

Abstract
Semisynthetic artemisinin-based therapies are the first-line treatment for P. falciparum malaria, but next-generation synthetic drug candidates are urgently required to improve availability and respond to the emergence of artemisinin-resistant parasites. Artemisinins are embryotoxic in animal models and induce apoptosis in sensitive mammalian cells. Understanding the cytotoxic propensities of antimalarial drug candidates is crucial to their successful development and utilization. Here, we demonstrate that, similarly to the model artemisinin artesunate (ARS), a synthetic tetraoxane drug candidate (RKA182) and a trioxolane equivalent (FBEG100) induce embryotoxicity and depletion of primitive erythroblasts in a rodent model. We also show that RKA182, FBEG100 and ARS are cytotoxic toward a panel of established and primary human cell lines, with caspase-dependent apoptosis and caspase-independent necrosis underlying the induction of cell death. Although the toxic effects of RKA182 and FBEG100 proceed more rapidly and are relatively less cell-selective than that of ARS, all three compounds are shown to be dependent upon heme, iron and oxidative stress for their ability to induce cell death. However, in contrast to previously studied artemisinins, the toxicity of RKA182 and FBEG100 is shown to be independent of general chemical decomposition. Although tetraoxanes and trioxolanes have shown promise as next-generation antimalarials, the data described here indicate that adverse effects associated with artemisinins, including embryotoxicity, cannot be ruled out with these novel compounds, and a full understanding of their toxicological actions will be central to the continuing design and development of safe and effective drug candidates which could prove important in the fight against malaria.
INTRODUCTION
Semisynthetic artemisinin-based therapies are the recommended first-line treatment for P. falciparum malaria, due to their high efficacy against blood-borne stages of multidrug-resistant forms of the parasite (1). Although relatively well tolerated in patients, artemisinins are reported to induce neurotoxicity (2) and embryotoxicity (3) in a number of animal species, with the latter risk prompting the contraindication of artemisinin-based therapies in the first trimester of pregnancy unless suitable alternatives are unavailable (4). At the cellular level, artemisinin embryotoxicity appears to involve the selective depletion of primitive erythroblasts during defined early periods of gestation (3).
At present there is a lack of consensus on the pharmacological mechanism of action of the artemisinins. It is clear, however, that the endoperoxide bridge within the 1,2,4-trioxane unit is essential for antimalarial activity of these compounds, as exemplified by the lack of antiparasitic activity associated with artemisinin counterparts in which the endoperoxide moiety is replaced with an ether linkage (5). It has been hypothesized that iron-catalyzed reductive cleavage of the endoperoxide bridge results in the generation of toxic carbon-centered radicals, which alkylate and disrupt macromolecules that are vital for parasite homeostasis (6). It also has been proposed that artemisinins are redox-active molecules and that the intrinsic activity of the endoperoxide moiety serves to exacerbate levels of oxidative stress within the parasite by interfering with the function of important redox-sensitive enzymes (7). Inhibition of the parasite sarco/endoplasmic reticulum calcium ATPase (SERCA/PfATP6) has been proposed as an alternative mechanism of antimalarial activity (8), although this has been contested (9).
In addition to its critical role in driving the antiparasitic activity of the artemisinins, the endoperoxide bridge appears to represent a toxicophore in sensitive mammalian cells (10). We have demonstrated that the selective activation of the endoperoxide bridge is the chemical basis for the differential cytotoxicity of the synthetic analogue 10β-(p-bromophenoxy)dihydroartemisinin toward sensitive HL-60 promyelocytic leukemia cells and insensitive peripheral blood mononuclear cells (PBMCs) (11). Bioactivation of the endoperoxide bridge is sensitive to pharmacological manipulation of cellular heme levels, and triggers the generation of reactive oxygen species (ROS), a process that is dependent upon the integrity of the mitochondrial electron transport chain (12). These biochemical events induce the onset of apoptotic cell death, characterized by mitochondrial membrane depolarization, activation of caspases 3 and 7, and DNA fragmentation (11,13).
The limited availability of the Chinese wormwood Artemisia annua, from which artemisinin is extracted, and the poor oral bioavailability of the semisynthetic artemisinins, have prompted concerted efforts to develop alternative next-generation synthetic endoperoxide-based drugs (14). Some of the most promising compounds to emerge from these efforts are the 1,2,4-trioxolane OZ439 (15) and our 1,2,4,5-tetraoxane drug candidate RKA182 (Figure 1), which has improved pharmacokinetic properties compared with established artemisinins (16). It is clear that, as for the artemisinins, the peroxidic moiety is essential for the pharmacological action of tetraoxanes and trioxolanes (17–19), yet little is known regarding the cytotoxic and embryotoxic propensity of these compounds. Indeed, this was highlighted as a key knowledge gap in a World Health Organization (WHO) report on the safety of endoperoxide-based antimalarials (4). To inform the design of safe and efficacious next-generation antimalarials, and with the ultimate aim of facilitating their optimal therapeutic utilization, we have evaluated the embryotoxic potential, as well as the chemical and molecular mechanisms of cytotoxicity, of RKA182 and a 1,2,4-trioxolane equivalent, FBEG100 (Figure 1).
Figure 1.
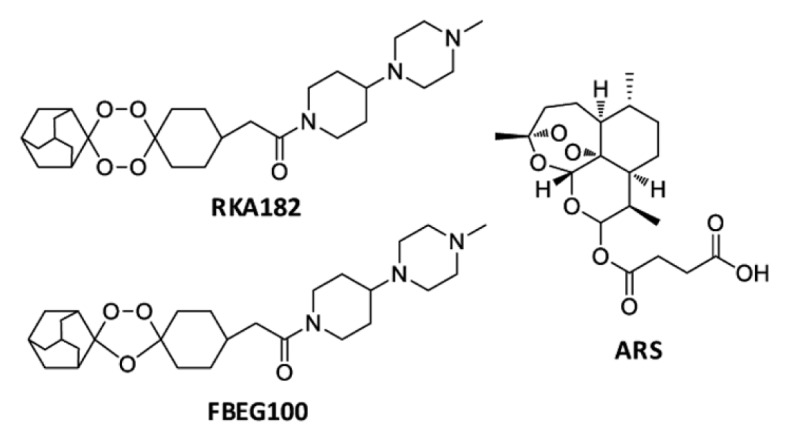
Chemical structures of the 1,2,4,5-tetraoxane RKA182, the 1,2,4-trioxolane FBEG100 and the established artemisinin ARS.
