

Abstract
The dedifferentiation agent ‘reversine’ (2-(4-morpholinoanilino)-N6-cyclohexyladenine 2) was found to be a moderately potent antagonist for the human A3 adenosine receptor (AR) with a Ki value 0.66 μM. This result prompted an exploration of the structure-activity relationship of related derivatives, synthesized via sequential substitution of 6-chloro-2-fluoropurine with selected nucleophiles. Optimization of substituents at these two positions identified 2-phenylamino-N6-(cyclohexyl)adenine 12, 2-phenylamino-N6-(cycloheptyl)adenine 19, and 2-phenylamino-N6-(endo-norbornyl)adenine 21 as potent A3 AR ligands with Ki values of 51, 42 and 37 nM, respectively, with 30 – 200-fold selectivity in comparison to A1 and A2A ARs. The most selective A3 AR antagonist (>200-fold) was 2-phenyloxy-N6-(cyclohexyl)adenine 22. 9-Methylation of 12, but not 19, was well tolerated in A3 AR binding. Extension of the 2-phenylamino group to 2-benzyl- and 2-(2-phenylethylamino) reduced affinity. In the series of 2-phenylamino, 2-phenyloxy, and 2-phenylthio substitutions, the order of affinity at the A3 AR was oxy ≥ amino > thio. Selected derivatives, including reversine (KB value of 466 nM in Schild analysis), competitively antagonized the functional effects of a selective A3 AR agonist, i.e. inhibition of forskolin-stimulated cAMP production in stably transfected Chinese hamster ovary (CHO) cells. These results are in agreement with other studies suggesting the presence of a lipophilic pocket in the AR binding site that is filled by moderately sized cycloalkyl rings at the N6 position of both adenine and adenosine derivatives. Thus, the compound series reported herein comprise an important new series of selective A3 AR antagonists. We were unable to reproduce the dedifferentiation effect of reversine, previously reported, or to demonstrate any connection between A3 AR antagonist effects and dedifferentiation.
Introduction
The A1, A2A, A2B and A3 adenosine receptors (ARs) are G protein-coupled receptors that are specifically distributed throughout various tissues and cell types of the human body.1 The ubiquitous nature of the natural ligand, adenosine, has prompted countless studies aimed to delineate the extent to which each subtype of the receptor could be selectively modulated and if such a modulation could be exploited for some therapeutic advantage. Over the past decade, it has become apparent that control of the four AR subtypes has a wide-ranging impact on stroke and other ischemic conditions, as well as inflammation, neurodegenerative diseases, diabetes, sleep regulation, and other therapeutic fronts.2
The structure-activity relationships (SAR) of adenosine derivatives as selective agonists at three of the four AR subtypes (except for A2B) is well developed.4–8 Among AR antagonists, several classes of small nonnucleoside heterocycles, including xanthines, deazaadenines, pyrazolopyrimidines and adenines, have varying degrees of potency and receptor subtype selectivity.1,9 Various 9-alkyl-adenine derivatives have displayed high selectivity for the A1 AR, and more modest selectivity for A2A and A3ARs.10–13 For example, 8-(N-methylisopropyl)amino-N6-(endo-2′-(endo-5′-hydroxy)norbornyl)-9-methyladenine (WRC-0571) 1a (Chart 1) displayed a Ki value of 1.7 nM at the human A1 AR.14 Compound 1b was 10-fold more potent at antagonizing the A2A AR in guinea pig coronary artery than a putative A2B AR in the aorta.15 An attempt to convert known A3 AR agonists, such as Cl-IB-MECA (2-chloro-N6-(3-iodobenzyl)-5′-N-methylcarboxamidoadenosine), into selective antagonists at the same subtype through truncation of the ribose moiety in stages greatly reduced receptor affinity.13 A series of 8-substituted adenine derivatives, such as 9-ethyl-8-phenylethynyl-9H-adenine 1c displayed selectivity as antagonists of the human A3 AR.11 A3 AR antagonists are of interest as antiglaucoma agents and in inflammatory diseases.32–34
Chart 1.

Structures of Selected Adenine Derivatives Reported as AR Antagonists (1a – c) and Reversine (2).
Adenine derivatives are also important pharmacological probes of other biochemical processes such as kinase inhibition,16 and the AR affinity of such derivatives has not been fully explored. An 9-alkyl adenine derivative SQ22536 that inhibits another signaling enzyme, adenylate cyclase, was also shown to antagonize the A1 AR.17 Recently, Schultz, Ding, and coworkers reported that ‘reversine’ (2-(4-morpholinoanilino)-N6-cyclohexyladenine) 2 (Chart 1), selected from a large chemical library, stimulated the dedifferentiation of lineage-committed cells (myoblasts) back to progenitor cells, which were capable of being redirected via osteogenic-inducing agents or adipogenic-inducing agents into osteoblasts or adipocytes, respectively.18 This remarkable discovery has vast consequences throughout biological and medical research. The mechanism by which this extraordinary transformation takes place is as yet unknown and subject to speculation and investigation. This project will no doubt be a formidable undertaking given the breadth of the cellular landscape and the complexity of the phenotypical response. We noted the similarity of the substitution pattern of 2 in relation to various known AR antagonists and considered that, while not knowing the significance of AR modulation on the cell cycle and/or dedifferentiation, reversine might selectively bind at one or more of the ARs.
Following the protocols set forth by Schultz and coworkers, we synthesized several 2,6-disubstituted purine analogues including reversine 2.19 Our initial results showed that 2 bound with moderate affinity to the A3 AR. Our selection of derivatives was motivated by a desire to initially explore the SAR for this series of compounds at the ARs rather than to expand on the theme of dedifferentiation. Accordingly, we diverged from the parent structure in several fundamental ways, i.e., the size of the cycloalkyl ring at N6, the substitution pattern of the aromatic ring at C2, heteroatom substitution of the 2-amino group and methylation at the 9 position.
Results and Discussion
Chemical Synthesis
The preparation of 2 and its analogues (4–24) was achieved via the established protocols of Schultz and coworkers (Scheme 1).19 Substitution of 6-chloro-2-fluoropurine (3) with a series of amines, including ethylamine and several cyclic amines of varying sizes from cyclopropyl to 2-endo-norbornyl, was accomplished in n-butanol with Hunig’s base at 80°C over 24 h. Following solvent removal the crude reaction mixture was treated with an excess of a second series of amines, including ethylamine and several amines with various aromatic groups (see Table 2), phenol or thiophenol in ethanol and heated in a sealed tube at 110°C for 48 h. Following flash chromatography the yields of the purified products ranged typically from 30–60% for the two-step procedure. We noted that the substitution of 2,6-dichloropurine resulted in the same transformation; however, the use of 6-chloro-2-fluoropurine was more facile and provided a convenient method to ensure that the correct 2,6-substitution pattern was achieved via mass analysis of the fluorinated intermediate. We also noted the success of microwave-assisted organic synthesis in the facile preparation of similar 2,6-disubstituted purine analogues (data not shown). Selected analogues were methylated at the N9 position in quantitative yield via treatment with methyl iodide in basic DMF at 0°C over 1–2 h.
Scheme 1.
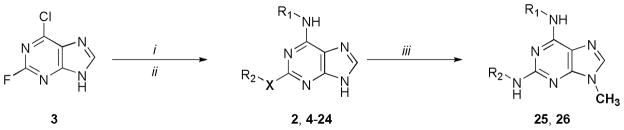
Synthesis of 2,6-Disubstituted Purine Derivatives. Reagents and conditions: (i) R-NH2 (0.9 equiv.), Hunig’s base, n-BuOH, 80°C, 24 h; (ii) R′-NH2, R′-OH, or R′-SH (2 equiv.), EtOH, 110°C, 48 h; (iii) MeI, K2CO3, DMF 0°C 1–2 h.
Table 2.
