCC cells resulted in increased sensitivity to apoptotic stimuli, reduced cell proliferation, and impaired tumor formation in a mouse xenograft transplantation model. Interestingly, analysis of gene regulation in these cells revealed that c-myc levels were not influenced by FBP1 in HCC cells. Instead, we identified the cell cycle inhibitor p21 as a direct target gene repressed by FBP1, and in addition, expression levels of the proapoptotic genes tumor necrosis factor α, tumor necrosis factor–related apoptosis-inducing ligand, Noxa, and Bik were elevated in the absence of FBP1.
Conclusion
Our data establish FBP1 as an important oncoprotein overexpressed in HCC that induces tumor propagation through direct or indirect repression of cell cycle inhibitors and proapoptotic target genes.
The human far upstream element (FUSE) binding protein 1 (FBP1) was originally identified as a factor that binds to the FUSE DNA sequence upstream of the c-myc proto-oncogene promoter.1,2 As the central domain of FBP1 binds single-stranded nucleic acids of a specific sequence, melting of the cis-regulatory FUSE DNA element is required for FBP1 binding. The driving force for unwinding of the AT-rich FUSE sequence is torsional stress occurring during basal c-myc transcription due to counter-rotation of DNA and transcription machinery.3 Through simultaneous interaction with transcription factor II H at the major P2 promoter,4 FBP1 action sharply raises c-myc transcription before the antagonistic FBP interacting repressor (FIR) binds to FBP1 and FUSE. FIR eventually replaces FBP1 in the DNA-containing complex and efficiently down-regulates c-myc transcription back to basal levels.5 As a result, FBP1, FIR, transcription factor II H, and FUSE serve as regulatory machinery monitoring the intensity of ongoing c-myc transcription by RNA polymerase-induced torsional stress and programming a pulse of c-Myc expression required for cell cycle entry.6,7 Apart from c-myc, no direct FBP1 target gene has been identified, although the protein binds to the promoters of several genes (unpublished data).
Recently, FBP1 was identified as one of many cellular factors associated with the 3′-nontranslated region of the hepatitis C virus genome.8 It has been suggested that FBP1 is required for efficient replication of hepatitis C virus,9 which is a chief cause for the formation of hepato-cellular carcinoma (HCC).
HCC is the fifth most common cancer worldwide and is the third leading cause of cancer mortality, with more than 500,000 deaths per year.10 Because the symptoms of HCC often arise late during disease development, tumor formation is usually detected too late and clinical prognosis is poor. HCC rarely responds to chemotherapeutic treatment, and without therapy, the median survival for patients with unresectable liver tumors of advanced and late stages is 3 to 6 months.11
In the search for the molecular changes associated with HCC development, no consistent pattern of oncogene activation and/or tumor suppressor gene inactivation has emerged so far.12 Therefore, the identification of molecular markers and targets for therapeutic approaches in HCC treatment remains an important challenge.
Materials and Methods
Additional experimental procedures are described in the Supporting Material.
Immunohistochemistry
Immunohistochemistry was performed on HCC Tissue Microarrays (BioCat, #BC03011) with a polyclonal antibody against FBP1 (Santa Cruz Biotechnology, sc-11098) at a concentration of 200 ng/mL. Slides were scanned with a ScanScope CS (Aperio).
Lentiviral Knockdown of FBP1 Expression
Short hairpin RNA (shRNA) against FBP1 was cloned into pSEW (provided by M. Grez), and the pSEW-shLuc control vector was provided by A. Weiss. Hep3B and HuH7 cells were transduced with lentivirus produced in 293T cells, and after three passages, transduction efficiencies were calculated by green fluorescent protein quantification using flow cytometry (see Supporting Material for details).
Tumor Xenograft Experiments
For analysis of tumorigenicity, 7 × 106 Hep3B cells were subcutaneously injected into the flanks of 5- to 6-week-old non-obese diabetic/severe combined immunodeficient mice. Tumor volumes were monitored over a period of 4 weeks using calipers. Tumors were isolated and analyzed by way of hematoxylineosin histology and microscopy.
Messenger RNA Quantification Using Real-Time Polymerase Chain Reaction
Total RNA was isolated from cells with the High Pure RNA Isolation Kit and transcribed into complementary DNA (cDNA) with the Transcriptor First Strand cDNA Synthesis Kit (Roche). Real-time polymerase chain reaction (PCR) analysis was performed on a LightCycler 480 (Roche) using Human Cell Cycle Regulation Panel 96 and Human Apoptosis Panel 384 (Roche). Relative messenger RNA (mRNA) levels were normalized to the expression of nine house keeping genes and calculated using the 2−ΔΔCt method.