MATERIALS AND METHODS
Materials
Freshly drawn venous blood was collected from healthy volunteers in heparinized tubes. Peripheral blood mononuclear cells (PBMCs) and red blood cells (RBCs) were isolated using Lymphoprep (Axis-Shield, Kimbolton, UK), as described previously (20). ARS was kindly donated by Dafra Pharma International (Turnhout, Belgium). The anti-caspase 3 antibody (#9662) was obtained from Cell Signaling Technology (Danvers, MA, USA). Unless noted, all other reagents were from Sigma-Aldrich (Poole, UK).
Chemical Synthesis of RKA182 and FBEG100
The synthesis of RKA182 (16) and FBEG100 (21) have been described previously.
Embryotoxicity Assessment
The protocols described here were undertaken in accordance with criteria outlined in a license granted under the Animals (Scientific Procedures) Act 1986 and approved by the Institution local animal ethics committee. Eleven-wk-old Crl:CD (Sprague Dawley) male and primiparous female rats were supplied by Charles River (Kent, UK). Rats were maintained under standard conditions (room temperature 21.5 ± 1.5°C, humidity 55 ± 5 %, artificial lighting from 6 AM to 6 PM, feed and tap water provided ad libitum). Paired female and male rats were left in cohabitation from 4 PM to 9 AM the next morning. Mating is assumed to have occurred at the midpoint of the dark cycle, hence noon of the next day is defined as gestation d 0.5. On gestation d 9.5, pregnant female rats were euthanized via increasing concentrations of CO2 and embryos were explanted. These were distributed randomly to experimental groups and exposed to the indicated concentrations of ARS, RKA182 or FBEG100 for 48 h. The vehicle was methanol, and the concentration of the solvent in the media was controlled to 0.05%, which is known to be nonembryotoxic in this model (22). Embryos were cultured essentially as described by New (23), in sterile 50 mL glass bottles containing four embryos per bottle in 100% serum (5 mL) collected from male Sprague Dawley rats. At the start of culture, the bottles were flushed for 1 min with a gas mixture of 5/5/90% O2/CO2/N2 and placed on a nonrocking roller mixer (60 rpm) at 37°C. The next morning the bottles were flushed again with a gas mixture of 20/5/75% O2/CO2/N2, and in the afternoon with 40/5/55% O2/CO2/N2. The total culture time was 48 h. At the end of the culture period the embryos were transferred into Tyrode salt solution and examined under a stereomicroscope. Yolk sac diameter, crown–rump length and head length were measured and total morphological scores were assigned using the method of Brown and Fabro (24).
Cull Culture and Treatments
HL-60 cells, RBCs and PBMCs were cultured in RPMI-1640 media supplemented with 25 mmol/L 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES), 300 mg/L l-glutamine, 10% fetal bovine serum (Biowest, Nuaillé, France), 100 U/mL penicillin and 100 μg/mL streptomycin. HEK293T, HeLa and HepG2 cells were cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 584 mg/L l-glutamine, 10 % fetal bovine serum (FBS), 100 U/mL penicillin and 100 μg/mL streptomycin. All cells were cultured at 37°C in a 5% CO2 atmosphere. For drug treatments, ARS, RKA182 and FBEG100 were dissolved in methanol, with the concentration of the solvent in the media controlled to 0.5%. For biochemical manipulation experiments, cells were pretreated for 1 h with succinyl acetone (SA; 1 mmol/L), δ-aminolevulinic acid (ALA; 1 mmol/L), protoporphyrin IX (PPIX; 1 μmol/L), desferroxamine (DFO; 10 μmol/L), holotransferrin (HTF; 10 μmol/L), tiron (1 mmol/L), N-acetylcysteine (NAC; 10 mmol/L) or Z.VAD.FMK (100 μmol/L), or for 16 h with buthionine sulphoximine (BSO; 30 μmol/L), before exposure to the indicated concentrations of RKA182, FBEG100 or ARS for a further 24 h.
Quantification of Cell Viability
Cell viability was measured using a CellTiter-Glo Luminescent Assay (Promega, Southampton, UK) for adenosine triphosphate (ATP) or a Cytotoxicity Detection Kit (Roche Applied Science, Burgess Hill, UK) for lactate dehydrogenase (LDH), both in accordance with the manufacturer’s instructions. The concentrations of each compound that induced a 50% loss of cellular viability (IC50) were calculated using GraFit (Erithacus Software, Horley, UK).
Live-Cell Imaging of Apoptosis/Necrosis
The ability of RKA182, FBEG100 and ARS to induce apoptosis/necrosis in adherent HepG2 cells was assessed using a live-cell fluorescent imaging assay, as previously described (25). In brief, the binding of annexin V Alexa Fluor 488 (Life Technologies Corporation, Grand Island, NY, USA) conjugate to phosphatidyl serine on the membranes of apoptotic cells was followed in real-time by imaging every 30 min after drug exposure with a BD Pathway 855 imager (Becton Dickinson, Erembodegem-Aalst, Belgium). At the same time points, the intercalation of propidium iodide (PI) with cellular DNA was quantified. The total fluorescent intensity per cell area was quantified using Image Pro (Media Cybernetics, Bethesda, MD, USA).
Liquid Chromatography Mass Spectrometry
HL-60 cells were exposed to RKA182 or FBEG100 (both 100 μmol/L) for 0–24 h. Cells and media (100 μL) were combined with ARS (10 nmol) and ice-cold acetonitrile (300 μL), vortexed and centrifuged at 18,000g for 10 min. Extracted material was filtered using MultiScreen Solvinert plates (Millipore, Watford, UK), in accordance with the manufacturer’s instructions, and analyzed by multiple reaction monitoring using an API 4000 QTRAP LC-MS/MS System (AB Sciex, Warrington, UK) interfaced to a Ultimate 3000 autosampler and pump (Dionex, Camberly, UK). The data were collected and analyzed using Analyst software version 1.5 (AB Sciex). Sample separation was achieved on an ACE C8 column (100 × 2.1 mm, 3 μm; Advanced Chromatography Technologies, Aberdeen, UK). The mobile phase consisted of acetonitrile with 10 mmol/L ammonium acetate (90:10, v/v), both supplemented with 0.1% formic acid, delivered at a flow rate of 0.2 mL/min. The mass spectrometer was operated in positive ion mode. General operating parameters and analyte-specific fragmentation transitions are detailed in Tables S1 and S2. Calibration curves for RKA182 and FBEG100 were generated by plotting the ratio of peak area for analyte versus internal standard (ARS), following extraction from media containing RKA182 or FBEG100 (1–300 μmol/L) as described above.