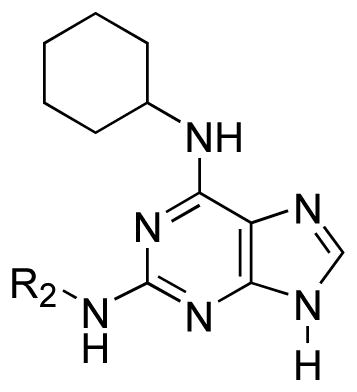
Binding affinities of 2-substituted N6-cyclohexyladenine derivatives at human A1, A2A and A3ARs expressed in CHO cells.a
| ||||
|---|---|---|---|---|
| Compound | R2 | Ki (hA3AR) μMa | Ki (hA1AR) μM, or % inhib., 10 μM | Ki (hA2AAR) μM, or % inhib., 10 μM |
| 11 | ethyl | 1.12 ± 0.61 | 51% | 42% |
| 12 | phenyl | 0.051 ± 0.017 | 2.73 ± 0.58 | 1.70 ± 0.95 |
| 13 | benzyl | 0.920 ± 0.121 | 42% | 34% |
| 14 | 2-phenylethyl | 1.46 ± 0.11 | 3.17 ± 1.02 | 46% |
| 15 | 2-naphthyl | 2.42 ± 1.44 | 39% | 52% |
| 16 | biphenyl-1-yl | 5.40 ± 1.00 | 14% | 22% |
| 17 | 4-piperidino-phenyl | 1.57 ± 0.56 | 16% | 50% |
| 18 | 4-dimethyl-aminophenyl | 0.070 ± 0.025 | 3.90 ± 0.96 | 6.1 ± 2.5 |
All A3AR experiments were performed using adherent CHO cells stably transfected with cDNA encoding one of the human adenosine receptor. Binding at human A1, A2A, and A3ARs in this study was carried out as described in Methods using [3H]DPCPX, [3H]ZM241,385 or [125I]AB-MECA as radioligand. Values from the present study are expressed as Ki values (mean±s.e.m., n = 3), or as percent displacement of radioligand.
Biological Characterization
Binding affinity at the human A3 AR, and in selected cases the rat A3 AR, was studied using the radioiodinated agonist [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide.20 Binding at the human A1 and A2A receptors utilized the selective antagonists [3H]8-cyclopentyl-1,3-dipropylxanthine (DPCPX) and [3H]4-2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol (ZM241,385), respectively. Functional assays of the human A3 receptor consisted of measuring inhibition of forskolin-stimulated cAMP production in intact Chinese hamster ovary (CHO) cells expressing the ARs.21
An initial screen of the dedifferentiation agent ‘reversine’ 2 at the ARs indicated binding at the A3 AR with a Ki value of 0.66 μM. At the A1, A2A ARs binding was much weaker with only 36% and 30% of radioligand binding inhibited, respectively, at 10 μM of 2. Following the identification of 2 as a moderately potent ligand in binding at the A3 AR we sought to explore the SAR of related 2,6-disubstituted purines in order to enhance potency and A3 AR selectivity. Our initial notion was to individually consider the two structurally distinct moieties on the purine ring system, i.e. the N6-cyclohexyl ring and the 2-(4-morpholinoanilino) moiety. The binding results for these two sets of analogues are displayed in Tables 1 and 2.
Table 1.
Binding affinities of N6-substituted derivatives of 2-(4-morpholinophenylamino)adenine at human A1, A2A and A3ARs expressed in CHO cellsa

| ||||
|---|---|---|---|---|
| Compound | R1 | Ki (hA3AR) μMa | Ki (hA1AR) μM, or % inhib., 10 μM | Ki (hA2AAR) μM, or % inhib., 10 μM |
| 4 | ethyl | 1.53 ± 0.47 | 23% | 16% |
| 5 | cyclopropyl | 1.87 ± 0.89 | 22% | 25% |
| 6 | cyclobutyl | 1.81 ± 0.50 | 29% | 18% |
| 7 | cyclopentyl | 1.39 ± 0.48 | 22% | 20% |
| 2b | cyclohexyl | 0.66 ± 0.14 | >10 (36%) | >10 (30%) |
| 8 | cycloheptyl | 0.42 ± 0.17 | 6% | 38% |
| 9 | cyclooctyl | 2.77 ± 0.83 | 0% | 17% |
| 10 | 2-endo-norbornyl | 0.122 ± 0.010 | 34% | 39% |
All A3AR experiments were performed using adherent CHO cells stably transfected with cDNA encoding one of the human adenosine receptor. Binding at human A1, A2A, and A3ARs in this study was carried out as described in Methods using [3H]DPCPX, [3H]ZM241,385 or [125I]AB-MECA as radioligand. Values from the present study are expressed as Ki values (mean±s.e.m., n = 3), or as percent displacement of radioligand.
Demonstrated to be antagonist of the human A3AR.
N6-Alkylation of both adenosine and adenine analogues is well established as a means to increase the potency and selectivity at the ARs.4–7,10,12 Clearly, as seen in Table 1, the size of the N6-cycloalkyl ring had important consequences for binding at the A3 AR. The A3 AR affinity increased in stages upon enlarging the ring size from the cyclopropyl derivative 5 (Ki = 1.9 μM) to the cycloheptyl derivative 8 (Ki = 0.42 μM) strongly suggesting that a bulky hydrophobic alkyl group was favored at the N6 position. The trend appeared to be limited to cycloalkyl rings smaller than 8 carbons in size, as demonstrated by a reduced tolerance for the cyclooctyl ring in 9. The most potent 2-(4-morpholinophenylamino)adenine derivative contained the rigid N6-(2-endo-norbornyl) substituent 10 (Ki = 0.122 μM).
Exploration of the moiety at the 2-NH suggested that smaller aromatic analogues were more apt to bind strongly at the A3 receptor subtype as demonstrated by the high affinity of the 2-phenylamino analogue 12 (Ki = 0.051 μM) and the 2-(4-dimethylamino)phenylamino analogue 18 (Ki = 0.070 μM) (Table 2). The 2-ethylamino derivative 11 was less potent than 2 at the A3 AR. In the series of phenylamino 12, benzylamino 13, and 2-phenylethylamino 14 substitutions, there was a pronounced preference for the shortest analogue 12. In comparison to substituted phenylamino derivatives 15 – 18, the unsubstituted 12 displayed the highest A3 AR affinity.
The compelling finding that 2-phenylamino analogues bound strongly at the A3 AR motivated us to further explore a series of analogues that maintained the 2-phenylamino moiety while optimizing the N6-cycloalkyl ring (Table 3). We also replaced the 2-NH with either oxygen or sulfur as the heteroatom. The results of these analogues supported the SAR regarding ring size at the N6 position; i.e. cyclohexyl 12 and cycloheptyl 19 rings provided analogues with strong binding properties while the cyclooctyl derivative 20 showed 7 – 8-fold weaker affinity (Table 3). The N6-endo-norbornyl analogue 21 appeared to be the most potent in the series in binding to the A3 AR. In the series of N6-cyclohexyl derivatives having phenylamino 12, phenyloxy 22, and phenylthio 24 substitutions, the order of affinity at the A3 AR was oxy ≥ amino > thio. This also matched the order of affinity of 2-substituted adenosine derivatives at the A3 AR, which was oxy > amino > thio.22 Thus, the replacement of the NH with other heteroatoms helped to optimize the SAR for the A3 AR only when NH was replaced with O; however, the phenylthio analogue maintained a moderate affinity for the A3 AR. We further altered analogues 12 and 19 by a simple methylation based upon numerous reports describing 9-alkyl adenine derivatives as AR antagonists.11,23 While 9-methylation of the N6-cyclohexyl analogue 12 to yield 25 (Ki = 0.070 μM) provided a derivative that maintained its affinity to the A1, A2A, and A3 ARs, N9 alkylation on the cycloheptyl analogue to yield 26 (Ki = 0.96 μM) was detrimental leading to an unanticipated 23-fold loss of A3 AR affinity in comparison to 19 with no significant change in affinity at the A1 and A2A ARs (Table 3).
Table 3.
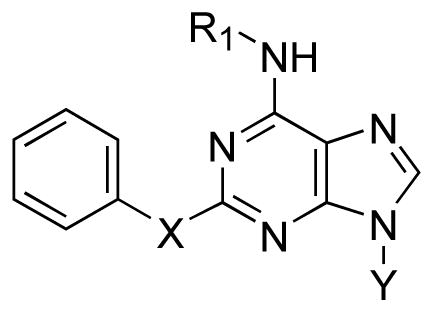
Binding affinities of 2,6-disubstituted adenine derivatives at human A1, A2A and A3ARs expressed in CHO cells.a
| ||||||
|---|---|---|---|---|---|---|
| Compound | X | Y | R1 | Ki (hA3AR) μMa | Ki (hA1AR) μM, or % inhib., 10 μM | Ki (hA2AAR) μM, or % inhib., 10 μM |
| 12 | NH | H | cyclohexyl | 0.051 ± 0.017 | 2.73 ± 0.58 | 1.70 ± 0.95 |
| 19 | NH | H | cycloheptyl | 0.042 ± 0.010 | 1.72 ± 0.02 | 4.12 ± 0.48 |
| 20 | NH | H | cyclooctyl | 0.340 ± 0.016 | 4.47 ± 0.01 | 46% |
| 21 | NH | H | 2-endo-norbornyl | 0.037 ± 0.009 | 2.22 ± 0.58 | 7.73 ± 0.13 |
| 22b | O | H | cyclohexyl | 0.047 ± 0.012 | 26% | 16% |
| 23 | O | H | 2-endo-norbornyl | 0.074 ± 0.007 | 1.47 ± 0.01 | 25% |
| 24 | S | H | cyclohexyl | 0.134 ± 0.007 | 3% | 14% |
| 25 | NH | methyl | cyclohexyl | 0.070 ± 0.021 | 1.88 ± 0.01 | 1.20 ± 0.15 |
| 26 | NH | methyl | cycloheptyl | 0.96 ± 0.51 | 1.94 ± 0.41 | 2.95 ± 1.00 |
All A3AR experiments were performed using adherent CHO cells stably transfected with cDNA encoding one of the human adenosine receptor. Bindingat human A1, A2A, and A3ARs in this study was carried out as described in Methods using [3H]DPCPX, [3H]ZM241,385 or [125I]AB-MECA as radioligand. Values from the present study are expressed as Ki values (mean±s.e.m., n = 3), or as percent displacement of radioligand.