Electrophoretic Mobility Shift Assay
Single-stranded oligonucleotide probes were generated from the p21 promoter sequence (Supporting Table 1) and tested using FBP1 proteins purified from Escherichia coli or insect cells as described.5
RNA Interference and Luciferase Reporter Assay
For transient down-regulation of FBP1, cells were transfected with small interfering RNAs (Stealth; Invitrogen) at 50 nM concentration using Lipofectamine 2000, and then transfected with the p21 promoter-firefly luciferase reporters (pGL3; Promega) by Lipofectamine reagent after 2 days. Cells were harvested after another 1 or 2 days and analyzed by luciferase assay.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism 5 software applying non-parametric Mann-Whitney test, two-way analysis of variance with Bonferroni posttest, or two-tailed t test.
Results
FBP1 Inhibits Apoptosis and Is Overexpressed in Human HCC
We developed a functional yeast survival screen to identify antiapoptotic proteins overexpressed in human tumors. In one of our screens, we transformed a Schizosaccharomyces pombe yeast strain that inducibly expresses the proapoptotic Caenorhabditis elegans protein CED413 with a human mamma carcinoma-derived cDNA library.14 One isolated cDNA that was capable of suppressing CED4-induced yeast cell death coded for a C-terminal fragment of the human FBP1 protein, and by overexpression in the human colon carcinoma cell line RKO, we confirmed that full-length FBP1 also inhibits apoptosis in mammalian cells (data not shown).
Because FBP1 was identified as a positive transcriptional regulator of the potent proto-oncogene c-myc1,15 and both c-myc expression and apoptosis inhibition contribute to tumor development, we speculated that FBP1 activity may be up-regulated in certain tumor entities. Therefore, we performed immunohistochemical stainings on multitumor tissue microarrays to investigate the expression levels of FBP1 in various tumor entities and corresponding normal tissues. Our data revealed that FBP1 was strongly expressed in 83% of the 109 HCC samples analyzed, whereas the protein was nearly undetectable in healthy liver tissue (Fig. 1).
Fig. 1.

FBP1 is overexpressed in human HCC. (A) Tissue microarrays were incubated with anti-FBP1 antibody (sc-11098; Santa Cruz Biotechnology). Representative pictures for normal liver tissue and HCC are shown. The brown areas indicate positive anti-FBP1 staining in the cell nuclei. (B) Summary of immunohistochemical quantification of FBP1 expression in all HCC and normal liver samples. FBP1 was strongly expressed in 83% of 109 HCC samples analyzed in total, whereas only one out of six (17%) normal liver slides showed weak antibody staining.
Loss of FBP1 Expression in the HCC Cell Line Hep3B Leads to Increased Apoptosis Sensitivity and to Decreased Proliferation
To test for functional implications of the observed FBP1 overexpression in HCC, we studied the function of FBP1 in the HCC cell line Hep3B. We stably transduced the cells with lentivirus containing an FBP1-specific shRNA, which resulted in efficient FBP1 knockdown compared to empty vector-transduced or luciferase control shRNA-transduced cells (Fig. 2A). The cells were then analyzed for their apoptosis sensitivity. Down-regulation of FBP1 expression led to increased cell death following ultraviolet irradiation or incubation with mitomycin C or doxorubicin compared to control cells (Fig. 2B).
Fig. 2.

Knockdown of FBP1 in HCC cells leads to increased apoptosis sensitivity and reduced cell proliferation. (A) FBP1 was efficiently down-regulated by lentiviral shRNA expression. Hep3B cells were transduced with empty control vector, lentivirus containing luciferase shRNA, or lentivirus encoding FBP1-targeting shRNA. Total cell lysates were analyzed for FBP1 expression by immunoblotting. (B) FBP1 knockdown increased sensitivity of HCC cells to apoptotic stimuli. We treated 3 × 105 stably transduced Hep3B cells with either doxorubicin (10 μM), mitomycin C (40 μg/mL), or ultraviolet irradiation (120 mJ), and apoptosis was quantified by flow cytometry after 24 hours. Three independent experiments were performed, and bars represent the standard errors. *P < 0.05. **P < 0.01. (C) We treated 2 × 105 stably transduced Hep3B cells with 10 μM BrdU for 2.5 hours. After staining with allophycocyanin-labeled anti-BrdU antibody and 7-AAD, cells were analyzed using flow cytometry. One representative experiment is presented. (D) Summary of BrdU labeling experiments with stably transduced Hep3B cells. In the absence of FBP1, proliferation is reduced significantly (**P < 0.01). Data are expressed as the mean ± standard error of the mean for three independent experiments.
