

Abstract
The Gi protein-associated A3 adenosine receptor (A3AR) is a member of the adenosine receptor family. Selective agonists at the A3AR, such as CF101 and CF102 were found to induce anti-inflammatory and anti-cancer effects. In this study, we examined the differential effect of CF102 in pathological conditions of the liver. The anti-inflammatory protective effect of CF101 was tested in a model of liver inflammation induced by Concanavalin A (Con. A) and the anti-cancer effect of CF102 was examined in vitro and in a xenograft animal model utilizing Hep-3B hepatocellular carcinoma (HCC) cells. The mechanism of action was explored by following the expression levels of key signaling proteins in the inflamed and tumor liver tissues, utilizing Western blot (WB) analysis. In the liver inflammation model, CF102 (100 μg/kg) markedly reduced the secretion of serum glutamic oxaloacetic transaminase and serum glutamic pyruvic transaminase in comparison to the vehicle-treated group. Mechanistically, CF102 treatment decreased the expression level of phosphorylated glycogen synthase kinase-3β, NF-κB, and TNF-α and prevented apoptosis in the liver. This was demonstrated by decreased expression levels of Fas receptor (FasR) and of the pro-apoptotic proteins Bax and Bad in liver tissues. In addition, CF102-induced apoptosis of Hep-3B cells both in vitro and in vivo via de-regulation of the PI3K-NF-κB signaling pathway, resulting in up-regulation of pro-apoptotic proteins. Taken together, CF102 acts as a protective agent in liver inflammation and inhibits HCC tumor growth. These results suggest that CF102 through its differential effect is a potential drug candidate to treat various pathological liver conditions.
The Gi protein-associated A3 adenosine receptor (A3AR) is a member of the adenosine receptor family. The A3AR was found to be highly expressed in a broad spectrum of cancerous and inflammatory tissues, as a result of over-expression of NF-κB and additional-related transcription factors, known to be up-regulated in these pathological conditions (Poulsen and Quinn, 1998; Ochaion et al., 2009). The natural ligand adenosine induces a direct anti-proliferative effect on various tumor cell types at micromolar (μM) concentrations. Indirectly, it affects tumor development via its capability to modulate cytokine release, cell migration, angiogenesis, and chemotaxis (Woodhouse et al., 1998; Barcz et al., 2000; Madi et al., 2004). Moreover, adenosine induces activation or suppression of T killer or natural killer cells, which affects tumor cell development (Harish et al., 2003). The anti-cancer effect of adenosine is partially mediated via the A3AR and is manifested by inhibition of some inflammatory cytokines such as TNF-α, interleukin-6, and interlukin-1 (Fishman et al., 2001, 2002a,b; Cronstein, 1994; Eigler et al., 1997; Mabley et al., 2003). Selective agonists at the A3AR such as CF101 (also referred to as IB-MECA) and CF102 (also referred to as Cl-IB-MECA) inhibit the development of a variety of tumors such as melanoma, lymphoma, prostate, and colon carcinoma (Kohno et al., 1996; Fishman et al., 2003, 2004; Lu et al., 2003; Madi et al., 2003; Bar-Yehuda et al., 2005, 2007, 2008). Recently, we demonstrated that CF102 has a robust anti-cancer effect towards hepatocellular carcinoma (HCC) in an orthotopic rat model. CF102 markedly inhibited HCC growth via a molecular mechanism that entailed up-regulation of Bax, Bad, and caspase-3, resulting in tumor cell apoptosis (Bar-Yehuda et al., 2008). CF102 was also found to induce apoptosis in lung cancer cells via down-regulation of cyclin D1, c-Myc, and CDK4 and up-regulation of caspase-3 (Kim et al., 2008).
The anti-inflammatory effects mediated via the A3AR were extensively reported and include the utilization of CF101, CF102, and CF502 (also referred to as MRS3558) in experimental animal models of arthritis, osteoarthritis, inflammatory bowel disease, and septic peritonitis (Mabley et al., 2003; Lee et al., 2006; Ochaion et al., 2008). The anti-inflammatory mechanism of action mediated via the A3AR involves de-regulation of the NF-κB signaling pathway and induction of apoptosis of inflammatory cells (Ochaion et al., 2008).
Interestingly, the effect of A3AR agonists on normal cells is different and includes stimulation of cell proliferation, for example, murine or human bone marrow cells and fibroblasts (Fishman et al., 2000; Ohana et al., 2001). This differential effect may be explained by the high versus low A3AR expression levels in the tumor and normal cells, respectively.
Furthermore, A3AR agonists are known to induce protective effects in normal body tissues and cells. These include cardio-, neuro-, and chemo-protective effects both in vitro and in vivo (Fishman et al., 2001; Black et al., 2002; Shneyvays et al., 2005; Chen et al., 2006; Ge et al., 2006; Matot et al., 2006; Xu et al., 2006). Cross et al. showed that A3AR over-expression leads to cardio-protection against ischemia/reperfusion injury preserving energy during ischemia. Moreover, transgenic mice that over-express the A3AR in cardiomyocytes demonstrate increased tolerance to ischemia/reperfusion injury (Black et al., 2002; Cross et al., 2002). This protective effect was further investigated in ischemic brain injuries in which systemic administration of CF102-reduced cerebral infarction and improved locomotor activity in a rat model of stroke (Chen et al., 2006). CF102 also induced proliferation of murine bone marrow cells via the stimulation of granulocyte colony-stimulating factor production, thus counteracting chemotherapy-induced myelotoxicity via an increase of white blood cell and neutrophil counts (Fishman et al., 2001).
The aim of the present study was to examine the ability of CF102 to induce a differential effect namely, anti-cancer and protection, in two pathological conditions originating in the liver. The anti-cancer effect of CF102 was demonstrated in in vitro and in vivo HCC models and the protective effect was exhibited in a liver inflammation model induced by Con. A. Interestingly, induction of apoptosis contributed to the anti-tumor effect, whereas prevention of apoptosis conferred the protective effect of CF102. The dual and differential effects of CF102 render it as an attractive drug candidate to be developed to combat liver pathological conditions.
