

Abstract
The intramolecular asymmetric cyclization of aldehydes has been accomplished using singly occupied molecular orbital (SOMO) catalysis. Selective oxidation of chiral enamines (formed by the condensation of an aldehyde and a secondary amine catalyst) leads to the formation of a 3π-electron radical species. These chiral SOMO-activated radical cations undergo enantioselective cyclization with an array of pendent allylsilanes thus efficiently providing a new approach to the construction of five-, six- and seven-membered carbocycles and heterocycles.
Introduction
The enantioselective formation of stereochemically complex carbocyclic and heterocyclic ring systems remains a longstanding goal within the field of chemical synthesis.1 The Diels–Alder reaction remains perhaps the archetypal example (eq 1), a powerful technology that builds molecular complexity from simple dienes and dienophiles via a routine and predictable intermolecular or intramolecular pathway.1a Likewise, ring-closing metathesis has emerged as a robust approach for the intramolecular cyclization of simple diene systems, a transformation that has now found widespread utility for the construction of natural products and medicinal agents, broadly defined.1b Recently, we hypothesized that SOMO-catalysis (singly occupied molecular orbital) might provide an additional approach to the enantioselective construction of complex ring systems via the asymmetric intramolecular α-allylation of aldehydes using formyl-tethered allylsilanes (eq 2).2–4 Enantioselective SOMO-catalysis is a unique and versatile mode of organocatalytic activation that features the transient generation of a 3π-radical cation species, which can participate in asymmetric bond construction with a variety of π-rich nucleophiles or electron-neutral SOMO-philes. Since its introduction in 2007, we have successfully utilized this activation mode to overcome a series of elusive challenges in asymmetric catalysis, including the α-allylic alkylation, α-enolation, α-vinylation, α-chlorination, and α-arylation of aldehydes and ketones.5 On this basis, we envisioned that various formyl tethered allylsilanes should readily undergo asymmetric cyclization in the presence of an oxidant and a chiral amine catalyst to stereoselectively generate five-, six- and seven-membered rings. Herein, we describe the successful execution of these ideals and outline a facile, intramolecular aldehyde α-allylation protocol, which, to our knowledge, is not known in any previous format (racemic or asymmetric).6 This new approach to enantioenriched carbocycles and heterocycles of various size and substitution pattern should be of utility to practitioners of both natural product and medicinal agent synthesis.
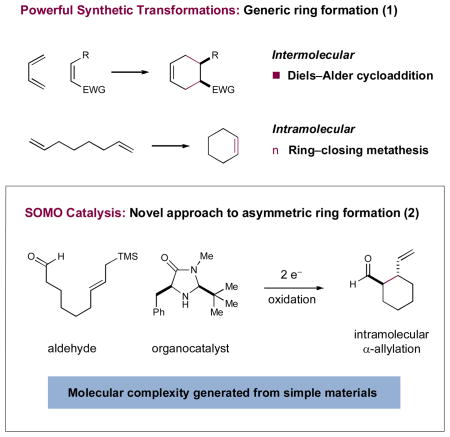
Design Plan
In accord with our previous SOMO-catalysis studies,5 we reasoned that exposure of an allylsilane-tethered aldehyde7–8 to amine catalyst 1 in the presence of a suitable oxidant would render the radical-cation 2 (Scheme 1). Rapid and enantioselective cyclization of the pendent olefin onto the 3π-electron species would then provide a β-silyl radical 3 that, upon further oxidation, would generate a stabilized cation (not shown). At this stage, we presumed that β-silyl elimination would be facile prior to hydrolysis of the iminium species to reconstitute catalyst 1 and deliver the desired 1,2-disubstituted ring system in enantioenriched form. In alignment with previous studies, we anticipated that the activated radical 2 would position the 3π-electron system away from the bulky tert-butyl moiety, while the benzyl group would effectively shield the Re-face,5 such that internal cyclization of the pendent allylsilane moiety would occur from the less shielded Si-face.
Scheme 1.

Proposed mechanism of organo-SOMO allylic cyclization.