The influence of FBP1 knockdown on proliferation of Hep3B cells was tested by bromodeoxyuridine (BrdU) pulse labeling and quantification of BrdU incorporation by way of fluorescence-activated cell sorting. FBP1 deficiency led to a significant decrease of BrdU-positive cells due to diminished proliferation compared with control cells with normal FBP1 expression (Fig. 2C,D).
FBP1 Knockdown Abrogates Tumor Growth in an HCC Xenograft Transplantation Model
Because our in vitro studies suggested a functional role for FBP1 in HCC proliferation and apoptosis resistance, we investigated the contribution of FBP1 to HCC growth in vivo. For this purpose, Hep3B cells transduced with either empty vector control or FBP1 shRNA-expressing lentivirus were subcutaneously injected into both flanks of immunocompromised non-obese severe combined immunodeficient (SCID) mice, and tumor growth was then monitored over a period of 25 days. Although control tumors reached a volume of 1 cm3, corresponding FBP1 knockdown tumors displayed an average volume of 250 mm3 and often had stopped expanding (Fig. 3A,B). Histological analysis of the SCID mouse tumors 25 days after injection exhibited significant differences in the extent of tumor cell proliferation and differentiation. Cells undergoing mitosis were quantified on 30 different paraffin-fixed and hematoxylin-eosin–stained tissue slides obtained from 10 different SCID mouse tumors. Strikingly, the mitotic index was twice as high in empty vector-transduced control tumors compared with FBP1 shRNA-expressing Hep3B tumors (Fig. 3C). In addition, FBP1 shRNA tumors displayed significantly fewer cells with an increased size ratio of nucleus and cytoplasm and a changed shape/appearance of the nucleus (Fig. 3D). These morphometric indices have been linked with a dedifferentiated (highly malignant) phenotype of HCC cells.16
Fig. 3.

FBP1 is required for HCC tumor growth in a xenograft transplantation model. Hep3B cells were stably transduced with sh-FBP1 or empty vector constructs. We injected 7 × 106 cells subcutaneously into the flanks of immunodeficient NOD/SCID mice. (A) Tumor volumes were measured over a period of 4 weeks using calipers (n = 10 for each cell type). Tumors arising from FBP1-deficient cells expanded significantly slower than control tumors. Data are expressed as the median ± standard error. **P < 0.01. ***P < 0.001. (B) Twenty-five days after injection, tumors were excised for microscopic analysis. Representative bright field and green fluorescent protein micrographs of one empty vector-transduced and one sh-FBP1–transduced tumor are shown. (C) Knockdown of FBP1 significantly reduces the mitotic index of experimental Hep3B tumors. Mitotic cells were counted on 30 hematoxylin-eosin–stained tumor tissue slides for both empty vector-transduced and sh-FBP1–transduced tumors (3 slides/tumor). (D) FBP1-deficient tumors contain reduced numbers of dedifferentiated cells. Cells with an increased size ratio of nucleus and cytoplasm and a changed shape/appearance of the nucleus were counted on 30 hematoxylineosin–stained tumor tissue slides (3 slides/tumor). Statistical significance in (C) and (D) was calculated with a nonparametric Mann-Whitney t test. ***P < 0.001.
FBP1 Regulates the Expression of Cell Cycle Inhibitors and Proapoptotic Genes
To understand the mechanisms of FBP1-dependent HCC cell proliferation and survival, we compared the expression of 84 selected cell cycle–relevant and 350 apoptosis-relevant genes in Hep3B cells in the presence or absence of FBP1 using real-time PCR. FBP1 was initially described as a positive regulator of c-myc transcription in various cell lines.2,7,17 Surprisingly, knockdown of FBP1 in Hep3B cells did not significantly influence the amount of c-myc mRNA (Fig. 4A) and protein (data not shown). Furthermore, we could not detect any positive correlation between FBP1 and c-Myc expression in HCC biopsies, suggesting that the regulation of the c-myc promoter might be uncoupled from FBP1 activity in HCC cells (data not shown). These results suggest that other unidentified FBP1 target genes may be responsible for the functions of FBP1 in HCC cells.
Fig. 4.