12, MRS3767; 19, MRS3769; 22, MRS3777.
ND not determined.
The affinity at human A1 and A2A ARs was low with most derivatives binding only weakly at 10 μM. Various 2-phenylamino derivatives displayed measurable affinity at the A1 AR with Ki values in the range of 2–3 μM, i.e. 12 (N6-cyclohexyl), 19 (N6-cycloheptyl), 21 (N6-endo-norbornyl), 25 (N6-cyclohexyl-9-methyl), and 26 (N6-cycloheptyl-9-methyl). Ki values of the same derivatives in μM at the A2A AR were: 12 (1.70), 18 (6.1), 19 (4.12), 21 (7.73), 25 (1.20), and 26 (2.95). Thus, the most selective A3 AR antagonists in this study was 22, with selectivity ratios of >200 in comparison to the A1 and A2A AR subtypes. The very potent compounds 12 and 21 were 54 and 60-fold selective, respectively, in comparison to the A1 AR. A comparison of N6-cyclohexyl derivatives 12 and 22 indicated that the 2-aryloxy group was more advantageous for A3 AR selectivity than the corresponding 2-arylamino group. Thus, within this series compound 22 displayed the most favorable combination of high affinity (Ki 47 nM) and selectivity for the human A3 AR.
Selected derivatives (2, 12, and 18) were tested in binding to the rat A3 AR and were shown to be inactive at 10 μM. Therefore, the previously noted species dependence of the affinity of most known A3 AR antagonists (human ≫ rat)1,7,32 also applied to this series of A3 AR antagonists.
Functional antagonism of the human A3 AR by this series of adenine derivatives was demonstrated in CHO cells. A functional assay consisting of human A3 AR-mediated inhibition of forskolin-stimulated adenylate cyclase stably expressing the receptor was used. A Schild analysis of inhibition by 2 of the effects of an A3 AR agonist Cl-IB-MECA at the human A3 AR confirmed that this adenine derivative was a competitive antagonist with a KB value of 466 nM (Figure 1). This value was in close agreement with the Ki value determined in the binding assay.
Figure 1.
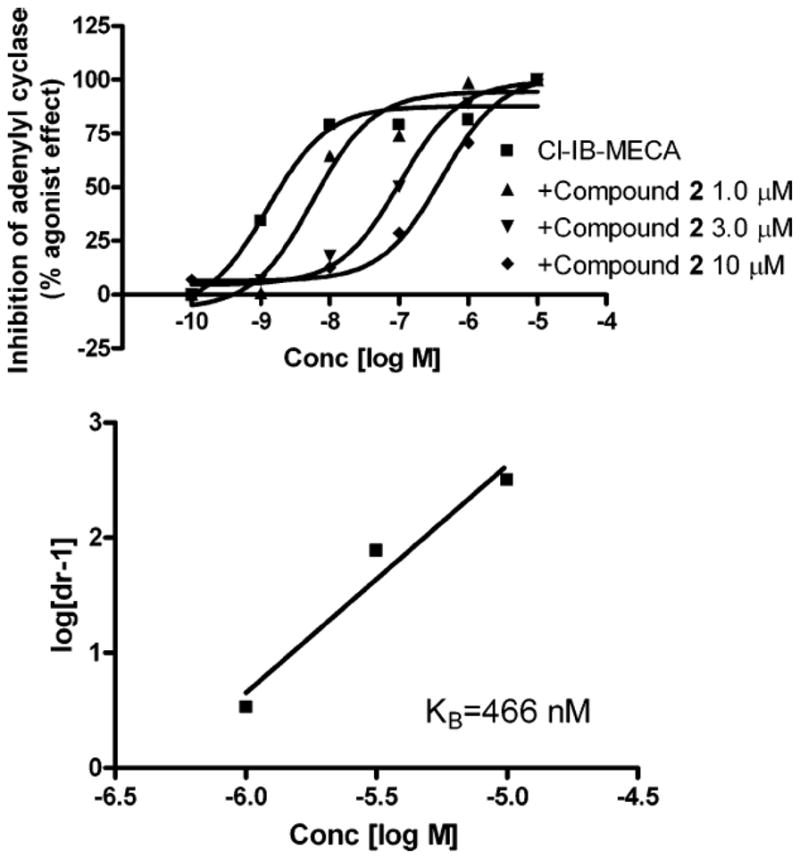
Schild Analysis for Antagonism of the Human A3 AR in Stably Transfected CHO Cells by Compound 2. Cl-IB-MECA is used as a potent A3 AR agonist. The experiment was performed in the presence of 10 μM rolipram. Forskolin (10 μM) was used to stimulate cyclic AMP levels. The level of cAMP corresponding to 100% was 220 ± 30 pmol ml−1.
Selected compounds were examined for antagonism of the human A2B AR. As with the A3 AR, the A2B AR was stably expressed in CHO cells for a standard adenylate cyclase assay.6 5′-N-Ethylcarboxamidoadenosine (NECA, 100 nM) was used as agonist, and each adenine derivative was tested at a concentration of 10 μM. Results are reported for each of the following compounds as percent inhibition of the NECA-elicited production of cAMP: 2 and 12 <10%; 21, 31 ± 4%; 22, 13 ± 2%; and 25, 12 ± 3%. The adenine derivatives in the absence of agonist had no effect on cAMP levels in the A2B AR-expressing CHO cells.
We attempted to investigate whether other AR antagonists might also display the dedifferentiation effect of 2 according to the reported procedure.18 However, the experiments were inconclusive since there were difficulties obtaining a definitive effect with 2, itself, which displayed toxic effects. Similar results were obtained with compound 12 (5 μM). Mouse C2C12 myogenic cells in culture normally fuse to form myotubes. We found that exposure of the cells for 4 days to 5 μM 2 and then for 7 days to a differentiating medium, consisting of 0.1 μM dexamethasone, 50 μg/mL ascorbic acid-2-phosphate and 10 mM β-glycerophosphate under the conditions reported,18 produced extensive cell death. The remaining live cells did not proliferate, and some cells still fused to form myotubes (Figure 2B), although to a lesser degree than control cells (Figure 2A). Only a few percent of the mononucleated cells stained positive for alkaline phosphatase (not shown), which is one indicator of osteogenic differentiation,18 but this staining was also obtained in the cells not treated with differentiating medium (Figure 2C). Other AR antagonists of low selectivity,1 including XAC (8-[4-(carboxymethyloxy)phenyl]-1,3-di-(n-propyl)xanthine), CGS15943 (N-[9-chloro-2-(2-furanyl)[1,2,4]triazolo[1,5-c]quinazolin-5-amine), and MRS1523 (5-propyl-2-ethyl-4-propyl-3-(ethylsulfanylcarbonyl)-6-phenylpyridine-5-carboxylate), each at 5 μM, were not toxic to the cells and did not show dedifferentiating effects using the same protocol.18 When, immediately after treatment with 2, the cells were transferred to medium containing 2 – 5% horse serum, which is a condition normally used for their differentiation (myogenesis), the cells differentiated to myotubes to a similar extent as control cells.
Figure 2.

Micrograph of the effects of differentiating medium and AR antagonists on mouse C2C12 myogenic cells. Shown are: (A) control differentiated myotubes and (B) cells after 4 day treatment with 5 μM of the adenine derivative 2 followed by maintaining the cells for an additional 7 days in differentiating medium containing 0.1 μM dexamethasone, 50 μg/mL ascorbic acid-2-phosphate and 10 mM β-glycerophosphate (see Materials and Methods). Cells treated identically to (B), except lacking differentiating medium are shown in (C). Arrows indicate cells that stained positive for alkaline phosphatase activity using a semiquantitative method. Bar in (A) indicates 20 μm.