Results and discussion
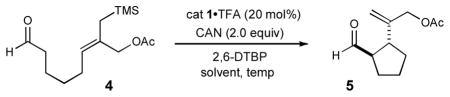
The feasibility of this proposed intramolecular cyclization was first examined using aldehyde 4, catalyst 1•TFA and CAN (ceric ammonium nitrate) in DME at −20 °C. As shown in Table 1, the desired cyclization could be accomplished using these initial conditions, albeit with modest yield and enantiocontrol along with poor diastereoinduction (entry 1, 53% yield, 3:1 dr, 82% ee). Changing the reaction medium from DME to acetone served to increase both reaction efficiency and stereoselectivity (entry 2, 71% yield, 88% ee), and further improvements could be realized through the addition of water (entry 3, 80% yield, 91% ee). We presume that the addition of water allows for rapid capture of the putative silyl-cation that is generated at the final step in the catalytic cycle (see Scheme 1), a species that is often found to be detrimental to organocatalytic systems.5h As might be expected, lowering the reaction temperature to −30 °C provided a modest enhancement in diastereoselectivity, however, the accompanying decrease in reaction efficiency rendered these conditions less favorable (entry 4). The superior levels of induction and efficiency exhibited by amine 1 in acetone at −20 °C to afford (1 R, 2S)-1-formyl-2-(3-isopropenyl acetate)-cyclopentane in 91% ee, 15:1 dr and 80% yield, prompted us to select these catalytic conditions for further exploration.
Table 1.
Enantioselective organo-SOMO cyclization: Initial studies.
| ||||||
|---|---|---|---|---|---|---|
| entry | solvent | H2O | temp, °C | % yielda | drb | % eec |
| 1 | DME | --- | −20 | 53 | 3 : 1 | 82 |
| 2 | acetone | --- | −20 | 70 | 8 : 1 | 88 |
| 3 | acetone | 2 equiv | −20 | 80 | 15 : 1 | 91 |
| 4 | acetone | 2 equiv | −30 | 72 | >20 : 1 | 92 |
Isolated yield.
Diastereomeric ratio determined by 1H NMR analysis.
Enantiomeric excess determined by GC analysis. 2,6-DTBP: 2,6-di-tert-butylpyridine; DME: 1,2-dimethoxyethane.
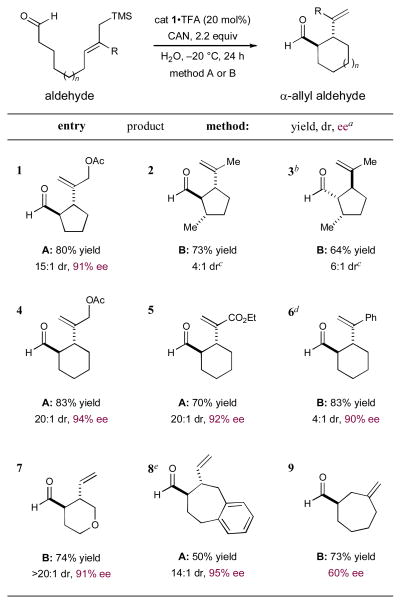
As revealed in Table 2, this new enantioselective α-formyl cyclization reaction can be employed to build a diverse range of carbo- and heterocyclic ring systems. With respect to five-membered rings, this amine-catalyzed protocol is tolerant to electronically diverse allylsilane terminators (entries 1–3). Moreover, enantiopure β-chiral aldehydes can also be used for the diastereoselective construction of 1,2-anti, 2,3-anti as well as 1,2-syn, 2,3-anti cyclopentane adducts (entries 2 and 3, 64–73%, 4:1 and 6:1 dr, respectively). It is interesting to note that the induced stereochemistry in both examples is dominated by the catalyst architecture rather than a matched and mismatched pairing with the specific catalyst antipode employed. Cyclohexyl systems can also be accessed with high levels of efficiency and enantiocontrol (entries 4–6, 70–83% yield, ≥90% ee). In this series, dramatic changes in the inherent π-nucleophilicity of the allylsilane unit can also be accommodated (c.f. α,β-unsaturated esters and styrenyl olefins, entries 5 and 6 respectively). Moreover, heteroatoms can be readily incorporated into the formyl-allylic silane linker thereby allowing direct access to heterocycles such as tetrahydropyranyl rings (entry 7, 74% yield, >20:1 dr, 91% ee). Finally, synthetically challenging enantioenriched seven-membered systems may be accessed using this new organocatalytic cyclization protocol. Efforts to accomplish 7-exo-cyclization resulted in formation of the desired adduct with high levels of enantiocontrol (entry 8, 95% ee), while the corresponding 7-endo cyclization is also possible, albeit with modest levels of enantiocontrol (entry 9, 60% ee). Importantly, for all cases examined in this manuscript, the potentially competitive intramolecular Sakurai-allylation pathway is almost completely suppressed.8
Table 2.
Enantioselective organo-SOMO cyclization: Reaction scope.
|
Isolated yields. Diastereomeric ratio and enantiomeric excess determined by 1H NMR or GC analysis. Method A: 2,6-DTBP in acetone. Method B: NaHCO3 in DME.
Employed ent-cat 1•TFA.
Major diastereomer shown, remaining two diastereomers are observed in only trace quantity by 1H NMR and GC analysis.
Employed (2S,5S)-2-tert-butyl-3,5-dimethylimidazolidin-4-one as catalyst.