FBP1 down-regulates cell cycle inhibitors and proapoptotic genes. Expression analysis of (A) cell cycle–regulating and (B) apoptosis-regulating genes after FBP1 knockdown. Total RNA prepared from sh-FBP1–transduced Hep3B cells was compared with RNA from sh-luciferase–transduced cells for expression of target genes by real-time PCR using the Cell Cycle Panel 96 (Roche) or the Apoptosis Panel 384 (Roche). Expression levels were normalized to nine housekeeping genes. Data are expressed as the mean ± standard error of the mean.
Our analysis revealed that the mRNA expression levels of several other known cell cycle regulators were affected by FBP1. The cell cycle inhibitors p15 and p21 were both up-regulated in the absence of FBP1 (Fig. 4A). In addition, Cyclin D2 mRNA levels were diminished in the FBP1 knockdown cells, whereas the amount of Cyclin D1 mRNA remained unaffected.
Interestingly, we also observed the up-regulation of proapoptotic genes following FBP1 knockdown in Hep3B cells (Fig. 4B). In particular, elevated expression of the Bcl-2 family members Bik and Noxa was detected. In addition, mRNA levels of the death ligands tumor necrosis factor (TNF) α and tumor necrosis factor–related apoptosis-inducing ligand (TRAIL) were significantly increased.
Using an antibody-spotted cytokine array, we quantified the amount of TNF-α secreted into the supernatant by Hep3B cells stably transduced with FBP1 shRNA, and compared it with that of empty vector-transduced parental Hep3B cells. Consistent with the real-time PCR data, a significant increase in TNF-α production was observed in FBP1-deficient cells also on protein level, and secretion of TNF-β/LTa3 was similarly increased (Supporting Fig. 1).
Our data demonstrate that in the absence of FBP1, an antiproliferative and proapoptotic status of the cell is established by up-regulation of several cell cycle inhibiting and apoptosis-inducing genes.
FBP1 Directly Regulates the p21 Promoter
Because p21 mRNA expression was increased 5.5-fold following stable FBP1 knockdown in our experiments (Fig. 4A), we aimed to investigate the process by which this important cell cycle inhibitor is regulated by FBP1. Closer inspection of the human p21 promoter (based on sequence similarity with the c-myc FUSE element) revealed four potential FBP1 binding sites within 3.2 kb upstream of the transcription start point (Fig. 5A). We designed short DNA oligonucleotides containing these potential FBP1 binding sites (Supporting Table 1) and tested for binding of recombinant FBP1. Oligonucleotide P3, located 2.7 kb upstream of the p21 transcription start site, interacted strongly with FBP1, suggesting direct binding of FBP1 to this region of the p21 promoter (Fig. 5A). Boundary mapping of the P3 FBP1 binding region using an electrophoretic mobility-shift assay allowed us to define a DNA sequence of 50 bp, the deletion of which completely abolished FBP1 binding activity in the P3 promoter region (Fig. 5B).
Fig. 5.

FBP1 directly regulates p21 promoter activity. (A) Location of the putative FBP1 binding sites within the p21 promoter. The p21 promoter sequence, up to 3.2 kb upstream from the transcriptional start site, was examined for potential FBP1 binding sites. Four regions selected based on the FBP1 consensus25,31 are shown on the top (P1) and the bottom strands (P2–P4). Single-stranded oligonucleotide probes were generated for the selected four regions and tested with the purified recombinant FBP1 DNA binding domain fused to GST. 0.5 pmole of each probe were incubated per reaction with 1 or 4 μg of GST-FBP KH3 + 4 (lanes 1 and 2, respectively). (B) Boundary mapping of the P3 FBP binding site on the p21 promoter. Nine nested oligonucleotides covering the potential FUSE region (P31-P39) were tested for FBP1 binding using an electrophoretic mobility-shift assay. 0.5 pmole of each radioactively labeled oligonucleotide probe was incubated without (bottom) or with (top) 0.1 μg of the purified full-length FBP1 protein. Strong FBP1 binding was detected with the probes P32 to P36, which cover a region of 50 bp in total. (C) Effect of P3 FUSE deletion and FBP1 knockdown on the p21 promoter activity. Luciferase activities driven by the p21 promoter with or without the 50 bp P3 FBP binding region (+FUSE or −FUSE, respectively) are shown; each is accompanied by FBP1 or control knockdown (si-FBP1 or si-Control, respectively). Data points are the mean from triplicate transfections, normalized to the total protein concentration, and error bars show the standard error of the mean. One set of the triplicate cell lysates was analyzed by western blotting to confirm efficient FBP1 knockdown. C and F represent si-Control and si-FBP1, respectively.