Materials and Methods
Reagents
The A3AR agonist CF102 (2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyl-uronamide, also known as Cl-IB-MECA), was synthesized for Can-Fite BioPharma by Albany Molecular Research, Inc., Albany, NY. A stock solution of 5 mg/ml was prepared in DMSO and further diluted in PBS.
Con. A (Canavalia ensiformis, Jack Bean Hemagglutinin) was purchased from CalBioChem (San Diego, CA). Rabbit polyclonal antibodies against mouse Fas receptor (CD95), anti-glycogen synthase kinase-3β (GSK-3β) total and phosphorylated, anti-Bax, anti-Bad, anti-caspase-8, anti-cytochrome c, and anti-β-actin (Santa Cruz Biotechnology, Santa Cruz, CA), anti-NF-κB (NF-κB p65 RelA; Chemicon, Temecula, CA), anti-PKB/Akt (PKB/Akt phosphorylated at pSer; Sigma, Rehovot, Israel, MI), anti-TNF-α (R&D Systems, Minneapolis, MN), anti-caspase-3 (cleaved caspase-3 [Asp175]; Cell Signaling Technology, Beverly, MA) were used.
Animals
Male C57BL/6J mice 8 weeks of age and male Balb/c nude 7 weeks of age were obtained from Harlan Laboratories (Jerusalem, Israel). The animals were maintained on a standardized pelleted diet and supplied with tap water. Experiments were performed in accordance with the guidelines established by the Institutional Animal Care and Use Committee at Can-Fite BioPharma, Petach-Tikva, Israel.
Liver inflammation
Male C57BL/6J mice were injected intravenously in the tail vein with Con. A (20 mg/kg). CF102 was orally administered by gavage, in a dose of 100 μg/kg, twice daily starting 8 h after Con. A administration. The control group received only DMSO in a dilution corresponding to a 100 μg/kg dose of CF102. Blood samples were collected 21 h after Con. A administration from the retro-orbital vein and serum levels of liver enzymes (serum glutamic oxaloacetic transaminase [SGOT] and serum glutamic pyruvic transaminase [SGPT]) were determined. The livers were subjected to Western blot and pathological analysis. Each group included 8–10 mice and the study was repeated three times.
Hep-3B in vitro assays
3[H]-thymidine incorporation assay was used to evaluate cell proliferation. Hep-3B cells (2.5 × 104 cells/ml) were starved in 1% FBS medium for 24 h and then incubated for 48 h with CF102 at the concentrations of 1 and 10 nM, in 96-well microtiter plates for 48 h. For the last 18 h of incubation, each well was pulsed with 1 mCi 3[H]-thymidine. Cells were harvested and the 3[H]-thymidine uptake was determined in an LKB liquid scintillation counter (LKB, Piscataway, NJ). The experiments were repeated three times. To study the modulations in the expression levels of key signaling proteins, Hep-3B (cells 3 × 106 cells/ml) were starved in 1% FBS medium for 24 h and then incubated for 24 h with CF102 at the concentration of 10 nM. At the end of the study the cells were collected, protein extract were prepared and subjected to Western blot (WB) analysis.
Hep-3B xenograft model
Hep-3B hepatoma cells (2.5 × 106) were subcutaneously injected into the right flank of balb/c nude mice. Tumor size was measured twice weekly in two dimensions using a caliper, and the volume was expressed in mm3, according to the formula: V =½ × A × B2 where A and B are the long and short diameters of the tumor, respectively. When the tumor reached ~50–100 mm3 in size, the animals were randomly assigned into vehicle or CF102-treated groups. CF102 (100 μg/kg) was orally administered by gavaouge, three times a day until termination of the study; the control group received only DMSO in a dilution corresponding to the 100 μg/kg dose of CF102.
At the end of the experiment, the mice were sacrificed and the tumors were excised. Protein extract were prepared as describe below and analyzed for the expression profile of apoptosis proteins (FasR, caspase-8, Bax, Bad, cytochrome-c, and caspase-3). Each group included five to eight mice and the study was repeated three times.
Histopathological analysis
The liver tissue were fixed in 4% buffered formalin and embedded in paraffin blocks. Sections of 5 μm in thickness were cut and stained with hematoxylin and eosin (H&E) for histological evaluation.
Protein extract preparation
Hep-3B cells, liver or tumor tissue were withdrawn and pressed through a mesh screen. The cells were extracted using RIPA assay extraction buffer containing 150 mM NaCl, 50 mM Tris, 1% NP40, 0.5% deoxycholate, and 0.1% sodium dodecyl sulfate (SDS). Cell debris was removed by centrifugation for 10 min at 7,500g. Protein concentrations were determined using the Bio-Rad protein assay dye reagent (BioRad Laboratories, Hercules, CA).
Western blot analysis
WB analyses were carried out according to the following protocol. Equal amounts of protein (50 μg) were separated by SDS–PAGE, using 10% polyacrylamide gels (Invitrogen, Carlsbad, CA). The resolved proteins were then electro-blotted onto nitrocellulose membranes (Schleicher & Schuell, Keene, NH). Membranes were blocked with 5% BSA and incubated with the desired primary antibody (dilution 1:1,000) in 1% BSA for overnight at 4°C. Blots were then washed and incubated with a secondary antibody (1:10,000) for 1 h at room temperature. Bands were recorded using BCIP/NBT color development kit (Promega, Madison, WI).
Statistical analysis
The results were evaluated using the Student’s t-test, with statistical significance set at P <0.05. Comparison between the mean values of different experiments was carried out. All data were reported as mean ± SE. Each figure presenting data of WB analysis includes a representative blot and a column figure summarizing three different studies.
Results
Anti-inflammatory effect of CF102 in Con. A-induced hepatitis
A3AR expression levels were up-regulated in liver tissue derived from animals with Con. A-induced hepatitis in comparison to liver tissue derived from naïve animals (Fig. 1). This result is in line with former findings showing A3AR over-expression in inflamed tissues.
Fig. 1.

Modulation of A3AR expression levels in liver tissue derived from Con. A-induced hepatitis in mice. Acute hepatitis was induced in mice by I.V. injection of Concanavalin A(Con.A). Liver tissue was collected 21 h later and protein extracts derived from naïve-and Con. A-induced hepatitis mice were subjected to WB analysis. A3AR expression levels were found to be up-regulated in the liver tissue derived from the mice suffering from liver inflammation in comparison to the naïve animals.