Performed in 5% MeCN/DME.
Finally, to further demonstrate the value of this new α-allylation cyclization strategy, we focused upon the development of a unique and expeditious route to enantioenriched 3,4-disubstituted piperidine rings, a structural motif that is prevalent in drug design and commercial medicines (e.g. Paxil). As shown in detail in Figure 1, implementation of our standard cyclization conditions with N-tosyl-linked formyl-allylsilanes enabled the rapid production of the desired trans-substituted 3-vinyl, 4-formyl piperidine rings with excellent levels of enantiocontrol (78–86% yield, 89–91% ee, 11–20:1 dr). Intriguingly, an examination of the impact of olefin geometry on product diastereoselectivity suggests that E-allylsilanes cyclize to form products with slightly higher levels of diastereocontrol than those observed with the corresponding Z-allylsilane substrates, while the reverse trend is observed in enantioselectivity.
Figure 1.

Catalyst-controlled stereoselective piperidine formation.a
aIsolated yields. Diastereomeric ratio determined by 1H NMR analysis. Enantiomeric excess determined by GC analysis. Reactions performed using Method B.
Conclusions
In summary, we have applied the mechanistic paradigm of organo-SOMO catalysis to the successful development of a novel cyclization reaction based upon the enantioselective intramolecular α-allylation of aldehydes. This protocol can be successfully employed to construct five-, six-, and seven-membered carbocycles as well as delivering a new route to tetrahydropyran and piperidine heterocyclic ring systems. We expect this new bond forming technology will find application in the construction of both natural products and medicinal agents. Further studies into the scope of this new transformation are ongoing in our laboratory.
Supplementary Material
Acknowledgments
Financial support was provided by NIHGMS (R01 GM078201-05) and kind gifts from Merck, Amgen and Abbott. P. V. P. thanks the Vietnamese government for a predoctoral fellowship, and K. A. thanks Merck for a postdoctoral fellowship.
Footnotes
Electronic Supplementary Information (ESI) available: Experimental procedures, structural proofs, and spectral data for all new compounds are provided (XX pages) (PDF). See DOI: 10.1039/b000000x/
Notes and references
- 1.(a) Kagan HB, Riant OM. Chem Rev. 1992;92:1007–1019. [Google Scholar]; (b) Monfette S, Fogg DEM. Chem Rev. 2009;109:3783–3816. doi: 10.1021/cr800541y. [DOI] [PubMed] [Google Scholar]
- 2.For a review of transition metal-catalyzed, asymmetric allylic alkylations, see: Trost BM, Van Vranken DLM. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804.and references therein; and for a recent example of a π-allyl palladium approach which employs an achiral organocatalyst, see: Bihelovic F, Matovic R, Vulovic B, Saicic RNM. Org Lett. 2007;9:5063–5066. doi: 10.1021/ol7023554.
- 3.For lead references concerning radical cyclizations, see: Snider BB. Manganese(III)-Mediated Radical Reactions. In: Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Vol. 2. Wiley-VCH; 2000. and references therein; Dhimane A-L, Fensterbank L, Malacria M. Polycyclic Compounds via Radical Cascade Reactions. In: Renaud P, Sibi MP, editors. Radicals in Organic Synthesis. Vol. 2. Wiley-VCH; 2000. and references therein; Togo H. Advanced Free Radical Reactions for Organic Synthesis. Vol. 3 Springer; New York: 1999. and references therein. For recent examples of radical cyclizations, see: Alabugin IV, Timokhin VI, Abrams JN, Manoharan M, Abrams R, Ghiviriga IM. J Am Chem Soc. 2008;130:10984–10995. doi: 10.1021/ja801478n.Servais A, Azzouz M, Lopes D, Courillon C, Malacria M. Angew Chem, Int Ed. 2007;46:576–579. doi: 10.1002/anie.200602940.de Turiso FG-L, Curran DPM. Org Lett. 2007;7:151–154. doi: 10.1021/ol0477226.Trost BM, Shen HC, Surviet J-PM. J Am Chem Soc. 2004;126:12565–12579. doi: 10.1021/ja048084p.Toyota M, Yokota M, Ihara M. J Am Chem Soc. 2001;123:1856–1861. doi: 10.1021/ja0035506.Tokuyama H, Yamashita T, Reding MT, Kaburagi Y, Fukuyama TM. J Am Chem Soc. 1999;121:3791–3792.(j) for reviews of enantioselective radical processes, see: Yoshioka E, Kohtani S, Miyabe HM. Heterocycles. 2009;79:229–242.and references therein; Sibi MP, Manyem S, Zimmerman JM. Chem Rev. 2003;103:3263–3295. doi: 10.1021/cr020044l.and references therein; for recent examples of enantioselective radical processes, see: Bauer A, Westkämper F, Grimme S, Bach TM. Nature. 2005;436:1139–1140. doi: 10.1038/nature03955.and references therein; Yang D, Zheng B-F, Gao Q, Gu S, Zhu N-YM. Angew Chem, Int Ed. 2006;45:255–258. doi: 10.1002/anie.200503056.Yang D, Gu S, Yan Y-L, Zhu N-Y, Cheung K-KM. J Am Chem Soc. 2001;123:8612–8613. doi: 10.1021/ja016383y.for reviews of oxidative cyclizations, see: Snider BBM. Chem Rev. 1996;96:339–363. doi: 10.1021/cr950026m.and references therein; for examples of oxidative cyclizations, see: Snider BB, Kiselgof JY, Foxman BMM. J Org Chem. 1998;63:7945–7952.Yang D, Ye X-Y, Gu S, Xu MM. J Am Chem Soc. 1999;121:5579–5580.Yang D, Ye X-Y, Xu M, Pang K-W, Cheung K-KM. J Am Chem Soc. 2000;122:1658–1663.Yang D, Xu M, Bian M-YM. Org Lett. 2001;3:111–114. doi: 10.1021/ol0068243.Huang Y, Moeller KDM. Org Lett. 2004;6:4199–4202. doi: 10.1021/ol048450+.