To test whether FBP1 regulates p21 promoter activity, we constructed a luciferase reporter containing 3 kb of the wild-type p21 promoter sequence, and a second construct in which this promoter region is lacking the 50 bp P3 FBP1 binding site. Luciferase assays were performed in HCC cells transiently transfected with FBP1 small interfering RNA or with an unrelated control small interfering RNA. The results displayed in Fig. 5C reveal that knockdown of FBP1 expression led to a three- to four-fold decrease of luciferase activity of the reporter construct containing the p21 wild-type promoter sequence. The corresponding p21 promoter construct lacking the P3 FBP1 binding site revealed a greatly reduced luciferase activity in the presence of FBP1 compared with the wild-type promoter construct. We observed similar outcomes in Hep3B and HuH7 HCC cell lines stably transduced with FBP1 shRNA (data not shown). Our results demonstrate that FBP1 not only binds to the p21 promoter, but indeed directly regulates its activity.
Discussion
Our study revealed strong overexpression of the single-stranded nucleic acid binding protein FBP1 in more than 80% of human HCC, whereas normal liver does not express detectable amounts of FBP1. Only one of six normal liver samples exhibited a weak positive immunohistochemical staining with the anti-FBP1 antibody and was scored positive.
Down-regulation of endogenous FBP1 in the HCC cell line Hep3B led to decreased proliferation and increased sensitivity for induction of apoptosis which are both oncogenic characteristics required for tumorigenesis.18 Using a xenograft transplantation model in immunocompromised SCID mice, we found that tumor formation by injected Hep3B cells was nearly abolished upon FBP1 down-regulation. Histological analysis revealed that the mitotic index of tumor cells was reduced by 50% upon FBP1 knockdown. In addition, the number of dedifferentiated tumor cells transduced with FBP1 shRNA was decreased to one-third of those observed in tumors with high endogenous FBP1 levels. Reduced proliferation and tumor cell dedifferentiation are both criteria of decreased malignancy of FBP1-deficient HCC cells, and these results clearly show a tumor-promoting potential for FBP1 in vivo.
FBP1 was initially described as a positive regulator of c-myc transcription in various cell lines.2,7,17 However, we could not detect significant changes in c-myc mRNA and protein levels following FBP1 knockdown in Hep3B cells, implying that the oncogenic antiapoptotic and pro-proliferative effect of FBP1 in HCC is mediated by target genes other than c-myc.
We found that increased apoptosis sensitivity of HCC cells in the absence of FBP1 correlates with up-regulation of the proapoptotic genes TNF-α, TRAIL, Bik, and Noxa. TRAIL and TNF-α both belong to the TNF superfamily and are capable of inducing apoptosis by activating their respective death receptors.19 For TNF-α, complete regression of established murine HCC by in vivo TNF-α gene transfer was described.20
The influence of the TRAIL ligand on HCC cells is less clear. Although TRAIL potently induces apoptosis in many tumor cells, normal hepatocytes and HCC cells are relatively resistant to TRAIL-mediated cytotoxicity. However, adoptive transfer of TRAIL-expressing natural killer cells prevented recurrence of HCC after partial hep-atectomy,21 and TRAIL-induced apoptosis in HCC cells was vastly augmented when the cells were simultaneously treated with chemotherapeutic drugs such as doxorubicin and camptothecin.22
Bik/Nbk and Noxa are members of the BH3 domain-only subgroup of the Bcl-2 family. As loss of Bik is a common feature of clear-cell renal cell carcinoma23 and Noxa-induced apoptosis is defective in all p53-deficient tumors, both, Bik and Noxa, have been implicated as tumor suppressor molecules. In correlation with our results, c-Jun prevents apoptosis in chemically induced murine HCC by antagonizing p53-dependent Noxa activity.24
Our data demonstrate that down-regulation of FBP1 in liver cancer cells induces a proapoptotic milieu by up-regulating proteins that trigger the extrinsic and intrinsic apoptosis pathways.
In addition, we identified the three cell cycle–regulating genes p21, p15, and CyclinD2 as differently regulated in HCC cells depending on the presence or absence of FBP1. In our analysis of FBP1 shRNA-transduced Hep3B cells, we found that p21 mRNA levels increase following FBP1 knockdown, suggesting that FBP1 functions as a repressor of p21. Accordingly, when we hybridized the HCC tissue microarrays used for the original quantification of FBP1 expression by immunohistochemistry (IHC) with an anti-p21 antibody, we observed that the majority (65%) of tumors with high FBP1 expression displayed low p21 expression levels (data not shown).