One of the first signs of liver injury is an increase in liver enzymes. When a hepatocyte is damaged, SGTP, and SGOT enzymes leak into the blood stream. To evaluate whether CF102 has a protective effect in the liver, we utilized a murine model of Con. A-induced hepatitis, resulting in a specific liver injury, followed by high levels of SGTP and SGOT.
CF102 treatment protected the liver from acute damage as was shown by a significant inhibition of the liver enzymes SGOT and SGPT levels in comparison to the vehicle-treated group (Fig. 2).
Fig. 2.
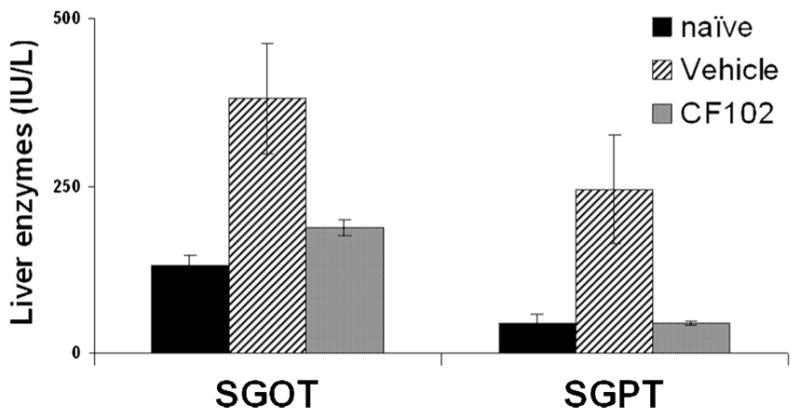
Effect of CF102 on the development of Con. A-induced hepatitis in mice. Acute hepatitis was induced in mice by I.V. injection of Con.A. CF102 (100 μg/kg) was administered orally twice daily, starting 8 h after Con. A injection. Serum levels of liver enzymes were measured 21 h after Con. A injection. CF102 markedly decreased SGOT and SGPT levels in comparison to the vehicle-treated group (P < 0.05).
Pathological analysis utilizing H&E staining of the vehicle-treated group revealed several morphological changes characterized by extensive area of necrosis at the parenchyma in a geographic map shape with no clear borders towards the liver tissue, whereas no sign of necrosis was observed in the CF102-treated group (Fig. 3).
Fig. 3.
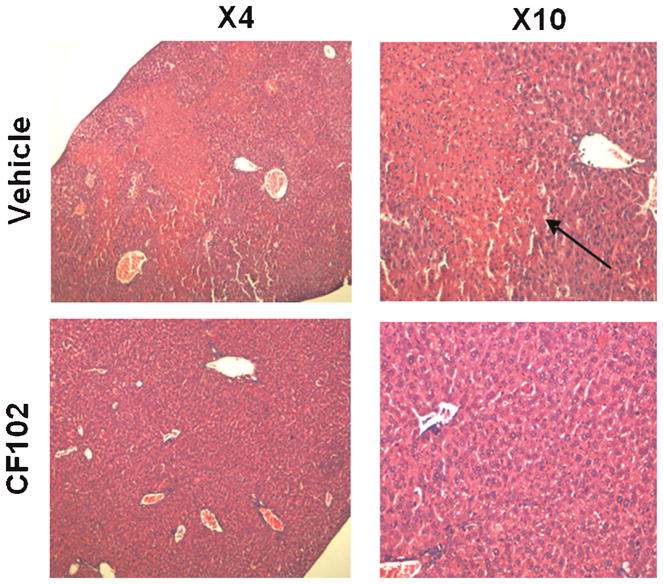
CF102 protected the liver tissue from Con. A-induced damage. Acute hepatitis was induced in mice by I.V. injection of Con.A. Tissue sections of the livers were withdrawn from the mice 21 h after Con. A injection, fixed in formalin and subjected to H&E staining. In the vehicle-treated group, an extensive area of necrosis was observed while in the CF102-treated group no sign of necrosis was noted.
It is well-established that during the course of hepatitis the NF-κB-TNF-α signaling pathway is maintained in an active state (Sun and Karin, 2008). TNF-α is one of the main cytokines mediating the inflammatory response in the Con. A hepatitis model (Tiegs, 1997). In this study, we looked at the expression levels of GSK-3β, known to control the expression and activity of NF-κB and TNF-α in the liver tissues.
A significant decrease in the expression levels of the phosphorylated GSK-3β (the inactive form of the protein) was noted upon treatment with CF102, while the expression level of the total GSK-3β remained almost the same as was observed in the vehicle-treated group (Fig. 4a). Consequently, the expression levels of NF-κB and TNF-α were down-regulated in liver tissue derived from the CF102-treated animals in comparison to the vehicle-treated ones (Fig. 4b), implicating the anti-inflammatory effect of CF102. Moreover, a decrease in the expression levels of the pro-apoptotic proteins, FasR, Bax, and Bad was observed in the liver tissue derived from the CF102-treated group, supporting the notion that the drug-prevented apoptosis of liver cells (Fig. 5).
Fig. 4.

CF102 acted as an anti-inflammatory agent in Con. A-induced hepatitis. Acute hepatitis was induced in mice by I.V. injection of Con.A. Liver tissues were collected 21 h later and protein extracts were derived from naïve- and Con. A-induced hepatitis mice were subjected to WB analysis. CF102 treatment induced (A) down-regulation in the expression levels of phosphorylated GSK-3β while the total GSK-3β expression levels remained almost unchanged in comparison to the vehicle-treated group. B: Down-regulation in the expression levels of the pro-inflammatory proteins NF-κB and TNF-α in comparison to the vehicle-treated group P < 0.05).
Fig. 5.

CF102 prevented apoptosis in the liver upon Con. A-induced hepatitis. Acute hepatitis was induced in mice by I.V. injection of Con.A. Liver tissue was collected 21 h later and protein extracts from derived from naïve- and Con. A-induced hepatitis mice were subjected to WB analysis. CF102 treatment induced down-regulation in the pro-apoptotic proteins FasR, Bax, and Bad in comparison to the vehicle-treated group (P = 0.05).