- 4.For recent examples of SOMO-catalyzed cyclizations, see: Nicolaou KC, Reingruber R, Sarlah D, Bräse SM. J Am Chem Soc. 2009;131:2086–2087. doi: 10.1021/ja809405c.Conrad JC, Kong J, Laforteza BN, MacMillan DWCM. J Am Chem Soc. 2009;131:11640–11641. doi: 10.1021/ja9026902.Um JM, Gutierrez O, Schoenebeck F, Houk KN, MacMillan DWCM. J Am Chem Soc. 2010;132:6001–6005. doi: 10.1021/ja9063074.
- 5.For examples of our group’s development of SOMO catalysis, see: Beeson TD, Mastracchio A, Hong J-B, Ashton K, MacMillan DWCM. Science. 2007;316:582–585.Jang H-Y, Hong J-B, MacMillan DWCM. J Am Chem Soc. 2007;129:7004–7005. doi: 10.1021/ja0719428.Kim H, MacMillan DWCM. J Am Chem Soc. 2008;130:398–399. doi: 10.1021/ja077212h.Amatore M, Beeson TD, Brown SP, MacMillan DWCM. Angew Chem, Int Ed. 2009;48:5121–5124. doi: 10.1002/anie.200901855.Wilson JE, Casarez AD, MacMillanM DWC. J Am Chem Soc. 2009;131:11332–11334. doi: 10.1021/ja904504j.Rendler S, MacMillan DWCM. J Am Chem Soc. 2010;132:5027–5029. doi: 10.1021/ja100185p.Jui NT, Lee ECY, MacMillan DWCM. J Am Chem Soc. 2010;132:10015–10017. doi: 10.1021/ja104313x.Devery JJ, III, Conrad JC, MacMillan DWC, Flowers RAM., II Angew Chem, Int Ed. 2010;49:6106–6110. doi: 10.1002/anie.201001673.Mastracchio A, Warkentin AA, Walji AM, MacMillan DWCM. Proc Nat Acad Sci, USA. 2010;107:20648–20651. doi: 10.1073/pnas.1002845107.
- 6.An enamine catalysis cyclization strategy was applied by the List group to construct 3- to 5-membered rings: Vignola N, List B. J Am Chem Soc. 2004;126:450–451. doi: 10.1021/ja0392566.
- 7.For the synthesis of allylsilanes, see: Sarkar TKM. Synthesis. 1990:969–983.Sarkar TK. Synthesis. 1990:1011–1111.Crowe WE, Goldberg DR, Zhang ZJM. Tetrahedron Lett. 1996;37:2117–2120.Tsuji Y, Funato M, Ozawa M, Ogiyama H, Kajita S, Kawamura TM. J Org Chem. 1996;61:5779–5787.
- 8.For examples of intramolecular Sakurai reactions with similar starting materials, see: Jervis PJ, Kariuki BM, Cox RLM. Org Lett. 2006;8:4649–4652. doi: 10.1021/ol061957v.Schlosser M, Franzini L, Bauer C, Leroux FM. Chem Eur J. 2001;7:1909–1914. doi: 10.1002/1521-3765(20010504)7:9<1909::aid-chem1909>3.0.co;2-h.(c) Identical substrates to those in Table 2, entries 2 and 3 are employed in Ref 8b. In our reaction, less than 20% of the Sakurai by-product is observed in either case. We presume this arises from an acid co-catalyst activation pathway.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.