In a recent study, a novel SELEX procedure was applied to define the optimal DNA binding site for FBP1.25 According to these sequence criteria for an authentic FBP1 binding site, the p21 FUSE element is a considerably better suited candidate for strong FBP1 binding than the c-myc FUSE element. The proof for binding of FBP1 to the p21 promotor as well as the regulation of p21 by FBP1 on the transcriptional level was provided by electrophoretic mobility-shift and luciferase reporter assays. Our results identify the tumor suppressor p21 as the second direct FBP1 target gene in addition to the proto-oncogene c-myc.
Surprisingly, FBP1 acted as an activator of the p21 promoter sequence that was tested in our luciferase assays. There are several possible explanations for this apparent contradiction. The reporter construct used in our assays consisted of the appropriate p21 promoter sequences without any p21 coding sequences. Such sequences, including additional binding sites for further FBP1-interacting proteins/repressors, may be important for the physiological p21 repressor function of FBP1. In addition, the p21 promoter might be similarly regulated by FBP1 as c-myc transcription. Here, additional binding of the repressor FIR (and ultimately, replacement of FBP1 by FIR) alters FBP1 activity from activation to repression.26
Several reports describe an inverse correlation between p21 expression and HCC tumorigenesis. p21 was identified as an independent survival prognostic factor for HCC patients after resection.27 Notch 1 signaling inhibits HCC tumor growth through induction of apoptosis and cell cycle arrest, whereby the latter is mediated by up-regulation of p21 and down-regulation of positive cell cycle regulators.28
The cell cycle inhibitor p15 was also up-regulated upon FBP1 knockdown. A decrease in p15 levels in about 50% of all HCC tumor samples has been described as a consequence of 5′CpG island methylation in the p15 promoter.29 Our analysis of HCC cells after FBP1 knockdown suggests that p15 mRNA levels may also (directly or indirectly) depend on FBP1 activity.
A positive cell cycle regulator that we found down-regulated in Hep3B cells upon FBP1 inactivation was cyclin D2. While gene amplification was reported as a mechanism of aberrant Cyclin D1 activation in HCC,30 no such data exist for Cyclin D2. Numerous studies have classified D-type cyclins as cell cycle–promoting oncoproteins important for cellular transformation, and our results suggest that cyclin D2 is a candidate FBP1-regulated oncoprotein in HCC.
Both proproliferative and antiapoptotic properties of FBP1 are mediated by (direct or indirect) regulation of specific target genes. Together with the inhibition of apoptosis, repression of cell cycle inhibitors like p21 contributes to the oncogenic potential of FBP1 (Fig. 6). As targeting of FBP1 induces cell cycle arrest and apoptosis, inhibition of FBP1 activity in HCC may represent a promising therapeutic approach.
Fig. 6.
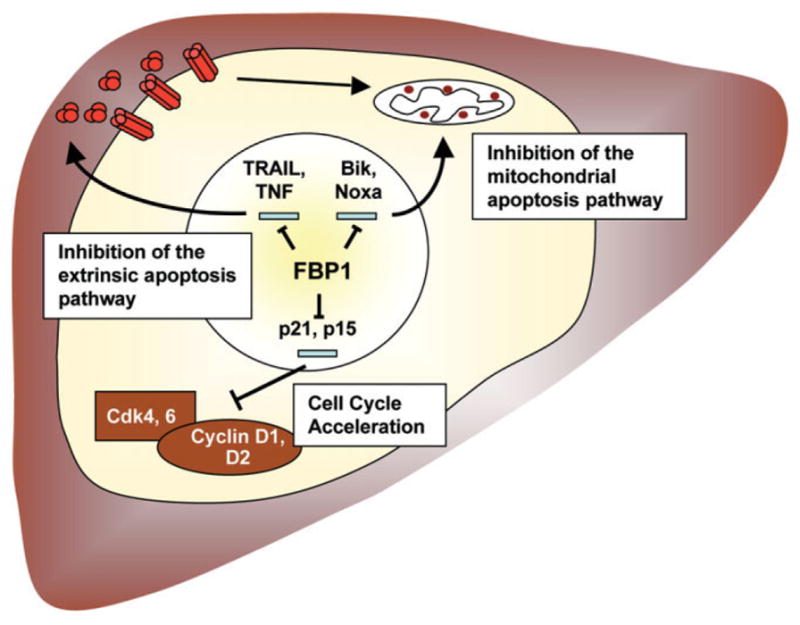
Model of the oncogenic FBP1 function in HCC.