Discussion
It was apparent from Tables 1 – 3 that 2-phenylamino-N6-cycloadenines are capable of binding to the AR family, in some cases with high selectivity toward the A3 subtype. Functional analysis of these compounds has demonstrated that they can produce a potent antagonistic effect at the human A3 AR. The initial SAR profile for this class of molecules suggested that among the variety of reported adenine derivatives, the 2-phenylamino-N6-cyclohexyl/cycloheptyl substitution pattern offered the highest affinity for binding at the A3 AR. While these adenine-based compounds demonstrated a reduced affinity compared to nucleoside-based ligands, they constitute an important advance, particularly with respect to A3 AR antagonists. A more extensive SAR study of this series, e.g. probing the effects of substitution pattern of the 2-phenylamino ring or the introduction of polar groups to increase aqueous solubility, is warranted.
More extensive SAR studies have been carried out for adenosine derivatives as agonists at the ARs than for adenine derivatives as antagonists. In general, a comparison of the SAR within these two classes provides parallels in some aspects (e.g. N6-cycloalkyl groups at the A1 AR) but not all characteristics.13 These results are in agreement with other studies suggesting the presence of a lipophilic pocket in the A1 and A3 AR binding sites that is filled by moderately sized cycloalkyl or other hydrophobic rings at the N6 position of adenosine derivatives.8 Data described herein demonstrated an important commonality between the SAR of the N6 cycloalkyl ring seen within this study of adenine derivatives and previous work9,12 on A1 AR antagonists. The dependence of A3 AR affinity on the ring size seemed to mirror the SAR profile for a class of N6-cycloalkyl-9-benzyl-2-phenyladenines reported to antagonize the A1 receptor by Lucacchini and coworkers.9 Within that study it was shown that the N6-cycloalkyl ring size was optimized at either cyclohexyl or cycloheptyl rings while the cyclooctyl analogues showed a marked decrease in affinity. These findings might represent a common feature of the ligand-receptor interaction characteristic of both receptor subtypes. Curiously, the N6-(2-endo-norbornyl) substituent led to high A3 AR affinity and selectivity in the present series of 2-substituted adenines, while it led to high affinity and selectivity at the A1 AR subtype for adenines not substituted at the 2 position14 and for adenosine derivatives.22 Integration of novel 2 position side-chains provided an initial SAR. The discovery of the seemingly favorable effect on the A3 AR of inclusion of either phenyl or 4-dimethylaminophenyl moieties at the 2 position represents a new finding.
On the theme of dedifferentiation by reversine 2, ARs are known to have effects on growth and proliferation; thus, we considered a possible connection between dedifferentiation ascribed to 2 and its activity at one or more of the ARs. Each of the four ARs is coupled to mitogen-activated protein kinases (MAPK).17 Specific activation of one receptor subtype versus another has been shown to initiate a myriad of biological events including alterations to the cell cycle.17 For example, expression of the A1 receptor within ob17 preadipocytes stimulated differentiation while slowing proliferation relative to unaltered cells.24 Stimulation of the A2B receptors by known agonists 2-chloroadenosine and 5′-N-methylcarboxamidoadenosine was shown to downregulate DNA synthesis and slow cellular proliferation.25 Treatment of rat lymphoma cells with adenosine and the specific A3 agonist IB-MECA was shown to down-regulate DNA synthesis, proliferation, and telomeric signaling.26 A3 AR agonists appear to have a net cytostatic effect triggered through activation of the ERK 1/2 pathway.17 Even from this small sampling of studies it is apparent that the function of the ARs in cell signaling is complex and still in the process of being elucidated.
A relationship between the antagonistic effects on the A3 AR demonstrated by 2 and the observed cellular dedifferentiation reported by Chen et al.18 could not be demonstrated in the present study. The analogues of 2 reported in the present study might be of use in further delineating the role that the ARs play during the dedifferentiation process; however, we were unsuccessful in our attempts to recreate an appreciable degree of myoblast dedifferentiation with various AR antagonists, including 2. Obviously, further study is needed to advance this series of compounds as modulators of the function of the ARs and within the delineation of the individual roles of the ARs on the cell cycle including dedifferentiation. However, A3 adenosine antagonists are not to be considered general dedifferentiation reagents. Even the preliminary information about the SAR of reversine congeners presented by Chen et al.18 did not appear to match the structural requirements for binding to the A3 receptor.
In conclusion, based on the relatively simple 2,6-disubstituted purine template, we have designed highly selective A3 AR antagonists reaching high affinity. Among the most potent and selective A3 AR antagonists in comparison to A1 and A2A ARs were compounds 12, 19 and 22. Compound 19 and other analogues were also demonstrated to be nearly inactive at the A2B AR. It will now be worthwhile to examine such antagonists as potential antiglaucoma agents, although the species selectivity of these antagonists will limit the experimental models that may be used.32 Although reversine was previously identified as a dedifferentiating agent and we have presently shown it to be a moderately potent AR antagonist, we were unable to demonstrate a link between AR antagonism and dedifferentiation.
Experimental Section
Reagents and instrumentation
6-Chloro-2-fluoropurine (3) was purchased from Oakwood Products, Inc. [125I]N6-(4-amino-3-iodobenzyl)adenosine-5′-N-methyluronaminde (I-AB-MECA, 2000 Ci/mmol), [3H]DPCPX, 80–130 Ci/mmol) and [3H]cyclic-AMP (40 Ci/mmol) were purchased from Amersham Biosciences Corp. (Piscataway, NJ). [3H]ZM241385 (4-2-[7-amino-2-(2-furyl)-1,2,4-triazolo[1,5-a][1,3,5]triazin-5-yl-amino]ethylphenol; 17 Ci/mmol) was purchased from Tocris Cookson Inc. (Ellisville MO). All other reagents and solvents were purchased from Sigma-Aldrich. Selected intermediates and products were analyzed on an Agilent 1100 series LC/MSD SL using a Zorbax® Eclipse XDB-C8 reverse phase column (4.6 × 150 mm, 5 μm). High-resolution mass spectroscopy measurements were performed on a Micromass/Waters LCT Premier Electrospray TOF mass spectrometer. 1H NMR data was recorded on a Varian Gemini300, and spectra were recorded in d6-DMSO and/or CDCl3 and were referenced to the residual solvent peaks at 2.50 and 7.26, respectively. Elemental analyses (CHN) were performed by Atlantic Microlabs, Inc. (Norcross, GA).
General procedure for the preparation of 2, and 4–21
Diisopropylethylamine (1.2 eq) and the appropriate primary amine for N6 substitution (0.9 eq) were added to a solution of 6-chloro-2-fluoropurine (3) (1 eq) in n-butanol (2–4 mL), and the mixture was heated for 15–24 h at 80°C. The solution was allowed to cool, and the solvent was removed under reduced pressure to yield a yellow solid. An aliquot was removed for LC-MS analysis. The remaining solid was transferred to a sealed reaction vessel containing EtOH (2–4 mL) and the appropriate amine (2 eq) for substitution at the 2 position, where the mixture was heated for 48 h at 110°C. The solution was allowed to cool, and the solvent was removed under reduced pressure. The resulting solid was purified via column chromatography (EtOAc) to yield compounds 2, 4 – 21 as white or off-white solids.
2-(4-Morpholinoanilino)-6-(cyclohexylamino)purine (2)
Yield 63 mg (55%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 10 min, linear gradient from 10% – 30% CH3CN; 10 – 15 min, linear gradient from 30% – 90% CH3CN at a flow rate of 1 mL/min: Rt 14.6 min). 1H NMR (d6-DMSO) δ 1.11–1.43 (m, 6H), 1.58–1.80 (m, 3H), 1.82–1.99 (m, 2H), 2.90–3.28 (m, 4H), 3.71–3.78 (m, 4H), 4.08 (bs, 1H), 6.81 (d, JHH = 9.0 Hz, 2H), 7.02 (bs, 1H), 7.63 (d, JHH = 9.0 Hz, 2H), 7.72 (s, 1H), 8.51 (s, 1H); (TOFMS) m/z (M+H+) 394.2355 (calculated for C18H23N6+) 394.2350
2-(4-Morpholinoanilino)-6-(ethylamino)purine (4)
Yield 93 mg (95%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 15 min, linear gradient from 25% – 90% CH3CN at a flow rate of 1 mL/min: Rt 3.1 min). 1H NMR (d6-DMSO) δ 1.29 (t, JHH = 7.2 Hz, 3H), 3.08–3.11 (m, 4H), 3.23–2.29 (m, 2H), 3.63 (bs, 1H), 3.81–3.84 (m, 4H), 6.93 (d, JHH = 8.4 Hz, 2H), 7.76 (d, JHH = 8.7 Hz, 2H), 7.82 (s, 1H), 7.39 (bs, 1H), 8.60 (s, 1H); (TOFMS) m/z 340.1886 (M+H+) (calculated for C17H22N7O+) 340.1880.