Data Analysis
Data are expressed as mean ± standard deviation of the mean from at least three independent experiments. The significance of differences within the data was assessed by one-way analysis of variance (ANOVA; with the Tukey post hoc test) or unpaired t test. A P value of =0.05 was considered to be statistically significant.
Supplementary Methods
The synthesis of deoxy-artesunate (deoxy-ARS), and procedures for determination of in vitro antiparasitic activity, flow cytometry, Western blot, DNA fragmentation and glutathione content are described in Supplementary Material.
All supplementary materials are available online at www.molmed.org.
RESULTS
Embryotoxicity of RKA182, FBEG100 and ARS
To assess the embryotoxic propensity of the model tetraoxane and trioxolane compounds, RKA182 and FBEG100, we utilized the whole embryo culture (WEC) model, which provides controlled conditions for exposure of live rat embryos to drugs in the absence of maternal factors. RKA182, FBEG100 and the artemisinin ARS were all toxic toward cultured embryos, as indicated by concentration-dependent decreases in total morphological score (incorporating several indices of embryonic morphological development; Figure 2A), visceral yolk sac diameter, and crown-rump and head lengths (Supplementary Figure S1). The IC50 values for RKA182, FBEG100 and ARS were 4.4, 3.7 and 2.9 μmol/L, respectively. Notably, and in keeping with previous reports of artemisinin embryotoxicity (3), there was a marked paleness to the circulatory system within the visceral yolk sacs of embryos exposed to each of the three compounds (Figure 2B), at both nontoxic and toxic (as judged by decreases in total morphological score) drug concentrations. Therefore, these data indicate that the depletion of primitive erythroblasts is a common cellular mechanism that precedes overt embryotoxicity induced by artemisinins, tetraoxanes and trioxolanes.
Figure 2.

Embryotoxicity of RKA182, FBEG100 and ARS. (A) Sprague Dawley rat embryos (gestation d 9.5) were cultured ex vivo for 48 h in the absence or presence of RKA182, FBEG100 or ARS at the indicated concentrations. Total morphological scores (TMS) were assigned according to the method of Brown and Fabro (24). Data are expressed relative to the TMS of embryos exposed only to vehicle (methanol, 0.05%). Data represent mean + SD, n = 3. (B) Representative stereomicroscope images of embryos with intact visceral yolk sacs, following exposure to vehicle, RKA182 (3 μmol/L), FBEG100 (3 μmol/L) or ARS (1 μmol/L) for 48 h. Bar = 1 mm.
Cytotoxicity of RKA182, FBEG100 and ARS in a Panel of Human Cells
To examine the cytotoxic propensity of RKA182 and FBEG100, we examined their effects on the viability of a panel of human cell lines originating from distinct organs. In HL-60 promyelocytic leukemia cells, which we have used previously to examine the chemical and molecular mechanisms of artemisinin toxicity (11,12), the IC50 values for RKA182 and FBEG100 were 5- and 18-fold greater, respectively, than ARS (Table 1, Figure 3A). We also examined the time-dependency of RKA182, FBEG100 and ARS toxicity. Equimolar concentrations of RKA182 and FBEG100 both induced rapid (= 1 h) concentration-dependent decreases in cellular ATP and LDH contents in HL-60 cells, whereas ARS elicited loss of cellular viability only at =16 h (Figures 3B–C, Supplementary Figure S2). RKA182 and FBEG100 also induced concentration-dependent loss of cell viability in HepG2 hepatocellular carcinoma cells, HeLa cervical adenocarcinoma cells and primary PBMCs, in all of which ARS demonstrated lesser toxicity (Table 1, Supplementary Figure S3). In each of the cell types within the panel, deoxy-ARS had an IC50 value >100 μmol/L (Table 1, Supplementary Figure S3), demonstrating the role of the endoperoxide moiety in the toxicity induced by ARS. Taken together, all three compounds have cytotoxic potential in mammalian cells, yet exhibit differential timecourses of cell death and degrees of cell-specificity.
Table 1.
Cytotoxicity of RKA182, FBEG100, ARS and deoxy-ARS toward a panel of human cells and antimalarial activities versus P. falciparum.
| Cell type | RKA182 | FBEG100 | ARS | Deoxy-ARS |
|---|---|---|---|---|
| HL-60 | 11.6 ± 4.0a | 40.6 ± 3.0 | 2.2 ± 0.2 | >100 |
| HEK293T | 14.7 ± 0.2 | 37.1 ± 2.5 | 20.1 ± 3.4 | >100 |
| HepG2 | 25.5 ± 5.3 | 58.8 ± 10.8 | 77.2 ± 17.2 | >100 |
| HeLa | 37.7 ± 5.5 | 55.2 ± 5.6 | 87.8 ± 11.4 | >100 |
| RBC | 28.0 ± 7.3 | >100 | 18.9 ± 3.7 | >100 |
| PBMC | 46.6 ± 0.7 | 56.8 ± 2.1 | >100 | >100 |
| P. falciparum (3D7) | 4.9 ± 1.2 nmol/Lb,c | 1.4 ± 0.3 nmol/L | 1.8 ± 0.6 nmol/Lc | ND |
ND, not determined.
IC50 values (μmol/L) were calculated from ATP assay concentration-response curves (Supplementary Figure S3) following incubation with the indicated compound for 24 h. Data represent mean ± SD, n = 3.
In vitro antimalarial activities were calculated against the chloroquine-sensitive 3D7 strain of P. falciparum.
See reference (16).
Figure 3.

Concentration and time-dependence of cytotoxicity induced by RKA182, FBEG100 and ARS in HL-60 cells. (A) Cells were exposed to the indicated concentrations of RKA182 (black circles), FBEG100 (white circles) or ARS (gray circles) for 24 h, and cytotoxicity was determined by quantification of cellular ATP content. Data are expressed relative to the ATP content of cells exposed only to vehicle (methanol, 0.5%). (B–C) Cells were exposed to 100 μmol/L of RKA182, FBEG100 or ARS for the indicated times, and cytotoxicity was determined by quantification of cellular ATP (B) or LDH (C) contents. Data are expressed relative to the ATP or LDH content of cells exposed only to vehicle at the corresponding timepoints. All data represent mean + SD, n = 3.