Supplementary Material
Acknowledgments
We thank Susanne Bösser for excellent technical assistance, Manuel Grez for providing lentiviral vector technology, Astrid Weiss for the luciferase shRNA lentiviral control construct, and Winfried Wels for helpful discussions and critical reading of the manuscript.
Supported in part by grants from the Research training group 1172 (Graduiertenkolleg GK-1172 to U. R. and M. Z.), the German Federal Ministry of Education and Research (NGFN2, SMP-DNA 01GR0417 to S. J., F. D., and P. L.), and the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research (to H.-J. C. and D. L. L.).
Abbreviations
- BrdU
bromodeoxyuridine
- cDNA
complementary DNA
- FBP1
far upstream element binding protein 1
- FIR
far upstream element binding protein interacting repressor
- FUSE
far upstream element
- HCC
hepatocellular carcinoma
- mRNA
messenger RNA
- PCR
polymerase chain reaction
- SCID
severe combined immunodeficient
- shRNA
short hairpin RNA
- TNF
tumor necrosis factor
- TRAIL
tumor necrosis factor–related apoptosis-inducing ligand
Footnotes
Potential conflict of interest: Nothing to report.
Additional Supporting Information may be found in the online version of this article.
References
- 1.Avigan MI, Strober B, Levens D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J Biol Chem. 1990;265:18538–18545. [PubMed] [Google Scholar]
- 2.Duncan R, Bazar L, Michelotti G, Tomonaga T, Krutzsch H, Avigan M, et al. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994;8:465–480. doi: 10.1101/gad.8.4.465. [DOI] [PubMed] [Google Scholar]
- 3.Chung HJ, Levens D. c-myc expression: keep the noise down! Mol Cells. 2005;20:157–166. [PubMed] [Google Scholar]
- 4.Liu J, Akoulitchev S, Weber A, Ge H, Chuikov S, Libutti D, et al. Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell. 2001;104:353–363. doi: 10.1016/s0092-8674(01)00223-9. [DOI] [PubMed] [Google Scholar]
- 5.Liu J, He L, Collins I, Ge H, Libutti D, Li J, et al. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol Cell. 2000;5:331–341. doi: 10.1016/s1097-2765(00)80428-1. [DOI] [PubMed] [Google Scholar]
- 6.Chung HJ, Liu J, Dundr M, Nie Z, Sanford S, Levens D. FBPs are calibrated molecular tools to adjust gene expression. Mol Cell Biol. 2006;26:6584–6597. doi: 10.1128/MCB.00754-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Kouzine F, Nie Z, Chung HJ, Elisha-Feil Z, Weber A, et al. The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J. 2006;25:2119–2130. doi: 10.1038/sj.emboj.7601101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harris D, Zhang Z, Chaubey B, Pandey VN. Identification of cellular factors associated with the 3′-nontranslated region of the hepatitis C virus genome. Mol Cell Proteomics. 2006;5:1006–1018. doi: 10.1074/mcp.M500429-MCP200. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Z, Harris D, Pandey VN. The FUSE binding protein is a cellular factor required for efficient replication of hepatitis C virus. J Virol. 2008;82:5761–5773. doi: 10.1128/JVI.00064-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vainer GW, Pikarsky E, Ben-Neriah Y. Contradictory functions of NF-kappaB in liver physiology and cancer. Cancer Lett. 2008;267:182–188. doi: 10.1016/j.canlet.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 11.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 12.Llovet JM, Bruix J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology. 2008;48:1312–1327. doi: 10.1002/hep.22506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan J. Divergence from a dedicated cellular suicide mechanism: exploring the evolution of cell death. Mol Cell. 2006;23:1–12. doi: 10.1016/j.molcel.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 14.Brezniceanu ML, Volp K, Bosser S, Solbach C, Lichter P, Joos S, et al. HMGB1 inhibits cell death in yeast and mammalian cells and is abundantly expressed in human breast carcinoma. FASEB J. 2003;17:1295–1297. doi: 10.1096/fj.02-0621fje. [DOI] [PubMed] [Google Scholar]