2-(4-Morpholinoanilino)-6-(cyclopropylamino)purine (5)
Yield 58 mg (57%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 5 min, linear gradient from 25% – 50% CH3CN; 5 – 10 min, linear gradient from 50% – 90% CH3CN at a flow rate of 1 mL/min: Rt 4.7 min). 1H NMR (d6-DMSO) δ 0.45–0.50 (m, 2H), 0.61–0.68 (m 2H), 0.70–0.78 (m, 1H), 2.97–3.08 (m, 4H), 3.69–3.76 (m, 4H), 7.48 (bs, 1H), 7.75 (d, JHH = 9.6 Hz, 2H), 7.92 (d, JHH = 9.0 Hz, 2H), 8.35 (s, 1H), 9.18 (s, 1H); (TOFMS) m/z 352.1885 (M+H+) (calculated for C18H22N7O+) 352.1880.
2-(4-Morpholinoanilino)-6-(cyclobutylamino)purine (6)
Yield 64 mg (60%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 15 min, linear gradient from 25% – 90% CH3CN at a flow rate of 1 mL/min: Rt 2.9 min). 1H NMR (d6-DMSO) δ 1.57–1.74 (m, 2H), 2.06–2.16 (m, 2H), 2.20–2.32 (m, 3H), 2.99–3.02 (m, 4H), 3.71–3.74 (m, 4H), 4.65 (bs, 1H), 6.85 (d, JHH = 9.0 Hz, 2H), 7.58 (bs, 1H), 7.66 (d, JHH = 9.0 Hz, 2H), 7.75 (s, 1H), 8.45 (s, 1H); (TOFMS) m/z 366.2042 (M+H+) (calculated for C19H24N7O+) 366.2037.
2-(4-Morpholinoanilino)-6-(cyclopentylamino)purine (7)
Yield 77 mg (70%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 12 min, linear gradient from 5% – 25% CH3CN; 12 – 20 min, linear gradient from 25% – 90% CH3CN at a flow rate of 1 mL/min: Rt 16.5 min). 1H NMR (d6-DMSO) δ 1.42–1.77 (m, 6H), 1.81–2.03 (m, 3H), 2.97–3.06 (m, 4H), 3.70–3.75 (m, 4H), 4.05 (bs, 1H), 6.83 (d, JHH = 9.3 Hz, 2H), 7.26 (bs, 1H), 7.67 (d, JHH = 9.0 Hz, 2H), 8.45 (s, 1H), 9.04 (s, 1H); (TOFMS) m/z 380.2199 (M+H+) (calculated for C20H26N7O+) 380.2193.
2-(4-Morpholinoanilino)-6-(cycloheptylamino)purine (8)
Yield 33 mg (28%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 5 min, linear gradient from 10% – 50% CH3CN; 5 – 20 min, linear gradient from 50% – 70% CH3CN at a flow rate of 1 mL/min: Rt 8.6 min). 1H NMR (d6-DMSO) δ 1.46–1.72 (m, 10H), 1.87–1.98 (m, 3H), 2.98–3.01 (m, 4H), 3.72–3.75 (m, 4H), 4.22 (bs, 1H), 6.83 (d, JHH = 9.0 Hz, 2H), 7.25 (bs, 1H), 7.66 (d, JHH = 9.0 Hz, 2H), 7.73 (s, 1H), 8.45 (s, 1H); (TOFMS) m/z 408.2512 (M+H+) (calculated for C22H30N7O+) 408.2506.
2-(4-Morpholinoanilino)-6-(cyclooctylamino)purine (9)
Yield 87 mg (71%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 15 min, linear gradient from 30% – 90% CH3CN at a flow rate of 1 mL/min: Rt 8.5 min). 1H NMR (d6-DMSO) δ 0.78–0.92 (m, 1H), 1.08–1.35 (m, 8H), 1.45–1.88 (m, 6H), 2.95–3.06 (m, 4H), 3.67–3.79 (m, 4H), 4.38 (bs, 1H), 6.83 (d, JHH = 9.0 Hz, 2H), 7.19 (bs, 1H), 7.65–7.72 (m, 3H), 8.48 (s, 1H); (TOFMS) m/z 422.2668 (M+H+) (calculated for C23H32N7O+) 422.2662.
2-(4-Morpholinoanilino)-6-((2-endo-norbornyl)amino)purine (10)
Yield 32 mg (27%); 1H NMR (CDCl3) δ 0.77–0.95 (m, 2H), 1.18–1.78 (m, 8H), 2.50–2.66 (m, 2H), 3.11 (dd, JHH= 4.8, 4.5 Hz, 4H), 3.86 (dd, JHH= 4.8, 4.5 Hz, 4H), 4.38 (bs, 1H), 5.70 (bs, 1H), 6.68 (s, 1H), 6.90 (d, JHH= 8.7 Hz, 2H), 7.45 (d, JHH= 8.7 Hz, 2H); (TOFMS) m/z 406.2369 (M+H+) (calculated for C22H28N7O+) 406.2355.
2-Ethylamino-6-(cyclohexylamino)purine (11)
Yield 72 mg (95%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 12 min, linear gradient from 5% – 30% CH3CN; 12 – 20 min, linear gradient from 30% – 90% CH3CN at a flow rate of 1 mL/min: Rt 16.6 min). 1H NMR (d6-DMSO) δ 1.09 (t, JHH = 7.2 Hz, 3H), 1.12–1.36 (m, 6H), 1.58–1.63 (m, 1H), 1.65–1.76 (m, 2H), 1.83–1.91 (m, 2H), 3.18–3.27 (m, 2H), 3.42 (bs, 1H), 4.02 (bs, 1H), 7.65 (s, 1H), 6.81 (bs, 1H); (TOFMS) m/z 261.1827 (M+H+) (calculated for C13H21N6+) 261.1822.
2-Phenylamino-6-(cyclohexylamino)purine (12)
Yield 43 mg (60%); 1H NMR (CDCl3) δ 1.20–1.50 (m, 6H), 1.60–1.85 (m, 4H), 2.05–2.20 (m, 2H), 4.10 (bs, 1H), 5.60 (bs, 1H), 6.95 (s, 1H), 7.06 (t, JHH= 7.8 Hz, 1H), 7.33 (t, JHH= 7.8 Hz, 2H), 7.56 (d, JHH= 7.8 Hz, 2H); (TOFMS) m/z 309.1823(M+H+) (calculated for C17H21N6+) 309.1828.
2-Benzylamino-6-(cyclohexylamino)purine (13)
Yield 35 mg (47%); 1H NMR (CDCl3) δ 1.10–1.50 (m, 6H), 1.56–1.82 (m, 4H), 2.00–2.12 (m, 2H), 4.66 (d, JHH= 6.0 Hz, 2H), 4.82 (bs, 1H), 5.58 (bs, 1H), 7.09 (s, 1H), 7.22–7.40 (m, 5H); (TOFMS) m/z 323.1975 (M+H+) (calculated for C18H23N6+) 323.1984.
2-(2-Phenylethylamino)-6-(cyclohexylamino)purine (14)
Yield 51 mg (65%); 1H NMR (CDCl3) δ 1.10–1.50 (m, 6H), 1.60–1.85 (m, 4H), 2.04–2.15 (m, 2H), 2.97 (t, JHH= 6.9 Hz, 2H), 3.69 (m, JHH= 6.9, 6.0 Hz, 2H), 4.88 (bs, 1H), 5.50 (bs, 1H), 7.22–7.36 (m, 5H), 7.40 (s, 1H); (TOFMS) m/z 337.2126 (M+H+) (calculated for C19H25N6+) 337.2141
2-(2-Naphthylamino)-6-(cyclohexylamino)purine (15)
Yield 37 mg (45%); 1H NMR (d6-DMSO) δ 1.10–1.50 (m, 6H), 1.60–1.85 (m, 4H), 1.94–2.04 (m, 2H), 4.20 (bs, 1H), 7.28 (t, JHH= 8.1 Hz, 1H), 7.42 (t, JHH= 8.1 Hz, 1H), 7.65–7.70 (m, 4H), 7.85 (s, 1H), 8.53 (s, 1H), 9.05 (bs, 1H); (TOFMS) m/z 359.1994 (M+H+) (calculated for C21H23N6+) 359.1984.