Anti-cancer effect of CF102
CF102 at a concentration of 1 and 10 nM induced a linear inhibitory effect on the proliferation of Hep-3B cells (34.5 ± 2.5% and 46.87 ± 3.72%, respectively; Fig. 6).
Fig. 6.
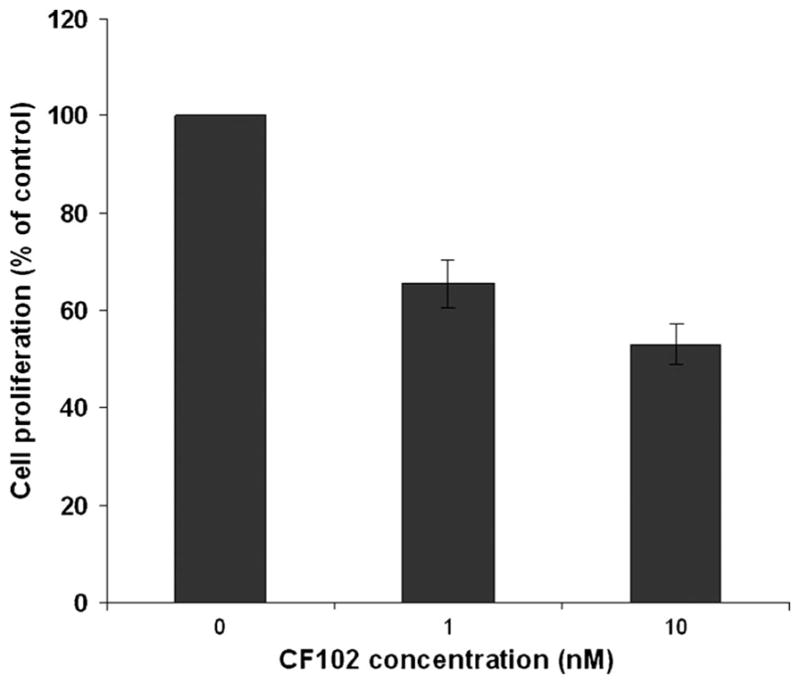
CF102 inhibits the proliferation of human HCC Hep-3B cells. Hep-3B cells were incubated for 48 h with CF102 at concentrations of 1 and 10 nM. 3[H]-thymidine incorporation assay revealed that CF102 inhibited linearly the proliferation of the Hep-3B cells.
WB analysis showed that Hep-3B cells highly express A3AR which was down-regulated upon CF102 treatment (Fig. 7). Down-regulation of PI3K, PKB/Akt, and NF-κB was also found upon CF102 treatment whereas up-regulation of the pro-apoptotic protein caspase-3 was noted (Fig. 8).
Fig. 7.

Modulation of A3AR expression levels in Hep-3B treated with CF102. Hep-3B cells were incubated for 48 h with CF102 at a concentration of 10 nM. Protein extracts derived from the Hep-3B cells were subjected to WB analysis. CF102 treatment resulted in down-regulation of A3AR expression levels.
Fig. 8.

Modulation of down-stream signaling proteins expression levels in Hep-3B treated with CF102. Hep-3B cells were incubated for 48 h with CF102 at a concentration of 10 nM. Protein extracts derived from the Hep-3B cells were subjected to WB analysis. CF102 treatment induced down-regulation of PI3K, PKB/Akt, and NF-κB expression levels and increased the expression levels of the pro-apoptotic protein caspase-3.
CF102 induced a marked inhibition of HCC tumor growth in vivo. A difference in tumor size between the CF102- and vehicle-treated groups was noticed on day 45 after tumor inoculation, and was maintained until study termination (Fig. 9A–C).
Fig. 9.

CF102 inhibited the development of Hep-3B tumors in a xenograft model. Hep-3B cells were injected subcutaneously into the flank of balb/c nude mice. Oral treatment with CF102 (100 μg/kg, three times per day) was initiated when the tumor reached ~50–100 mm3 in size and lasted until study termination. A: CF102 treatment inhibited tumor growth in comparison to the vehicle-treated group. B,C: At the end of the study an inhibition of 46% in tumor volume was observed in the CF102-treated group (P < 0.05).
The tumor tissues were collected and protein extracts were subjected to WB. CF102 treatment induced up-regulation of the apoptotic proteins FasR, caspase-8, Bax, Bad, cytochrome-c, and caspase-3 in comparison to the vehicle-treated group (Fig. 10).
Fig. 10.

CF102 up-regulated the apoptotic pathway in Hep-3B tumor cells. Hep-3B cells were injected subcutaneously into the flank of Balb/c nude mice. Oral treatment with CF102 (100 μg/kg, three times per day) was initiated when the tumor reached ~50–100 mm3 in size and lasted until study termination. Upon study termination, the tumors were excised and subjected to WB analysis. CF102 treatment induced up-regulation in the expression levels the pro-apoptotic proteins FasR, caspase-8, Bax, Bad, cytochrome-c, and caspase-3 in comparison to the vehicle-treated group (P = 0.05).
Discussion
In this study, we show that CF102 induces a protective anti-inflammatory effect and an anti-cancer effect in the liver via modulation of the PI3K-NF-κB signal transduction pathway.
The anti-inflammatory effect of CF102 was tested in a liver inflammation model known to be mediated via interlukin-2 release followed by CD4+ T-cell proliferation and migration to the liver (Tiegs, 1997). In addition, the activation of natural killer cells and macrophages as well as the release of a variety of pro-inflammatory cytokines such as TNF-α, interlukin-4, and interferon-γ takes place within the inflamed liver (Tiegs et al., 1992; Tiegs, 1997; Kaneko et al., 2000). This chain of events leads to severe liver injury within 8 h after challenge manifested by an increase in the levels of SGOT and SGPT, the latter being secreted only from hepatocytes (Gantner et al., 1995).
We first showed that A3AR was found to be over-expressed in the inflammatory liver tissue, a phenomenon described earlier in other inflammatory conditions such as rheumatoid arthritis and osteoarthritis (Fishman et al., 2006; Rath-Wolfson et al., 2006; Bar-Yehuda et al., 2009). A3AR over-expression is considered as a disease manifestation rather than a disease cause and is induced by up-regulation of some inflammatory cytokines which are involved with A3AR transcription. A3AR over-expression is also the basis for the utilization of the receptor as an anti-inflammatory target (Madi et al., 2007; Ochaion et al., 2009).