- 15.Lutz W, Leon J, Eilers M. Contributions of Myc to tumorigenesis. Biochim Biophys Acta. 2002;1602:61–71. doi: 10.1016/s0304-419x(02)00036-7. [DOI] [PubMed] [Google Scholar]
- 16.Kawai Y, Takeshige K, Nunome M, Kuroda H, Suzuki H, Banno K, et al. Prognosis after hepatic resection in patients with hepatocellular carcinoma, estimated on the basis of the morphometric indices. Cancer Chemother Pharmacol. 1994;33(Suppl):S24–S28. doi: 10.1007/BF00686663. [DOI] [PubMed] [Google Scholar]
- 17.He L, Liu J, Collins I, Sanford S, O’Connell B, Benham CJ, et al. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 2000;19:1034–1044. doi: 10.1093/emboj/19.5.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 19.Sheikh MS, Huang Y. Death receptors as targets of cancer therapeutics. Curr Cancer Drug Targets. 2004;4:97–104. doi: 10.2174/1568009043481597. [DOI] [PubMed] [Google Scholar]
- 20.Cao G, Kuriyama S, Du P, Sakamoto T, Kong X, Masui K, et al. Complete regression of established murine hepatocellular carcinoma by in vivo tumor necrosis factor alpha gene transfer. Gastroenterology. 1997;112:501–510. doi: 10.1053/gast.1997.v112.pm9024304. [DOI] [PubMed] [Google Scholar]
- 21.Ohira M, Ohdan H, Mitsuta H, Ishiyama K, Tanaka Y, Igarashi Y, et al. Adoptive transfer of TRAIL-expressing natural killer cells prevents recurrence of hepatocellular carcinoma after partial hepatectomy. Transplantation. 2006;82:1712–1719. doi: 10.1097/01.tp.0000250935.41034.2d. [DOI] [PubMed] [Google Scholar]
- 22.Yamanaka T, Shiraki K, Sugimoto K, Ito T, Fujikawa K, Ito M, et al. Chemotherapeutic agents augment TRAIL-induced apoptosis in human hepatocellular carcinoma cell lines. Hepatology. 2000;32:482–490. doi: 10.1053/jhep.2000.16266. [DOI] [PubMed] [Google Scholar]
- 23.Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, et al. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ. 2006;13:619–627. doi: 10.1038/sj.cdd.4401782. [DOI] [PubMed] [Google Scholar]
- 24.Eferl R, Ricci R, Kenner L, Zenz R, David JP, Rath M, et al. Liver tumor development. c-Jun antagonizes the proapoptotic activity of p53. Cell. 2003;112:181–192. doi: 10.1016/s0092-8674(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 25.Benjamin LR, Chung HJ, Sanford S, Kouzine F, Liu J, Levens D. Hierarchical mechanisms build the DNA-binding specificity of FUSE binding protein. Proc Natl Acad Sci U S A. 2008;105:18296–18301. doi: 10.1073/pnas.0803279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Crichlow GV, Zhou H, Hsiao HH, Frederick KB, Debrosse M, Yang Y, et al. Dimerization of FIR upon FUSE DNA binding suggests a mechanism of c-myc inhibition. EMBO J. 2008;27:277–289. doi: 10.1038/sj.emboj.7601936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kao JT, Chuah SK, Huang CC, Chen CL, Wang CC, Hung CH, et al. P21/WAF1 is an independent survival prognostic factor for patients with hepatocellular carcinoma after resection. Liver Int. 2007;27:772–781. doi: 10.1111/j.1478-3231.2007.01499.x. [DOI] [PubMed] [Google Scholar]
- 28.Qi R, An H, Yu Y, Zhang M, Liu S, Xu H, Guo Z, et al. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003;63:8323–8329. [PubMed] [Google Scholar]
- 29.Roncalli M, Bianchi P, Bruni B, Laghi L, Destro A, Di Gioia S, et al. Methylation framework of cell cycle gene inhibitors in cirrhosis and associated hepatocellular carcinoma. Hepatology. 2002;36:427–432. doi: 10.1053/jhep.2002.34852. [DOI] [PubMed] [Google Scholar]
- 30.Zhang YJ, Jiang W, Chen CJ, Lee CS, Kahn SM, Santella RM, et al. Amplification and overexpression of cyclin D1 in human hepatocellular carcinoma. Biochem Biophys Res Commun. 1993;196:1010–1016. doi: 10.1006/bbrc.1993.2350. [DOI] [PubMed] [Google Scholar]
- 31.Braddock DT, Louis JM, Baber JL, Levens D, Clore GM. Structure and dynamics of KH domains from FBP bound to single-stranded DNA. Nature. 2002;415:1051–1056. doi: 10.1038/4151051a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.