Pathways of Cell Death Induced by RKA182, FBEG100 and ARS
To explore the pathways of cell death induced by RKA182 and FBEG100, HL-60 cells were stained with tetramethylrhodamine ethyl ester (TMRE) following exposure to the compounds for 24 h, and mitochondrial membrane depolarization was measured by flow cytometry. Both RKA182 and FBEG100 induced concentration-dependent mitochondrial membrane depolarization (Figure 4A). In addition, exposure of cells to RKA182 and FBEG100, as well as ARS, resulted in a concentration-dependent increase in the activity of caspases 3 and 7 (Figure 4B), as well as concentration-dependent (Figure 4C) and time-dependent (Figure 4D) increases in the processing of caspase 3, symptomatic of activation of the mitochondrial/intrinsic apoptotic pathway. In all cases, the induction of caspase activity was consistent with the respective profiles of ATP and LDH depletion (Figures 3B–C), and was abrogated by the caspase inhibitor Z.VAD.FMK (Figure 4B, Supplementary Figure S4). Cell cycle analysis, performed on PI-stained cells using flow cytometry, revealed that RKA182 and FBEG100 induced the concentration-dependent formation of a sub-G0/G1 population of cells (Figure 4E), representative of DNA fragmentation, which was confirmed by electrophoresis of genomic DNA isolated from drug-exposed cells (Figure 4F). The latter effect was inhibited by Z.VAD.FMK (Figure 4F), demonstrating the functional significance of caspase processing in the evolution of drug-induced apoptosis. Taken together, these data indicate that RKA182, FBEG100 and ARS induce classical hallmarks of apoptosis in HL-60 cells.
Figure 4.

Induction of apoptosis by RKA182, FBEG100 and ARS in HL-60 cells. (A) Cells were exposed to the indicated concentrations of RKA182 (black circles) or FBEG100 (white circles) for 24 h. The number of cells with reduced mitochondrial membrane potential, indicative of depolarization, was quantified by TMRE staining and flow cytometry, and expressed as percentage of total cells. Data represent mean + SD, n = 3. (B) Cells were pretreated (triangles) or not (circles) with the caspase inhibitor Z.VAD.FMK (100 μmol/L, 1 h) and then exposed to the indicated concentrations of ARS (gray symbols), RKA182 (black symbols) or FBEG100 (white symbols) for a further 24 h. As an index of caspase activity, cleavage of a luminogenic caspase 3/7 substrate was quantified, and expressed as a percentage of the activity in cells exposed only to vehicle (methanol, 0.5 %). Data represent mean + SD, n = 3. (C) Cells were exposed to methanol (MeOH, 0.5 %), RKA182 (3, 5, 7.5 or 10 μmol/L), FBEG100 (20, 30, 40, 50 μmol/L) or ARS (0.1, 0.3, 1, 3 μmol/L) for 24 h. The processing of caspase 3 from its inactive 32 kDa form to its proteolytically active 17 kDa form was determined in whole cell lysates by Western blotting. Staurosporine (STS; 1 μmol/L) was used as a positive control for caspase 3 activation. β-Actin was probed as a loading control. (D) Cells were exposed to RKA182 (55 μmol/L), FBEG100 (75 μmol/L) or ARS (10 μmol/L) for the indicated times. Caspase 3 processing was detected by Western blotting. (E) Cells were exposed to the indicated concentrations of RKA182 (black circles) or FBEG100 (white circles) for 24 h. The number of cells in sub-G0/G1 phase of the cell cycle was quantified by PI staining and flow cytometry, and expressed as percentage of total cells. Data represent mean + SD, n = 3. (F) Cells were pretreated or not with the caspase inhibitor Z.VAD.FMK (100 μmol/L, 1 h) and then exposed to STS (1 μmol/L), methanol (0.5 %), ARS (1 μmol/L), RKA182 (10 μmol/L) or FBEG100 (40 μmol/L) for a further 24 h. Extracted genomic DNA was separated by electrophoresis on an agarose gel stained with ethidium bromide. (G) Cells were pretreated (gray bars) or not (black bars) with the caspase inhibitor Z.VAD.FMK (100 μmol/L, 1 h) and exposed to RKA182, FBEG100 or ARS for a further 24 h. Cytotoxicity was determined by quantification of cellular ATP content. IC50 values were calculated from concentration-response curves. Unpaired t test, N.S. no statistically significant increase in IC50 value with Z.VAD.FMK pretreatment. Data represent mean + SD, n = 3.
Notably, Z.VAD.FMK was unable to protect against the loss of cellular ATP content induced by RKA182, FBEG100 and ARS in HL-60 cells (Figure 4G), indicating that these compounds also induce a form of caspase-independent cell death. Indeed, that all three compounds induced caspase-independent cell death, reminiscent of necrosis, was confirmed in adherent HepG2 cells, in spite of previous reports demonstrating that artemisinins induce hallmarks of apoptosis in these cells (26,27). In our hands, HepG2 cells exhibited a similar phenotype to HL-60 cells, in that RKA182, FBEG100 and ARS induced a concentration-dependent decrease in cellular ATP content (Figure 5A) that was not abrogated by pretreatment of cells with Z.VAD.FMK (Figure 5B). By use of live-cell fluorescent imaging, a time-dependent accumulation of annexin V–and PI-positive HepG2 cells was observed following exposure to IC50 concentrations of RKA182, FBEG100 or ARS over a 24 h timecourse (Figures 6A–D). In each case, PI staining increased prior to, or simultaneously with, the accumulation of annexin V–positive cells, while neither signal (Figures 6A–D), nor overall cytotoxicity (Figure 5B), was affected by Z.VAD.FMK. In contrast, the apoptosis-inducing compound cisplatin induced
Figure 5.

Cytotoxicity of RKA182, FBEG100 and ARS in HepG2 cells. (A) Cells were exposed to the indicated concentrations of RKA182 (black circles), FBEG100 (white circles) or ARS (gray circles) for 24 h, and cytotoxicity was determined by quantification of cellular ATP content. Data are expressed relative to the ATP content of cells exposed only to vehicle (methanol, 0.5%). (B) Cells were pretreated (gray bars) or not (black bars) with the caspase inhibitor Z.VAD.FMK (100 μmol/L, 1 h) and exposed to RKA182, FBEG100 or ARS for a further 24 h. Cytotoxicity was determined by quantification of cellular ATP content. IC50 values were calculated from concentration-response curves. Unpaired t test, N.S. no statistically significant increase in IC50 value with Z.VAD.FMK pretreatment. Data represent mean + SD, n = 3.
Figure 6.