CF102 treatment in the liver inflammation model resulted in a marked reduction in serum levels of SGOT and SGPT, showing that CF102 treatment counteracted the effect of Con. A and restored the steady state. This was further observed in H&E staining performed on the liver sections, demonstrating normal healthy liver segments in the CF102-treated group compared to the vehicle-treated group which exhibited an extensive area of necrosis.
Liver damage in this model is caused mainly by the secretion of TNF-α (Gantner et al., 1995), known to up-regulate NF-κB which acts via an autocrine pathway as a transcription factor of TNF-α (Hanada and Yoshimura, 2002; Fishman et al., 2006). TNF-α inhibition was reported to have clinical benefits in several immune diseases such as inflammatory bowel disease and rheumatoid arthritis (Targan et al., 1997; Lipsky et al., 2000). In a previous work published by our group, CF101, an additional A3AR agonist, was found to down-regulate TNF-α expression levels in the lymph node and synovial tissue derived from arthritic rats, resulting in marked improvement in the inflammatory state (Baharav et al., 2005). In this study, the expression levels of both TNF-α and NF-κB in the liver tissue derived from the CF102-treated animals were down-regulated, resulting in an anti-inflammatory effect. Gomez and Sitkovsky showed that in the same liver inflammation model, A3AR activation resulted in an anti-inflammatory effect, manifested by a decrease in the liver enzymes and low expression of TNF-α. This effect was abrogated in A3AR-deficient mice, where high levels of liver enzymes were measured. Moreover, when the agonist was given to mice, lacking the A2AR, the anti-inflammatory effect was maintained, indicating the importance of the A3AR in mediating the anti-inflammatory response (Gomez and Sitokovsky, 2003).
NF-κB expression level and activity are mediated by various signaling proteins, including GSK-3β. The latter is a serine/ threonine kinase playing a central role in metabolism, proliferation, and gene transcription in various cell types, due to its ability to phosphorylate key signaling proteins. Upon phosphorylation on its serine 9 residue GSK-3β becomes inactive. GSK-3β directly phosphorylate the p65 NF-κB subunit at Ser468 or Ser536, thereby reducing its activity. In addition, GSK-3β reduces the activation of NF-κB by preventing the association of the transcriptional co-activator CBP with p65. This in turn may result in a reduced formation of pro-inflammatory cytokines such as TNF-α (Buss et al., 2004; Dugo et al., 2005; Ougolkov et al., 2005; Whittle et al., 2006). In this study, the expression level of the phosphorylated GSK-3β, which is the inactive form of the kinase was found to be decreased upon CF102 treatment. This result may explain the down-regulation of NF-κB and TNF-α.
Down-regulation of the phosphorylated GSK-3β was also noted in an adjuvant-induced arthritis experimental model, in which rats were treated with another A3AR agonist, CF502. Oral administration of CF502 upon onset of arthritis markedly suppressed the clinical and pathological manifestations of the disease. The anti-inflammatory mechanism of action of CF502 entails down-regulation in the expression levels of phosphorylated GSK-3β, which was followed by a decrease in the expression levels of NF-κB and TNF-α in the inflammatory tissues (Ochaion et al., 2008).
CF102 treatment also down-regulated the expression levels of the death receptor FasR in the inflamed liver tissue. FasR regulation is partially mediated by NF-κB (Chan et al., 1999), which was also down-regulated upon CF102 treatment. In addition, the expression levels of the down-stream pro-apoptotic proteins Bax and Bad were decreased dramatically, suggesting that CF102 treatment prevents hepatocyte apoptosis in the liver inflammatory model and therefore protects the liver against injury.
Patients suffering from hepatitis are more likely to develop HCC tumors (Schafer and Sorrell, 1999), typically resistance to chemotherapy and irradiation (Okano et al., 2003). Cumulative data indicate that the A3AR plays an important role in controlling tumor growth (Fishman et al., 2002a,b, 2003; Madi et al., 2004). Our previous data demonstrate that CF102 inhibits the growth of N1S1 HCC tumors in a rat orthotopic model via de-regulation of the NF-κB and the Wnt signal transduction pathways, resulting in apoptosis of tumor cells (Bar-Yehuda et al., 2008). The results of the present study further show that CF102 treatment inhibits both in vitro and in vivo tumor growth via down-regulation of the PI3K-NF-κB signaling pathway. In addition, an increase in the expression levels of FasR caspase-8, Bax, Bad, cytochrome-c, and caspase-3 was noted, suggesting that inhibition of tumor growth took place via cell apoptosis. Interestingly, in our former studies we showed that another A3AR agonist, CF101 induced a decrease in phosphorylated-GSK-3β which subsequently induced degradation of β-catenin followed by inhibition of tumor cells growth (Fishman et al., 2004). The ability of A3AR agonists to induce down-regulation of phosphorylated-GSK-3β in both the inflammatory and tumor cells, resulting in inhibition of NF-κB and β-catenin may serve as an explanation for the anti-inflammatory and anti-cancer effects mediated via these compounds.
In addition, an increase in the expression levels of FasR, caspase-8, Bax, Bad, cytochrome-c, and caspase-3 was noted, suggesting that inhibition of tumor growth took place via cell apoptosis.
Additional suggested cellular pathways that could attribute to the effects of CF102 may include the MAP kinase and angiogenesis. It has been demonstrated that activation of A3AR inhibits ERK1/2 activity in cancer cells (Merighi et al., 2005; Jajoo et al., 2009) and modulates angiogenesis by the effect on down-stream signaling proteins that play a role in the expression and secretion of angiogenic factors (Feoktistov et al., 2003; Headrick and Peart, 2005).
The beneficial effect of CF102 in liver disease is presented in this study. CF102 was found to protect the liver against inflammation by preventing apoptosis of hepatocytes. The anti-cancer effect of CF102 was demonstrated by its ability to induce apoptosis of tumor cells which resulted in tumor growth inhibition. This differential effect was previously demonstrated by Ohana et al., showing that the A3AR agonist was able to inhibit tumor cell growth while stimulating normal cell proliferation such as bone marrow cells (Fishman et al., 2000).