2-(Biphen-1-ylamino)-6-(cyclohexylamino)purine (16)
Yield 46 mg (41%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 12 min, linear gradient from 5% – 25% CH3CN; 12 – 15 min, linear gradient from 25% – 60% CH3CN at a flow rate of 1 mL/min: Rt 12.4 min). 1H NMR (d6-DMSO) δ 1.15–1.39 (m, 6H), 1.59–1.78 (m, 3H), 1.92–2.01 (m, 2H), 3.71 (bs, 1H), 7.31–7.33 (m, 1H), 7.42–7.47 (m, 3H), 7.56–7.66 (m, 4H), 7.79 (s, 1H), 8.15 (d, JHH = 9.0 Hz, 2H), 9.42 (s, 1H); (TOFMS) m/z 385.2141 (M+H+) (calculated for C23H25N6+) 385.2135.
2-(4-Piperidinoanilino)-6-(cyclohexylamino)purine (17)
Yield 25 mg (27%); 1H NMR (CDCl3) δ 1.10–1.80 (m, 16H), 2.00–2.20 (m, 2H), 3.00–3.25 (m, 4H), 4.13 (bs, 1H), 5.70 (bs, 1H), 6.68 (s, 1H), 6.92 (d, JHH= 9.0 Hz, 2H), 7.41 (d, JHH= 9.0 Hz, 2H); (TOFMS) m/z 392.2576 (M+H+) (calculated for C22H30N7+) 392.2563.
2-(4-Dimethylaminophenylamino)-6-(cyclohexylamino)purine (18)
Yield 20 mg (25%); 1H NMR (CDCl3) δ 1.10–1.50 (m, 6H), 1.60–1.85 (m, 4H), 2.04–2.14 (m, 2H), 2.92 (s, 6H), 4.10 (bs, 1H), 5.53 (bs, 1H), 6.56 (s, 1H), 6.74 (d, JHH= 9.0 Hz, 2H), 7.38 (d, JHH= 9.0 Hz, 2H); (TOFMS) m/z 352.2247 (M+H+) (calculated for C19H26N7+) 352.2250
2-Phenylamino-6-(cycloheptylamino)purine (19)
Yield 36 mg (48%); 1H NMR (CDCl3) δ 1.40–1.80 (m, 12H), 2.00–2.20 (m, 2H), 4.30 (bs, 1H), 5.60 (bs, 1H), 6.90 (s, 1H), 7.05 (t, JHH= 7.5 Hz, 1H), 7.32 (t, JHH= 7.5 Hz, 2H), 7.57 (d, JHH= 7.5 Hz, 2H); (TOFMS) m/z 323.1982(M+H+) (calculated for C18H23N6+) 323.1984.
2-Phenylamino-6-(cyclooctylamino)purine (20)
Yield 78 mg (80%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 15 min, linear gradient from 30% – 90% CH3CN at a flow rate of 1 mL/min: Rt 10.1 min). 1H NMR (d6-DMSO) δ 1.49–1.87 (m, 15H), 3.22–3.44 (bs, 1H), 7.17–7.22 (m, 3H), 7.79–7.84 (m, 3H), 8.02 (s, 1H), 8.75 (s, 1H); (TOFMS) m/z 337.2141 (M+H+) (calculated for C19H25N6+) 337.2135.
2-Phenylamino-6-(2-endo-norbornylamino)purine (21)
Yield 31 mg (42%); 1H NMR (CDCl3) δ 0.77–0.95 (m, 2H), 1.18–1.78 (m, 8H), 2.50–2.66 (m, 2H), 4.38 (bs, 1H), 5.80 (bs, 1H), 6.90 (s, 1H), 7.07 (t, JHH= 7.5 Hz, 1H), 7.21 (dd, JHH= 7.8, 7.5 Hz, 2H), 7.54 (d, JHH= 7.8 Hz, 2H); (TOFMS) m/z 321.1828 (M+H+) (calculated for C18H21N6+) 321.1828.
2-Phenoxy-6-(cyclohexylamino)purine (22)
Cyclohexylamine (30 μL, 0.26 mmol) and diisopropylethylamine (61 μL, 0.35 mmol) were added to a solution of 6-chloro-2-fluoropurine (3)(50 mg, 0.29 mmol) in n-butanol (2 mL), and the mixture heated for 15 h at 80°C. The solution was allowed to cool, and the solvent was removed under reduced pressure to yield a yellow solid. An aliquot was removed for LC-MS analysis. The remaining solid was transferred to a sealed reaction vessel with EtOH (5 mL), and phenol (48 mg, 0.5 mmol) and potassium tert-butoxide (47 mg, 0.42 mmol) were added. The mixture was heated for 48 h at 110°C. The solution was allowed to cool, and the solvent was removed under reduced pressure. The resulting solid was purified via column chromatography (EtOAc) to yield 22 as a white powder. Yield: 6 mg (8%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 15 min, linear gradient from 25% – 90% CH3CN at a flow rate of 1 mL/min: Rt 7.3 min). 1H NMR (d6-DMSO) δ 1.14–1.46 (6H), 1.67–1.98 (m, 5H), 4.13 (bs, 1H), 7.22–7.30 (m, 3H), 7.46–7.51 (m, 2H), 7.61 (bs, 1H), 7.99 (s, 1H); (TOFMS) m/z 310.1668 (M+H+) (calculated for C17H20N5O+) 310.1662.
2-Phenoxy-6-(2-endo-norbornylamino)purine (23)
2-Aminonorborane hydrochloride (46 mg, 0.31 mmol) and diisopropylethylamine (73 μL, 0.42 mmol) were added to a solution of 6-chloro-2-fluoropurine (3) (60 mg, 0.35 mmol) in n-butanol (2 mL), and the mixture heated for 15 h at 80°C. The solution was allowed to cool, and the solvent was removed under reduced pressure to yield a yellow solid. An aliquot was removed for LC-MS analysis. The remaining solid was transferred to a sealed reaction vessel with EtOH (5 mL), and phenol (66 mg, 0.70 mmol) and postassium tert-butoxide (79 mg, 0.70 mmol) were added. The mixture was heated for 48 h at 110°C. The solution was allowed to cool, and the solvent was removed under reduced pressure. The resulting solid was purified via column chromatography (EtOAc) to yield 23 as a white powder. Yield: 4.0 mg (4.3 %); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing amounts of CH3CN (0–15 min, linear gradient from 25%–90% CH3CN at a flow rate of 1 mL/min: Rt 10.2 min). 1H NMR (d6-DMSO) δ 1.22–1.62 (m, 10H), 1.80–1.95 (m, 1H), 4.16 (bs, 1H), 7.13–7.20 (m, 3H), 7.36–7.41 (m, 2H), 7.77 (bs, 1H), 7.92 (s, 1H); (TOFMS) m/z (M+H+) 322.1668 (calculated for C18H20N5O+ 322.1662).
2-Phenylthio-6-(cyclohexylamino)purine (24)
Cyclohexylamine (30 μL, 0.26 mmol) and diisopropylethylamine (61 μL, 0.35 mmol) were added to a solution of 6-chloro-2-fluoropurine (3) (50 mg, 0.29 mmol) in n-butanol (2 mL), and the mixture heated for 15 h at 80°C. The solution was allowed to cool, and the solvent was removed under reduced pressure to yield a yellow solid. An aliquot was removed for LC-MS analysis. The remaining solid was transferred to a sealed reaction vessel where EtOH (2 mL), and thiophenol (51 μL, 0.5 mmol) were added, and the mixture was heated for 48 h at 110°C. The solution was allowed to cool, and the solvent was removed under reduced pressure. The resulting solid was purified via column chromatography (EtOAc) to yield 24 as a white powder. Yield: 43 mg (63%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 15 min, linear gradient from 25% – 90% CH3CN at a flow rate of 1 mL/min: Rt 9.6 min). 1H NMR (d6-DMSO) δ 1.12–1.30 (m, 6H), 1.50–1.89 (m, 5H), 3.66 (bs, 1H), 7.41–7.45 (m, 3H), 7.58–7.62 (m, 3H), 7.93 (s, 1H); (TOFMS) m/z 326.1439 (M+H+) (calculated for C17H20N5S+) 326.1434.
9-Methyl-2-phenylamino-6-(cyclohexylamino)purine (25)
12 (17 mg, 0.06 mmol) was added to a slurry of potassium carbonate (9.7 mg, 0.07 mmol) in DMF (0.5 mL). The mixture was cooled to 0°C, and methyl iodide (5.6 μL, 0.09 mmol) was added. The reaction was allowed to stir for 1 h at 0°C followed by the addition of water (1 mL) and EtOAc (1 mL). The mixture was extracted with EtOAc (3 × 5 mL). The organic layers were collected, dried (Na2SO4), and the solvent was removed under reduced pressure to yield 25 as a white solid. Yield: 18 mg (95%); Purity analysis was achieved by C8 reversed-phase LC-MS using a linear gradient of H2O containing increasing amounts of CH3CN (0 – 5 min, linear gradient from 25% – 50% CH3CN; 5 – 10 min, linear gradient from 50% – 90% CH3CN at a flow rate of 1 mL/min: Rt 9.8 min). 1H NMR (d6-DMSO) δ 1.12–1.81 (m, 6H), 1.24–1.43 (m, 3H), 1.91–1.99 (m, 2H), 3.64 (s, 3H), 4.10 (bs, 1H), 7.19–7.24 (m, 3H), 7.86 (d, JHH = 8.4 Hz, 2H), 7.96 (s, 1H); (TOFMS) m/z 323.1984 (M+H+) (calculated for C18H23N6+) 323.1979.