Induction of caspase-independent necrosis by RKA182, FBEG100 and ARS in HepG2 cells. Cells were pretreated (white symbols) or not (black symbols) with the caspase inhibitor Z.VAD.FMK (100 μmol/L, 1 h) and exposed to vehicle (methanol, 0.5%; A), RKA182 (20 μmol/L; B), FBEG100 (60 μmol/L; C), ARS (70 μmol/L; D) or cisplatin (10 μmol/L; E) for a further 24 h. To follow cell death in real-time, annexin V Alexa Fluor 488 (circles) and PI (triangles) staining was measured by live-cell fluorescent imaging at the indicated time points. The total fluorescent intensity of each signal is expressed per cell area. Representative images of annexin and PI signals at the indicated time points, in the absence and presence of Z.VAD.FMK, are presented below the relevant charts.
Role of Heme and Iron in the Mechanism of RKA182 and ARS Cytotoxicity
In light of the similar pathways of cell death induced by RKA182 and FBEG100 in sensitive mammalian cells, and previous reports demonstrating a role for heme and iron in the mechanism of artemisinin toxicity (6,12,28,29), we examined the importance of these biochemical factors in the cell death induced by RKA182 in comparison to ARS. In keeping with our previous report that inhibition of heme synthesis protects against artemisinin toxicity in mammalian cells (12), here the δ-aminolevulinate dehydratase inhibitor SA perturbed the ability of ARS and RKA182 to induce cytotoxicity (Figure 7A) and caspase 3/7 activation (Supplementary Figure S5) in HL-60 cells. Furthermore, the heme precursors ALA and PPIX augmented the toxic effects of ARS and, to a greater extent, RKA182 (Figure 7A). The ability of ALA to enhance the toxicity of ARS and RKA182 was dependent on its incorporation into heme, as SA reversed the ALA-induced exacerbation of toxicity, but had no effect on the ability of PPIX to enhance toxicity, for either compound (Figure 7A). We next probed the role of iron in the toxicity induced by ARS and RKA182. Pretreatment of HL-60 cells with the iron chelator DFO provided substantial protection against the toxic effects of ARS and RKA182 (Figure 7B), as well as inhibiting the ability of these compounds to induce caspase 3/7 activation (Supplementary Figure S5). Pretreatment of cells with HTF, an exogenous source of Fe(II), had no discernable effect on ARS toxicity (Figure 7B), in keeping with our previous report (12), but HTF did enhance RKA182 toxicity (Figure 7B). These results indicate that heme and iron play important roles in the mechanism of toxicity of ARS and RKA182 (Figure 7F), although the latter is more sensitive to exacerbation of normal cellular heme and iron levels.
Figure 7.

Role of heme, iron and oxidative stress in RKA182 and ARS toxicity. (A) HL-60 cells were pretreated or not for 1 h with the δ-aminolevulinate dehydratase inhibitor SA (1 mmol/L) and/or the heme precursors ALA (1 mmol/L) or PPIX (1 μmol/L), before a further 24-h exposure to ARS (2 μmol/L) or RKA182 (10 μmol/L). (B) HL-60 cells were pre-treated or not for 1 h with the iron chelator DFO (10 μmol/L) or the exogenous iron source HTF (10 μmol/L), before a further 24-h exposure to ARS (10 μmol/L) or RKA182 (15 μmol/L). (C) HL-60 cells were pretreated or not for 1 h with the superoxide scavenger tiron (1 mmol/L) or the thiol antioxidant NAC (10 mmol/L), before a further 24-h exposure to ARS (30 μmol/L) or RKA182 (20 μmol/L). (D–E) Cells were pretreated or not for 16 h with the glutathione synthesis inhibitor BSO (30 μmol/L), followed by a further 1-h exposure to NAC (10 mmol/L). (D) Total glutathione content was determined using the DTNB recycling assay, and is normalized to total protein content. (E) Cells were subsequently exposed to ARS (1 μmol/L) or RKA182 (2 μmol/L) for a further 24 h. In all cases, cytotoxicity was determined by quantification of cellular ATP content. Data are expressed relative to the ATP content of cells exposed only to vehicle (methanol, 0.5%). All data represent mean + SD, n = 3. One-way ANOVA, *P ≥ 0.05, **P ≥ 0.01, ***P ≥ 0.001 versus non-pretreated cells, or for the indicated comparison, N.S., no statistically significant change in ATP content with indicated pretreatment. (F) Proposed mechanisms underpinning cytotoxicity induced by artemisinins, trioxolanes and tetraoxanes.
Role of Oxidative Stress in the Mechanism of RKA182 and ARS Cytotoxicity
Previously, we have shown that artemisinins induces concentration and time-dependent elevations of ROS in mammalian cells, and that the superoxide scavenger tiron protects against the toxicity of these compounds (12). In keeping with these observations, here both ARS and RKA182 toxicity was diminished by tiron and the thiol antioxidant NAC in HL-60 cells (Figure 7C, Supplementary Figure S5). To further probe the involvement of oxidative stress in the toxicity induced by ARS and RKA182, we pre-treated HL-60 cells with BSO, an inhibitor of glutathione biosynthesis (30). BSO provoked glutathione depletion (Figure 7D) and augmented the cytotoxic action of ARS and RKA182, an effect that was reversed by coincubation with NAC (Figure 7E). Taken together, these data indicate that oxidative stress is an important mechanistic component of ARS and RKA182 -induced cell death (Figure 7F).
Role of Chemical Decomposition in RKA182 and FBEG100 Cytotoxicity
Previously, we have shown that the selective activation of the endoperoxide bridge is the chemical basis for the differential cytotoxicity of a model artemisinin toward sensitive HL-60 cells and insensitive PBMCs (11). Furthermore, we have reported =30% decomposition of the model artemisinin 10β-(p-fluorophenoxy)dihydroartemisinin in sensitive mammalian cells under conditions of marked cytotoxicity (12). In contrast, here there was no discernable change in the amount of RKA182 recovered following exposure of HL-60 cells for up to 24 h, despite induction of significant cytotoxicity within 1 h (Figure 8A). Furthermore, there was no evidence for general decomposition of RKA182 following exposure to HL-60 cells pretreated with ALA, PPIX or HTF (data not shown), despite each pretreatment enhancing the overall cytotoxicity of RKA182 (Figures 7A–B). The level of FBEG100 did decline steadily over a 24 h timecourse (Figure 8B), however this was attributed to spontaneous decomposition in cell culture media (Figure 8C), and importantly there was no change in the amount of FBEG100 recovered from cells at early timepoints in which significant losses of ATP content were recorded (Figure 8B). In the absence of a suitable chemical marker of bioactivation, these data do not exclude the possibility of a specific bioactivation process underlying the toxicity of trioxolanes and tetraoxanes. However, in contrast to model artemisinins, the ability of these compounds to induce cell death does appear to be relatively independent of general chemical decomposition.
Figure 8.