Taken together, the results of the present study suggest that CF102, through its differential effect, is a potential drug candidate in treating various pathological conditions of the liver.
Abbreviations
- A3AR
A3 adenosine receptor
- CF101
(N6-(3-iodobenzyl)-adenosine-5′-N-methyl-uronamide
- CF102
2-chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyl-uronamide (generically known as Cl-IB-MECA)
- Con.A
Concanavalin A
- HCC
hepatocellular carcinoma
- SGOT
serum glutamic oxaloacetic transaminase
- SGPT
serum glutamic pyruvic transaminase
- NF-κB
nuclear factor κB
- TNF-α
tumor necrosis factor-α
- FasR
Fas receptor
- CF502
[(1′R; 2′R; 3′R; 4′R; 5′S)-4-{2-chloro-6-[(3 chlorophenylmethyl)amino]purin-9-yl}-1-(methylaminocarbonyl)bicyclo[3.1.0]hexane-2,3-diol] (generically known as MRS3558)
- H&E
hematoxylin and eosin
- WB
Western blot
Literature Cited
- Baharav E, Bar-Yehuda S, Madim L, Silberman D, Rath-Wolfson L, Halpren M, Ochaion A, Weinberger A, Fishman P. The anti-inflammatory effect of A3 adenosine receptor agonists in murine autoimmune arthritis models. J Rheumatol. 2005;32:469–476. [PubMed] [Google Scholar]
- Barcz E, Sommer E, Janik P, Marianowski L, Skopinska-Rózewska E. Adenosine receptor antagonism causes inhibition of angiogenic activity of human ovarian cancer cells. Oncol Rep. 2000;7:1285–1291. doi: 10.3892/or.7.6.1285. [DOI] [PubMed] [Google Scholar]
- Bar-Yehuda S, Madi L, Silberman D, Slosman G, Shkapenuk M, Fishman P. CF101, an agonist to the A3 adenosine receptor enhances the chemotherapeutic effect of 5-flurouracil in a colon carcinoma murine model. Neoplasia. 2005;7:85–90. doi: 10.1593/neo.04364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Yehuda S, Silverman MH, Kerns WD, Ochaion A, Cohen S, Fishman P. The anti-inflammatory effect of A3 adenosine receptor agonists: A novel targeted therapy for rheumatoid arthritis. Expert Opin Investig Drugs. 2007;16:1601–1613. doi: 10.1517/13543784.16.10.1601. [DOI] [PubMed] [Google Scholar]
- Bar-Yehuda S, Stemmer SM, Madi L, Castel D, Ochaion A, Cohen S, Barer F, Zabutti A, Perez-Liz G, Del Valle L, Fishman P. The A3 adenosine receptor agonist CF102 induces apoptosis of hepatocellular carcinoma via de-regulation of the Wnt and NF-kappaB signal transduction pathways. Int J Oncol. 2008;33:287–295. [PubMed] [Google Scholar]
- Bar-Yehuda S, Rath-Wolfson L, Del Valle L, Ochaion A, Cohen S, Patoka R, Zozulya G, Barer F, Atar E, Piña-Oviedo S, Perez-Liz G, Castel D, Fishman P. Induction of an antiinflammatory effect and prevention of cartilage damage in rat knee osteoarthritis by CF101 treatment. Arthritis Rheum. 2009;60:3061–3071. doi: 10.1002/art.24817. [DOI] [PubMed] [Google Scholar]
- Black RG, Jr, Guo Y, Ge ZD, Murphree SS, Prabhu SD, Jones WK, Bolli R, Auchampach JA. Gene dosage-dependent effects of cardiac-specific overexpression of the A3 adenosine receptor. Circ Res. 2002;91:165–172. doi: 10.1161/01.res.0000028007.91385.ee. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, Kracht M. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem. 2004;279:49571–49574. doi: 10.1074/jbc.C400442200. [DOI] [PubMed] [Google Scholar]
- Chan H, Bartos DP, Owen-Schaub LB. Activation-dependent transcriptional regulation of the human Fas promoter requires NF-kappaB p50-p65 recruitment. Mol Cell Biol. 1999;19:2098–2108. doi: 10.1128/mcb.19.3.2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen GJ, Harvey BK, Shen H, Chou J, Victor A, Wang Y. Activation of A3 adenosine receptors reduces ischemic brain injury in rodents. J Neurosci Res. 2006;84:1848–1855. doi: 10.1002/jnr.21071. [DOI] [PubMed] [Google Scholar]
- Cronstein BN. Adenosine, an endogenous anti-inflammatory agent. J Appl Physiol. 1994;76:5–13. doi: 10.1152/jappl.1994.76.1.5. [DOI] [PubMed] [Google Scholar]
- Cross HR, Murphy E, Black RG, Auchampach J, Steenbergen C. Overexpression of A3 adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am J Physiol. 2002;283:1562–1568. doi: 10.1152/ajpheart.00335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugo L, Collin M, Allen DA, Patel NS, Bauer I, Mervaala EM. GSK 3beta inhibitors attenuate the organ injury/dysfunction caused by endotoxemia in the rat. Crit Care Med. 2005;33:1903–1912. doi: 10.1097/01.ccm.0000178350.21839.44. [DOI] [PubMed] [Google Scholar]
- Eigler A, Greten TF, Sinha B, Haslberger C, Sullivan GW, Endres S. Endogenous adenosine curtails lipopolysaccharide-stimulated tumour necrosis factor synthesis. Scand J Immunol. 1997;45:132–139. doi: 10.1046/j.1365-3083.1997.d01-377.x. [DOI] [PubMed] [Google Scholar]
- Feoktistov I, Ryzhov S, Goldstein AE, Biaggioni I. Mast cell-mediated stimulation of angiogenesis: Cooperative interaction between A2B and A3 adenosine receptors. Circ Res. 2003;92:485–492. doi: 10.1161/01.RES.0000061572.10929.2D. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Farbstein T, Barer F, Ohana G. Adenosine acts as a chemoprotective agent by stimulating G-CSF production: A role for A1 and A3 adenosine receptors. J Cell Physiol. 2000;183:393–398. doi: 10.1002/(SICI)1097-4652(200006)183:3<393::AID-JCP12>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Barer F, Madi L, Multani AF, Pathak S. The A3 adenosine receptor as a new target for cancer therapy and chemoprotection. Exp Cell Res. 2001;269:230–236. doi: 10.1006/excr.2001.5327. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Madi L, Cohn I. A3 adenosine receptor as a target for cancer therapy. Anticancer Drugs. 2002a;13:437–443. doi: 10.1097/00001813-200206000-00001. [DOI] [PubMed] [Google Scholar]