9-Methyl-2-phenylamino-6-(cycloheptylamino)purine (26)
19 (20 mg, 0.062 mmol) was added to a slurry of potassium carbonate (10.3 mg, 0.075 mmol) in DMF (0.5 mL). The mixture was cooled to 0°C, and methyl iodide (5.8 μL, 0.093 mmol). The reaction was allowed to stir for 1 h at 0°C followed by the addition of water (1 mL) and EtOAc (1 mL). The mixture was extracted with EtOAc (3 × 5 mL). The organic layers were collected and dried (Na2SO4), and solvent was removed under reduced pressure to yield 26 as a white solid. Yield 18 mg (86%); 1H NMR (CDCl3) δ 1.40–1.80 (m, 12H), 2.00–2.20 (m, 2H), 3.73 (s, 3H), 4.30 (bs, 1H), 6.97 (t, JHH= 7.5 Hz, 1H), 7.31 (t, JHH= 7.5 Hz, 2H), 7.72 (d, JHH= 7.5 Hz, 2H), 8.02 (s, 1H); (TOFMS) m/z 337.2148 (M+H+) (calculated for C19H25N6+) 337.2141.
Cell Culture and Membrane Preparation
CHO (Chinese hamster ovary) cells expressing the recombinant human ARs were cultured in Dulbecco’s modified Eagle medium (DMEM) and F12 (1:1) supplemented with 10% fetal bovine serum, 100 units/mL penicillin, 100 μg/mL streptomycin, 2 μmol/mL glutamine. After harvesting, cells were homogenized and suspended. Cells were then centrifuged at 500g for 10 min and the pellet was resuspended in 50 mM Tris-HCl buffer (pH 7.4) containing 10 mM MgCl2. The suspension was homogenized and was then recentrifuged at 20, 000g for 20 min at 4°C. The resultant pellets were resuspended in Tris buffer and the suspension was stored at −80°C until the binding experiments. The protein concentration was measured using the Bradford assay.27
Radioligand Binding Assays
Each tube in the A3 AR competitive binding assay20 contained 100 μL of membrane suspension (20 μg of protein), 50 μL of [125I]4-amino-3-iodobenzyl)adenosine-5′-N-methyluronamide (0.5 nM), and 50 μL of increasing concentrations of the test ligands in Tris-HCl buffer (50 mM, pH 7.4) containing 10 mM MgCl2 and 1 mM EDTA. Nonspecific binding was determined using 10 mM 5′-N-ethylcarboxamidoadenosine in the buffer. The mixtures were incubated at 25°C for 60 min. Binding reactions were terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester (Brandell, Gaithersburg, MD). Filters were washed three times with 9 mL of ice-cold buffer. Radioactivity was determined using a Beckman γ-counter, and the percent inhibition was calculated.
The antagonists [3H]DPCPX (0.5 nM) and [3H]ZM241,385 (2 nM) were used as radioligands for A1 and A2A ARs, respectively. Each assay tube used to measure competitive binding for human A1 and A2A ARs contained 100 μL of membrane suspension (20 μg of protein), 50 μL of radioligand, and 50 μL of 10 μM test ligand in the Tris-HCl buffer. Non-specific binding was measured using 10 μM NECA in the buffer. Each assay was incubated at 25°C for 60 min and was terminated by filtration through Whatman GF/B filters under reduced pressure using a MT-24 cell harvester. Filters were washed three times with ice-cold buffer and placed in scintillation vials. Each vial also contained 5 ml of Hydrofluor scintillation buffer. All compounds that gave more than 60% inhibition at 10 μM for either A1 or A2A ARs were also tested using a full concentration curve between 10−9 and 10−5 M in order to determine a Ki value.
Cyclic AMP Accumulation Assay
Intracellular cyclic AMP levels were measured with a competitive protein binding method.28,29 CHO cells expressing the recombinant human A2B AR or A3 AR were harvested by trypsinization. After centrifugation and resuspension in medium, cells were plated in 24-well plates in 0.5 mL of medium. After 24 h the medium was removed, and cells were washed three times with 1 mL of DMEM containing 50 mM HEPES, pH 7.4. Cells were then treated with the appropriate agonist, NECA (100 nM), for the A2B AR or variable concentrations of Cl-IB-MECA for the A3 AR, and/or adenine derivatives in the presence of rolipram (10 μM). For the A2B AR, incubation was carried out for 1 h. For the A3 AR, after an initial incubation of 45 min forskolin (10 μM) was added to the medium, and incubation was continued an additional 15 min. The reaction was terminated by removing the supernatant, and cells were lysed upon the addition of 200 μL of 0.1 M ice-cold HCl. The cell lysate was resuspended and stored at −20°C. For determination of cyclic AMP production, protein kinase A (PKA) was incubated with [3H]cyclic AMP (2 nM) in K2HPO4/EDTA buffer (K2HPO4, 150 mM; EDTA, 10 mM), 20 μL of the cell lysate, and 30 μL 0.1M HCl or 50 μL of cyclic AMP solution (0–16 pmol/200 μL for standard curve). Bound radioactivity was separated by rapid filtration through Whatman GF/C filters and washed once with cold buffer. Bound radioactivity was measured by liquid scintillation spectrometry. A Schild constant (KB) was calculated as described using the equation.30
Statistical Analysis
Binding and functional parameters were calculated using Prism 5.0 software (GraphPAD, San Diego, CA). IC50 values obtained from competition curves were converted to Ki values using the Cheng-Prusoff equation.31 Data were expressed as the mean ± standard error.
Studies of Cellular Differentiation
Mouse C2C12 myogenic cells (ATCC, Manassas, VA) were plated in 6-well plates in growth medium (DMEM with 10% fetal bovine serum) and after overnight incubation (during which time cells attach to the bottom of the plate) 5 μM of the adenine derivative or other AR antagonists were added. After 4 days, medium containing the antagonists was removed and replaced with differentiating medium containing 50 μg/mL ascorbic acid-2-phosphate, 0.1 μM dexamethasone, and 10 mM β-glycerophosphate in 10% fetal bovine serum (to direct the cells from myogenic differentiation to osteogenic). The culture was maintained for an additional 4 – 7 days. Cells were stained for alkaline phosphatase activity using a semiquantitative method (“Leukocyte” Alkaline Phosphatase kit, Procedure No. 85, Sigma) and observed using a Nikon microscope and compared with control cells subjected to the same treatment except not exposed to differentiating medium or AR antagonist.
Supplementary Material
Acknowledgments
The authors thank Dr. John Lloyd and Dr. Sonya Hess (NIDDK) for assistance in obtaining the HRMS data.