Cytotoxicity of RKA182 and FBEG100 is independent of chemical decomposition. HL-60 cells were exposed to 100 μmol/L RKA182 (A) or 100 μmol/L FBEG100 (B) for the indicated times. Cytotoxicity was determined by quantification of cellular ATP content (black circles). Data are expressed relative to the ATP content of cells exposed only to vehicle (methanol, 0.5%) at the corresponding timepoints. Following extraction with acetonitrile at the indicated timepoints, the amounts of RKA182 and FBEG100 recovered were quantified by LCMS (white circles), and are expressed relative to the amounts detected at 0 h. An expansion of the 0–3 h timeframe for FBEG100 is shown below the main chart. All data represent mean + SD, n = 3. One-way ANOVA, *P ≥ 0.05, ***P ≥ 0.001 versus vehicle-treated cells (ATP content) or 0 h (LCMS). (C) Cell-free media was supplemented with 100 μmol/L of RKA182 or FBEG100 and extracted with acetonitrile after 24 h. The amount of RKA182 or FBEG100 recovered was quantified by LCMS, and is expressed relative to the amount detected at 0 h. All data represent mean + SD, n = 3. One-way ANOVA, ***P ≥ 0.001 decomposition versus 0 h (LCMS).
DISCUSSION
Synthetic trioxolanes and tetraoxanes have shown promise as next-generation antimalarial drug candidates, particularly in terms of therapeutic efficacy and lack of synthetic constraints. Indeed, we recently have identified the tetraoxane drug candidate RKA182 that possesses improved pharmacokinetic properties and outstanding antimalarial activity (16). In addition, a number of trioxolane and tetraoxane compounds currently are being supported by the Medicines for Malaria Venture (MMV) and/or are in clinical trials (15,31), yet the continuing development and optimal therapeutic utilization of these compounds must be supported by an understanding of their potential to induce cytotoxicity in mammalian cells, and an appreciation of the underpinning chemical and molecular mechanisms. Here, we have revealed that, similarly to the established artemisinin ARS, both RKA182 and its trioxolane counterpart FBEG100 can induce embryotoxicity in the rodent WEC model, as well as cytotoxicity across a panel of primary and established human cells. These findings may have important implications for the continuing development and ultimate therapeutic utilization of these promising antimalarial drug candidates.
P. falciparum infection during pregnancy poses a significant health risk to both the mother and unborn child (32), yet artemisinin-based therapies are currently contraindicated in the first trimester of pregnancy due to an increasing body of evidence indicating that artemisinins are embryotoxic in laboratory animals (3). Here, we have demonstrated that RKA182 and FBEG100 are embryotoxic in the rodent WEC model. Both compounds, together with ARS, induced embryotoxicity at >1 μmol/L, in keeping with previous reports of embryotoxicity induced by artemisinin (lower limit for toxicity 0.9 μmol/L) (33), dihydroartemisinin (1.8 μmol/L) (34) and the trioxolane OZ277 (1.3 μmol/L) (33). It is thought that the selective depletion of primitive erythroblasts, and the anemia that ensues, is the underlying cause of artemisinin embryotoxicity in rodents (3). Indeed, the sensitive period for artemisinin embryotoxicity in the rat (gestation d 10–14) correlates with the timecourse of dependence upon primitive, rather than definitive, erythroblasts for embryonic oxygen requirements (3). In keeping with this notion, we observed that overt embryotoxicity induced by ARS, RKA182 and FBEG100 was preceded by a marked paleness within the visceral yolk sac circulation, indicating that the targeting of primitive erythroblasts is a common basis for the embryotoxicity of artemisinins, tetraoxanes and trioxolanes. The ultimate risk of embryotoxicity in humans can only be established through improved translation of animal data together with robust monitoring of inadvertent exposures in pregnant patients.
Although the mechanism by which artemisinins deplete embryonic erythroblasts has yet to be elucidated, it is well documented that these compounds induce hallmarks of apoptotic cell death in sensitive mammalian cells in vitro. Here, we have demonstrated that RKA182, FBEG100 and ARS induce concentration, time and caspase-dependent apoptosis in sensitive cells, while also invoking a form of caspase-independent cell death that bears the hallmarks of necrosis. As such, the ultimate mechanism of cytotoxicity appears to be similar for semi-synthetic artemisinins and synthetic tetraoxanes and trioxolanes (Figure 7F). Recently, we and others have demonstrated the role of heme, iron and ROS in the pharmacological and toxicological actions of the artemisinins (6,12,28,29). Here, oxidative stress was shown to underpin the cytotoxic action of both RKA182 and ARS, indicating a convergence of mechanism between novel tetraoxanes and established artemisinins. Although not tested here, the generation of oxidative stress could perturb critical, but as yet poorly defined, cellular processes, or provoke the onset of lipid peroxidation, contributing to the induction of cell death (7,36–38). We also have demonstrated that the heme synthesis inhibitor SA and the iron chelator DFO diminish the toxic effects of ARS and RKA182, although the latter was relatively less sensitive to these interventions. In contrast, RKA182 toxicity was relatively more sensitive to elevation of cellular heme and iron to nonphysiological levels.
The above data indicate that tetraoxanes are less reliant, relative to artemisinins, on interaction with basal levels of heme and iron for the induction of cytotoxicity. However, under conditions of elevated heme and/or iron, which are notably associated with malaria infection (39), tetraoxane toxicity appears to be augmented to a greater extent than that of the artemisinins. That such subtle differences should exist in the biochemical mechanisms that underlie the toxic effects of artemisinins compared with tetraoxanes and trioxolanes is consistent with our inability to detect appreciable chemical decomposition of RKA182 or FBEG100 under conditions of substantial cytotoxicity. However, it has been shown that the peroxidic moiety is essential for the pharmacological action of tetraoxanes and trioxolanes (17–19). Therefore, it is possible that a relatively minor and/or specific bioactivation, prompted by interaction with an as yet undefined cellular target(s), may contribute to the chemical mechanism of toxicity of these novel compounds. Further work is therefore required to identify suitable chemical markers of trioxolane and tetraoxane bioactivation, to fully elucidate the latter’s role in cytotoxicity.