- Fishman P, Madi L, Bar-Yehuda S, Barer F, Del Valle L, Khalili K. Evidence for involvement of Wnt signaling pathway in IB-MECA mediated suppression of melanoma cells. Oncogene. 2002;21:4060–4064. doi: 10.1038/sj.onc.1205531. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Rath-Wolfson L, Ardon E, Barrer F, Ochaion A, Madi L. Targeting the A3 adenosine receptor for cancer therapy: Inhibition of prostate carcinoma cell growth by A3AR agonist. Anticancer Res. 2003;23:2077–2083. [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Ohana G, Ochaion A, Engelberg A, Barer F, Madi L. An agonist to the A3 adenosine receptor inhibits colon carcinoma growth in mice via modulation of GSK-3β and NF-κB. Oncogene. 2004;23:2465–2471. doi: 10.1038/sj.onc.1207355. [DOI] [PubMed] [Google Scholar]
- Fishman P, Bar-Yehuda S, Madi L, Rath-Wolfson L, Ochaion A, Cohen S, Baharav E. The PI3K-NF-kappaB signal transduction pathway is involved in mediating the anti-inflammatory effect of IB-MECA in adjuvant-induced arthritis. Arthritis Res Ther. 2006;8:R33. doi: 10.1186/ar1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner F, Leist M, Lohse AW, Germann PG, Tiegs G. Concanavalin A-induced T-cell-mediated hepatic injury in mice: The role of tumor necrosis factor. Hepatology. 1995;21:190–198. doi: 10.1016/0270-9139(95)90428-x. [DOI] [PubMed] [Google Scholar]
- Ge ZD, Peart JN, Kreckler LM, Wan TC, Jacobson MA, Gross GJ, Auchampach JA. Cl-IB-MECA [2-chloro-N6-(3-iodobenzyl)adenosine-5′-N-methylcarboxamide] reduces ischemia/reperfusion injury in mice by activating the A3 adenosine receptor. J Pharmacol Exp Ther. 2006;319:1200–1210. doi: 10.1124/jpet.106.111351. [DOI] [PubMed] [Google Scholar]
- Gomez G, Sitokovsky MV. Differential requirement for A2a and A3 adenosine receptors for the protective effect of inosine in vivo. Blood. 2003;102:4473–4478. doi: 10.1182/blood-2002-11-3624. [DOI] [PubMed] [Google Scholar]
- Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- Harish A, Hohana G, Fishman P, Arnon O, Bar-Yehuda S. A3 adenosine receptor agonist potentiates natural killer cell activity. Int J Oncol. 2003;23:1245–1249. [PubMed] [Google Scholar]
- Headrick JP, Peart J. A3 adenosine receptor-mediated protection of the ischemic heart. Vascul Pharmacol. 2005;42:271–279. doi: 10.1016/j.vph.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Jajoo S, Mukherjea D, Watabe K, Ramkumar V. Adenosine A(3) receptor suppresses prostate cancer metastasis by inhibiting NADPH oxidase activity. Neoplasia. 2009;11:1132–1145. doi: 10.1593/neo.09744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko Y, Harada M, Kawano T, Yamashita M, Shibata Y, Gejyo F, Nakayama T, Taniguchi M. Augmentation of Valpha14 NKT cell-mediated cytotoxicity by interleukin 4 in an autocrine mechanism resulting in the development of concanavalin A-induced hepatitis. J Exp Med. 2000;191:105–114. doi: 10.1084/jem.191.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Min HY, Chung HJ, Park EJ, Hong JY, Kang YJ, Shin DH, Jeong LS, Lee SK. Inhibition of cell proliferation through cell cycle arrest and apoptosis by thio-Cl-IB-MECA, a novel A3 adenosine receptor agonist, in human lung cancer cells. Cancer Lett. 2008;264:309–315. doi: 10.1016/j.canlet.2008.01.037. [DOI] [PubMed] [Google Scholar]
- Kohno Y, Sei Y, Koshiba M, Kim HO, Jacobson KA. Induction of apoptosis in HL-60 human promyelocytic leukemia cells by adenosine A3 receptor agonists. Biochem Biophys Res Commun. 1996;219:904–910. doi: 10.1006/bbrc.1996.0331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HT, Kim M, Joo JD, Gallos G, Chen JF, Emala CW. A3 adenosine receptor activation decreases mortality and renal and hepatic injury in murine septic peritonitis. Am J Physiol Regul Integr Comp Physiol. 2006;291:R959–R969. doi: 10.1152/ajpregu.00034.2006. [DOI] [PubMed] [Google Scholar]
- Lipsky PE, van der Heijde DM, St Clair EW, Furst DE, Breedveld FC, Kalden JR, Smolen JS, Weisman M, Emery P, Feldmann M, Harriman GR, Maini RN. Infliximab and methotrexate in the treatment of rheumatoid arthritis. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. N Engl J Med. 2000;343:1594–1602. doi: 10.1056/NEJM200011303432202. [DOI] [PubMed] [Google Scholar]
- Lu J, Pierron A, Ravid K. An adenosine analogue, IB-MECA, down-regulates estrogen receptor alpha and suppresses human breast cancer cell proliferation. Cancer Res. 2003;63:6413–6423. [PubMed] [Google Scholar]
- Mabley J, Soriano F, Pacher P, Haskó G, Marton A, Wallace R, Salzman A, Szabó C. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–329. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- Madi L, Bar-Yehuda S, Barer F, Ardon E, Ochaion A, Fishman P. A3 adenosine receptor activation in melanoma cells: Association between receptor fate and tumor growth inhibition. J Biol Chem. 2003;278:42121–42130. doi: 10.1074/jbc.M301243200. [DOI] [PubMed] [Google Scholar]