References
- 1.Fredholm BB, IJzerman AP, Jacobson KA, Klotz KN, Linden JM. International Union of Pharmacology. XXV. Nomenclature and Classification of Adenosine Receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 2.Yao L, Burbiel JC, Maass A, Müller CE. Adenosine receptor agonists: from basic medicinal chemistry to clinical development. Expert Opin Emerging Drugs. 2003;8:537–576. doi: 10.1517/14728214.8.2.537. [DOI] [PubMed] [Google Scholar]
- 3.Tchilibon S, Joshi BV, Kim S-Y, Duong HT, Gao Z-G, Jacobson KA. (N)-Methanocarba 2,N6-disubstituted adenine nucleosides as highly potent and selective A3 adenosine receptor agonists. J Med Chem. 2005;48:1745–1758. doi: 10.1021/jm049580r. and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mogensen JP, Roberts SM, Bowler AN, Thomsen C, Knutsen LJ. The synthesis of new adenosine A3 selective ligands containing bioisosteric isoxazoles. Bioorg Med Chem Lett. 1998;8:1767–1770. doi: 10.1016/s0960-894x(98)00302-3. [DOI] [PubMed] [Google Scholar]
- 5.Lee K, Ravi RG, Ji X-d, Marquez VE, Jacobson KA. Ring-constrained (N) methanocarba-nucleosides as adenosine receptor agonists: Independent 5′-uronamide and 2′-deoxy modifications. Bioorg Med Chem Lett. 2001;11:1333–1337. doi: 10.1016/s0960-894x(01)00213-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohno M, Gao ZG, Van Rompaey P, Tchilibon S, Kim SK, Harris BA, Blaustein J, Gross AS, Duong HT, Van Calenbergh S, Jacoboson KA. Modulation of adenosine receptor affinity and intrinsic efficacy in nucleosides substituted at the 2-position. Bioorg Med Chem. 2004;12:2995–3007. doi: 10.1016/j.bmc.2004.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tchilibon S, Kim SK, Gao ZG, Harris BA, Blaustein J, Gross AS, Melman N, Jacobson KA. Exploring distal regions of the A3 adenosine receptor binding site: Sterically constrained N6-(2-phenylethyl)adenosine derivatives as potent ligands. Bioorg Med Chem Lett. 2004;12:2021–2034. doi: 10.1016/j.bmc.2004.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao ZG, Blaustein JB, Gross AS, Melman N, Jacobson KA. N6-Substituted adenosine derivatives: selectivity, efficacy and species differences at A3 adenosine receptors. Biochem Pharm. 2003;65:1675–1684. doi: 10.1016/s0006-2952(03)00153-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biagi G, Giorgi I, Livi O, Nardi A, Pacchini F, Scartoni V, Lucacchini A. N6-Cycloalkyl-2-phenyl-3-deaza-8-azaadenines: a new class of A1 adenosine receptor ligands. A comparison with the corresponding adenines and 8-azaadenines. Eur J Med Chem. 2003;38:983–990. doi: 10.1016/j.ejmech.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 10.Volpini R, Costanzi S, Lambertucci C, Taffi S, Vittori S, Klotz KN, Cristalli G. N6-Alkyl-2-alkynyl derivatives of adenosine as potent and selective agonists at the human adenosine A3 receptor and a starting point for searching A2b ligands. J Med Chem. 2002;45:3271. doi: 10.1021/jm0109762. [DOI] [PubMed] [Google Scholar]
- 11.Klotz KN, Kachler S, Lambertucci C, Vittori S, Volpini R, Cristalli G. 9-Ethyladenine derivatives as adenosine receptor antagonist: 2- and 8-substitution results in distinct selectivities. Naunyn-Schmiedeberg’s Arch Pharmacol. 2003;367:629–634. doi: 10.1007/s00210-003-0749-9. [DOI] [PubMed] [Google Scholar]
- 12.Thompson RD, Secunda S, Daly JW, Olsson RA. N6,9-disubstituted adenines: potent, selective antagonists at the A1 adenosine receptor. J Med Chem. 1991;34:2877–82. doi: 10.1021/jm00113a029. [DOI] [PubMed] [Google Scholar]
- 13.Jacobson KA, Siddiqi SM, Olah ME, Ji XD, Melman N, Bellamkonda K, Meshulam Y, Stiles GL, Kim HO. Structure-activity relationships of 9-alkyladenine and ribose-modified adenosine derivatives at rat A3 adenosine receptors. J Med Chem. 1995;38:1720–35. doi: 10.1021/jm00010a017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martin PL, Wysocki RJ, Jr, Barrett RJ, May JM, Linden J. Characterization of 8-(N-methylisopropyl)amino-N6-(5′-endohydroxy-endonorbornyl)-9-methyladenine (WRC-0571), a highly potent and selective non-xanthine antagonist of A1 adenosine receptors. J Pharmacol Exp Ther. 1996;276:490–9. [PubMed] [Google Scholar]
- 15.Martin PL, Ueeda M, Olsson RA. 2-Phenylethoxy-9-methyladenine: an adenosine receptor antagonist that discriminates between A2 adenosine receptors in the aorta and the coronary vessels from the guinea pig. J Pharmacol Exp Ther. 1993;265:248–53. [PubMed] [Google Scholar]
- 16.Laufer SA, Domeyer DM, Scior TR, Albrecht W, Hauser DR. Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. J Med Chem. 2005;48:710–22. doi: 10.1021/jm0408767. [DOI] [PubMed] [Google Scholar]
- 17.Schulte G, Fredholm BB. Signaling from adenosine receptors to mitogen-activated protein kinases. Cell Signal. 2003;15:813–827. doi: 10.1016/s0898-6568(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 18.Chen S, Zhang Q, Wu X, Schultz PG, Ding S. Dedifferentiation of lineage-committed cells by a small molecule. J Am Chem Soc. 2004;126:410–411. doi: 10.1021/ja037390k. [DOI] [PubMed] [Google Scholar]
- 19.Ding S, Gray NS, Wu X, Ding Q, Schultz PG. A combinatorial scaffold approach toward kinase-directed heterocycle libraries. J Am Chem Soc. 2002;124:1594–1596. doi: 10.1021/ja0170302. [DOI] [PubMed] [Google Scholar]
- 20.Olah ME, Gallo-Rodrigues C, Jacobson KA, Stiles GL. [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 21.Klotz KN, Hessling J, Hegler J, Owman C, Kull B, Fredholm BB, Lohse MJ. Comparative pharmacology of human adenosine receptor subtypes –characterization of stably transfected receptors in CHO cells. Naunyn-Schmiedeberg’s Arch Pharmacol. 1998;357:1–9. doi: 10.1007/pl00005131. [DOI] [PubMed] [Google Scholar]
- 22.Gao ZG, Mamedova L, Chen P, Jacobson KA. 2-Substituted adenosine derivatives: Affinity and efficacy at four subtypes of human adenosine receptors. Biochem Pharmacol. 2004;68:1985–1993. doi: 10.1016/j.bcp.2004.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yasuda SU, Nagashima S, Douyon E, Benton RE, Woosley RL, Barbey JT. Adenosine A1-receptor occupancy predicts A1-receptor antagonist effects of N-0861. Clin Pharmacol Ther. 1998;64:536–541. doi: 10.1016/S0009-9236(98)90136-9. [DOI] [PubMed] [Google Scholar]
- 24.Tatsis-Kotsidis I, Erlanger BF. Initiation of a process of differentiation by stable transfection of ob17 preadipocytes with the cDNA of human A1 adenosine receptor. Biochem Pharm. 1999;58:167–170. doi: 10.1016/s0006-2952(99)00069-6. [DOI] [PubMed] [Google Scholar]
- 25.Dubey RK, Gillespie DG, Shue H, Jackson EK. A2b Receptors mediate antimitogenesis in vascular smooth muscle cells. Hypertension. 2000;35:267–272. doi: 10.1161/01.hyp.35.1.267. [DOI] [PubMed] [Google Scholar]
- 26.Fishman P, Bar-Yehuda S, Ohana G, Pathak S, Wasserman L, Barer F, Multani AS. Adenosine acts as an inhibitor of lymphoma cell growth: a major role for the A3 adenosine receptor. Eur J Cancer. 2000;36:1452–1458. doi: 10.1016/s0959-8049(00)00130-1. [DOI] [PubMed] [Google Scholar]
- 27.Bradford MM. A rapid and sensitive method for the quatitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1990;189:231–234. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 28.Nordstedt C, Fredholm BB. A modification of a protein-binding method for the rapid quatification of cAMP in cell-culture supernatants and body fluid. Anal Biochem. 1990;189:231–234. doi: 10.1016/0003-2697(90)90113-n. [DOI] [PubMed] [Google Scholar]
- 29.Post SR, Ostrom RS, Insel PA. Biochemical methods for detection and measurement of cyclic AMP and adenylyl cyclase activity. Methods Mol Biol. 2000;126:363–374. doi: 10.1385/1-59259-684-3:363. [DOI] [PubMed] [Google Scholar]
- 30.Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. Br J Pharmacol Chemother. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50% inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 32.Yang H, Avila MY, Peterson-Yantorno K, Coca-Prados M, Stone RA, Jacobson KA, Civan MM. The cross-species A3 adenosine-receptor antagonist MRS 1292 inhibits adenosine-triggered human nonpigmented ciliary epithelial cell fluid release and reduces mouse intraocular pressure. Current Eye Res. doi: 10.1080/02713680590953147. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okamura T, Kurogi Y, Hashimoto K, Sato S, Nishikawa H, Kiryu K, Nagao Y. Structure-activity relationships of adenosine A3 receptor ligands: new potential therapy for the treatment of glaucoma. Bioorg Med Chem Lett. 2004;14:3775–3779. doi: 10.1016/j.bmcl.2004.04.099. [DOI] [PubMed] [Google Scholar]
- 34.Young HW, Molina JG, Dimina D, Zhong H, Jacobson M, Chan LN, Chan TS, Lee JJ, Blackburn MR. A3 adenosine receptor signaling contributes to airway inflammation and mucus production in adenosine deaminase-deficient mice. J Immunol. 2004;173:1380–9. doi: 10.4049/jimmunol.173.2.1380. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.