It is important to note that, at least based on the ex vivo and in vitro data presented here, the benefit:risk balance of these promising antimalarial drug candidates appears to be considerable, in terms of the differences between pharmacologically active and toxic concentrations of the compounds. Specifically, in terms of the ratio between their IC50 values for embryotoxicity (in the rodent WEC model) and antimalarial activity (in the P. falciparum 3D7 screen), RKA182 (904.1), FBEG100 (2619.3) and ARS (1630.0) each appear to have excellent therapeutic indices. However, in the absence of robust in vivo pharmacokinetic data for these and other novel synthetic endoperoxide-based antimalarials, adverse effects associated with artemisinins cannot yet be ruled out with next-generation tetraoxane and trioxolane drug candidates. Such knowledge is particularly important in light of the present focus on the development of synthetic drug candidates with enhanced stability and bioavailability, which will increase systemic exposure and the likelihood of a single-dose cure for malaria, but also the potential for toxicity. On the other hand, in light of proposals that the sensitivity of cancerous cells to artemisinin toxicity in vitro be exploited therapeutically (35), our finding that model tetraoxanes and trioxolanes are cytotoxic toward mammalian cells suggests that these compounds also may represent promising candidates for anti-cancer therapy.
CONCLUSION
In summary, we have revealed that model tetraoxane and trioxolane antimalarials are embryotoxic in the rodent WEC model and induce rapid cytotoxicity in mammalian cells. The subtle differences in mechanisms and timecourses of toxicity for these compounds and established artemisinins reported here serve to further emphasize the need to better understand pharmacological and toxicological mechanisms of action of next-generation antimalarial drug candidates to enable a more informed assessment of the ultimate risk of adverse effects in patients. In the continuing search for more potent endoperoxide-based antimalarials, it is vital that we explore and exploit differences in mechanisms of action within the parasite and mammalian cells to ensure the successful development of safe and efficacious new drugs.
Supplemental Data
ACKNOWLEDGMENTS
The authors thank Richard Amewu and Fatima Bousejra-El Garah (Department of Chemistry, The University of Liverpool, Liverpool, United Kingdom) for the synthesis and provision of RKA182 and FBEG100; Nuna Araújo (Departamento de Química e Farmácia, Universidade do Algarve, Portugal) for helpful suggestions on the synthesis of deoxy-ARS; Claire Taylor and Victoria Daniels (Liverpool School of Tropical Medicine, Liverpool, United Kingdom) for technical assistance with the WEC experiments; Anahi Santoyo Castelazo and James Maggs (Centre for Drug Safety Science, The University of Liverpool. Liverpool, United Kingdom) for assistance with mass spectrometry.
This work was supported by the European Union Seventh Framework Programme (ARTEMIP, 200805) and the UK Medical Research Council (MRC), as part of the Centre for Drug Safety Science (G0700654). Work at Leiden University was facilitated by the award of a UK Royal Society International Travel Grant (TG102742) and the British Toxicology Society’s Norman Aldridge Travelling Fellowship to IM Copple. J Firman’s PhD is supported by the MRC Integrative Toxicology Training Partnership.
Footnotes
Online address: http://www.molmed.org
DISCLOSURES
The authors declare that they have no competing interests as defined by Molecular Medicine, or other interests that might be perceived to influence the results and discussion reported in this paper.
REFERENCES
- 1.WHO. Guidelines for the treatment of malaria [Internet] 2nd ed. Geneva: WHO; 2010. [cited 2011 Jun 1]. Available from: http://www.who.int/malaria/publications/atoz/9789241547925/en/index.html. [Google Scholar]
- 2.Toovey S. Safety of artemisinin antimalarials. Clin. Infect. Dis. 2006;42:1214–5. doi: 10.1086/502658. [DOI] [PubMed] [Google Scholar]
- 3.Clark RL. Embryotoxicity of the artemisinin antimalarials and potential consequences for use in women in the first trimester. Reprod. Toxicol. 2009;28:285–96. doi: 10.1016/j.reprotox.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 4.WHO. Assessment of the safety of artemisinin compounds in pregnancy: report of two joint informal consultations convened in 2006 by: the Special Programme for Research and Training in Tropical Diseases (TDR) sponsored by UNICEF/UNDP/World Bank/WHO and the Global Malaria Programme of the World Health Organization [Internet] Geneva: WHO; 2007. [cited 2011 Jun 1]. http://www.who.int/malaria/publications/atoz/9789241596114/en/index.html. [Google Scholar]
- 5.Beekman AC, et al. Stereochemistry-dependent cytotoxicity of some artemisinin derivatives. J Nat Prod. 1997;60:325–30. doi: 10.1021/np9605495. [DOI] [PubMed] [Google Scholar]
- 6.O’Neill PM, Barton VE, Ward SA. The molecular mechanism of action of artemisinin—the debate continues. Molecules. 2010;15:1705–21. doi: 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haynes RK, et al. Facile oxidation of leucomethylene blue and dihydroflavins by artemisinins: relationship with flavoenzyme function and antimalarial mechanism of action. ChemMedChem. 2010;5:1282–99. doi: 10.1002/cmdc.201000225. [DOI] [PubMed] [Google Scholar]
- 8.Krishna S, Pulcini S, Fatih F, Staines H. Artemisinins and the biological basis for the PfATP6/SERCA hypothesis. Trends Parasitol. 2010;26:517–23. doi: 10.1016/j.pt.2010.06.014. [DOI] [PubMed] [Google Scholar]
- 9.Cardi D, et al. Purified E255L mutant SERCA1a and purified PfATP6 are sensitive to SERCA-type inhibitors but insensitive to artemisinins. J Biol Chem. 2010;285:26406–16. doi: 10.1074/jbc.M109.090340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fishwick J, McLean WG, Edwards G, Ward SA. The toxicity of artemisinin and related compounds on neuronal and glial cells in culture. Chem Biol Interact. 1995;96:263–71. doi: 10.1016/0009-2797(94)03597-2. [DOI] [PubMed] [Google Scholar]
- 11.Mercer AE, et al. Evidence for the involvement of carbon-centered radicals in the induction of apoptotic cell death by artemisinin compounds. J Biol Chem. 2007;282:9372–82. doi: 10.1074/jbc.M610375200. [DOI] [PubMed] [Google Scholar]