- Madi L, Ochaion A, Rath-Wolfson L, Bar-Yehuda S, Erlanger A, Ohana G, Harish A, Merimski O, Barer F, Fishman P. The A3 adenosine receptor is highly expressed in tumor versus normal cells: Potential target for tumor growth inhibition. Clin Cancer Res. 2004;10:4472–4479. doi: 10.1158/1078-0432.CCR-03-0651. [DOI] [PubMed] [Google Scholar]
- Madi L, Cohn S, Ochaion A, Bar-Yehuda S, Barer F, Fishman P. Over-expression of A3 adenosine receptor in PBMNC of rheumatoid arthritis patients: Involvement of NF-κB in mediating receptor level. J Rheumatol. 2007;34:20–26. [PubMed] [Google Scholar]
- Matot I, Weiniger CF, Zeira E, Galun E, Joshi BV, Jacobson KA. A3 adenosine receptors and mitogen-activated protein kinases in lung injury following in vivo reperfusion. Crit Care. 2006;10:R65. doi: 10.1186/cc4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merighi S, Benini A, Mirandola P, Gessi S, Varani K, Leung E, Maclennan S, Borea PA. A3 adenosine receptor activation inhibits cell proliferation via phosphatidylinositol 3-kinase/Akt-dependent inhibition of the extracellular signal-regulated kinase 1/2 phosphorylation in A375 human melanoma cells. J Biol Chem. 2005;280:19516–19526. doi: 10.1074/jbc.M413772200. [DOI] [PubMed] [Google Scholar]
- Ochaion A, Bar-Yehuda S, Cohen S, Amital H, Jacobson KA, Joshi BV, Gao ZG, Barer F, Patoka R, Del Valle L, Perez-Liz G, Fishman P. The A3 adenosine receptor agonist CF502 inhibits the PI3K, PKB/Akt and NF-kappaB signaling pathway in synoviocytes from rheumatoid arthritis patients and in adjuvant-induced arthritis rats. Biochem Pharmacol. 2008;76:482–494. doi: 10.1016/j.bcp.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochaion A, Bar-Yehuda S, Cohen S, Barer F, Patoka R, Amital H, Reitblat T, Reitblat A, Ophir J, Konfino I, Chowers Y, Ben-Horin S, Fishman P. The anti-inflammatory target A3 adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn’s disease. Cell Immunol. 2009;258:115–122. doi: 10.1016/j.cellimm.2009.03.020. [DOI] [PubMed] [Google Scholar]
- Ohana G, Bar-Yehuda S, Barer F, Fishman P. Differential effect of adenosine on tumor and normal cell growth: Focus on the A3 adenosine receptor. J Cell Physiol. 2001;186:19–23. doi: 10.1002/1097-4652(200101)186:1<19::AID-JCP1011>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Okano H, Shiraki K, Inoue H, Kawakita T, Yamanaka T, Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K, Murata K. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest. 2003;83:1033–1043. doi: 10.1097/01.lab.0000079328.76631.28. [DOI] [PubMed] [Google Scholar]
- Ougolkov AV, Fernandez-Zapico E, Savoym D, Nrutia RA, Billadeau DD. Glycogen synthase kinase-3beta participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076–2081. doi: 10.1158/0008-5472.CAN-04-3642. [DOI] [PubMed] [Google Scholar]
- Poulsen SA, Quinn RJ. Adenosine receptors: New opportunities for future drugs. Bioorg Med Chem. 1998;6:619–641. doi: 10.1016/s0968-0896(98)00038-8. [DOI] [PubMed] [Google Scholar]
- Rath-Wolfson L, Bar Yehuda S, Madi L, Ochaion A, Cohen S, Zabutti A, Fishman P. IB-MECA, an A3 adenosine receptor agonist prevents bone resorption in rats with adjuvant induced arthritis. Clin Exp Rheumatol. 2006;24:400–406. [PubMed] [Google Scholar]
- Schafer DF, Sorrell MF. Hepatocellular carcinoma. Lancet. 1999;353:1253–1257. doi: 10.1016/S0140-6736(98)09148-X. [DOI] [PubMed] [Google Scholar]
- Shneyvays V, Leshem D, Zinman T, Mamedova LK, Jacobson KA, Shainberg A. Role of adenosine A1 and A3 receptors in regulation of cardiomyocyte homeostasis after mitochondrial respiratory chain injury. Am J Physiol Heart Circ Physiol. 2005;288:H2792–H2801. doi: 10.1152/ajpheart.01157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Karin M. NF-kappaB signaling, liver disease and hepatoprotective agents. Oncogene. 2008;27:6228–6244. doi: 10.1038/onc.2008.300. [DOI] [PubMed] [Google Scholar]
- Targan SR, Hanauer SB, van Deventer SJ, Mayer L, Present DH, Braakman T, DeWoody KL, Schaible TF, Rutgeerts PJ. A short-term study of chimeric monoclonal antibody cA2 to tumor necrosis factor alpha for Crohn’s disease. Crohn’s Disease cA2 Study Group. N Engl J Med. 1997;337:1029–1035. doi: 10.1056/NEJM199710093371502. [DOI] [PubMed] [Google Scholar]
- Tiegs G. Experimental hepatitis and role of cytokines. Acta Gastroenterol Belg. 1997;60:176–179. [PubMed] [Google Scholar]
- Tiegs G, Hentschel J, Wendel A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J Clin Invest. 1992;90:196–203. doi: 10.1172/JCI115836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle BJ, Varga C, Posa A, Molnar A, Collin M, Thiemermann C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3beta. Br J Pharmacol. 2006;147:575–582. doi: 10.1038/sj.bjp.0706509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhouse EC, Amanatullah DF, Schetz JA, Liotta LA, Stracke ML, Clair T. Adenosine receptor mediates motility in human melanoma cells. Biochem Biophys Res Commun. 1998;246:888–894. doi: 10.1006/bbrc.1998.8714. [DOI] [PubMed] [Google Scholar]
- Xu Z, Jang Y, Mueller RA, Norfleet EA. IB-MECA and cardioprotection. Cardiovasc Drug Rev. 2006;24:227–238. doi: 10.1111/j.1527-3466.2006.00227.x. [DOI] [PubMed] [Google Scholar]