- 12.Mercer AE, Copple IM, Maggs JL, O’Neill PM, Park BK. The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials. J Biol Chem. 2011;286:987–96. doi: 10.1074/jbc.M110.144188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Disbrow GL, et al. Dihydroartemisinin is cytotoxic to papillomavirus-expressing epithelial cells in vitro and in vivo. Cancer Res. 2005;65:10854–61. doi: 10.1158/0008-5472.CAN-05-1216. [DOI] [PubMed] [Google Scholar]
- 14.Jefford CW. New developments in synthetic peroxidic drugs as artemisinin mimics. Drug Discov Today. 2007;12:487–95. doi: 10.1016/j.drudis.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 15.Charman SA, et al. Synthetic ozonide drug candidate OZ439 offers new hope for a single-dose cure of uncomplicated malaria. Proc Natl Acad Sci U S A. 2011;108:4400–5. doi: 10.1073/pnas.1015762108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Neill PM, et al. Identification of a 1,2,4,5-tetraoxane antimalarial drug-development candidate (RKA 182) with superior properties to the semisynthetic artemisinins. Angew Chem Int Ed Engl. 2010;49:5693–7. doi: 10.1002/anie.201001026. [DOI] [PubMed] [Google Scholar]
- 17.Fugi MA, Wittlin S, Dong Y, Vennerstrom JL. Probing the antimalarial mechanism of artemisinin and OZ277 (arterolane) with nonperoxidic isosteres and nitroxyl radicals. Antimicrob Agents Chemother. 2010;54:1042–6. doi: 10.1128/AAC.01305-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dong Y, et al. Spiro and dispiro-1,2,4-trioxolanes as antimalarial peroxides: charting a workable structure-activity relationship using simple prototypes. J Med Chem. 2005;48:4953–61. doi: 10.1021/jm049040u. [DOI] [PubMed] [Google Scholar]
- 19.Kaiser M, et al. Peroxide bond-dependent antiplasmodial specificity of artemisinin and OZ277 (RBx11160) Antimicrob Agents Chemother. 2007;51:2991–3. doi: 10.1128/AAC.00225-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams DP, Pirmohamed M, Naisbitt DJ, Maggs JL, Park BK. Neutrophil cytotoxicity of the chemically reactive metabolite(s) of clozapine: possible role in agranulocytosis. J Pharmacol Exp Ther. 1997;283:1375–82. [PubMed] [Google Scholar]
- 21.Bousejra-El Garah F, et al. Comparison of the reactivity of antimalarial 1,2,4,5-tetraoxanes with 1,2,4-trioxolanes in the presence of ferrous iron salts, heme, and ferrous iron salts/phosphatidylcholine. J Med Chem. 2011;54:6443–55. doi: 10.1021/jm200768h. [DOI] [PubMed] [Google Scholar]
- 22.Brown-Woodman PD, et al. In vitro assessment of the effect of methanol and the metabolite, formic acid, on embryonic development of the rat. Teratology. 1995;52:233–43. doi: 10.1002/tera.1420520409. [DOI] [PubMed] [Google Scholar]
- 23.New DA. Whole-embryo culture and the study of mammalian embryos during organogenesis. Biol Rev Camb Philos Soc. 1978;53:81–122. doi: 10.1111/j.1469-185x.1978.tb00993.x. [DOI] [PubMed] [Google Scholar]
- 24.Brown NA, Fabro S. Quantitation of rat embryonic development in vitro: a morphological scoring system. Teratology. 1981;24:65–78. doi: 10.1002/tera.1420240108. [DOI] [PubMed] [Google Scholar]
- 25.Puigvert JC, de Bont H, van de Water B, Danen EH. High-throughput live cell imaging of apoptosis. Curr. Protoc. Cell Biol. 2010;Unit 18.10:1–13. doi: 10.1002/0471143030.cb1810s47. [DOI] [PubMed] [Google Scholar]
- 26.Hou J, Wang D, Zhang R, Wang H. Experimental therapy of hepatoma with artemisinin and its derivatives: in vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin Cancer Res. 2008;14:5519–30. doi: 10.1158/1078-0432.CCR-08-0197. [DOI] [PubMed] [Google Scholar]
- 27.Gao X, et al. Dihydroartemisinin induces endoplasmic reticulum stress-mediated apoptosis in HepG2 human hepatoma cells. Tumori. 2011;97:771–80. doi: 10.1177/030089161109700615. [DOI] [PubMed] [Google Scholar]
- 28.Haynes RK, et al. The Fe2+-mediated decomposition, PfATP6 binding, and antimalarial activities of artemisone and other artemisinins: the unlikelihood of C-centered radicals as bioactive intermediates. ChemMedChem. 2007;2:1480–97. doi: 10.1002/cmdc.200700108. [DOI] [PubMed] [Google Scholar]
- 29.Stocks PA, et al. Evidence for a common non-heme chelatable-iron-dependent activation mechanism for semisynthetic and synthetic endoperoxide antimalarial drugs. Angew Chem Int Ed Engl. 2007;46:6278–83. doi: 10.1002/anie.200604697. [DOI] [PubMed] [Google Scholar]
- 30.Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982;257:13704–12. [PubMed] [Google Scholar]
- 31.Olliaro P, Wells TN. The global portfolio of new antimalarial medicines under development. Clin Pharmacol Ther. 2009;85:584–95. doi: 10.1038/clpt.2009.51. [DOI] [PubMed] [Google Scholar]
- 32.Desai M, et al. Epidemiology and burden of malaria in pregnancy. Lancet Infect Dis. 2007;7:93–104. doi: 10.1016/S1473-3099(07)70021-X. [DOI] [PubMed] [Google Scholar]
- 33.Longo M, et al. Comparative embryotoxicity of different antimalarial peroxides: in vitro study using the rat whole embryo culture model (WEC) Reprod Toxicol. 2010;30:583–590. doi: 10.1016/j.reprotox.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 34.Longo M, et al. Effects of the antimalarial drug dihydroartemisinin (DHA) on rat embryos in vitro. Reprod Toxicol. 2006;21:83–93. doi: 10.1016/j.reprotox.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 35.Firestone GL, Sundar SN. Anticancer activities of artemisinin and its bioactive derivatives. Expert Rev. Mol. Med. 2009;11:e32. doi: 10.1017/S1462399409001239. [DOI] [PubMed] [Google Scholar]
- 36.D’Alessandro S, et al. Hypoxia modulates the effect of dihydroartemisinin on endothelial cells. Biochem Pharmacol. 2011;82:476–84. doi: 10.1016/j.bcp.2011.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Efferth T, Giaisi M, Merling A, Krammer PH, Li-Weber M. Artesunate induces ROS-mediated apoptosis in doxorubicin-resistant T leukemia cells. PLoS One. 2007;2:e693. doi: 10.1371/journal.pone.0000693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Haynes RK, et al. Reactions of antimalarial peroxides with each of leucomethylene blue and dihydroflavins: flavin reductase and the cofactor model exemplified. ChemMedChem. 2011;6:279–91. doi: 10.1002/cmdc.201000508. [DOI] [PubMed] [Google Scholar]
- 39.Hunt NH, Stocker R. Heme moves to center stage in cerebral malaria. Nat Med. 2007;13:667–9. doi: 10.1038/nm0607-667. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.