

Abstract
Oxime Click chemistry was used to form hydrogels that support cell adhesion. Eight-armed aminooxy poly(ethylene glycol) (PEG) was mixed with glutaraldehyde to form oxime-linked hydrogels. The mechanical properties, gelation kinetics, and water swelling ratios were studied and found to be tunable. It was also shown that gels containing the integrin ligand arginine-glycine-aspartic acid (RGD) supported mesenchymal stem cell (MSC) incorporation. High cell viability and proliferation of the encapsulated cells demonstrated biocompatibility of the material.
Keywords: hydrogel, oxime, stem cells, PEG, biocompatible, tissue engineering
Hydrogels are a common class of biomaterials utilized in a wide range of applications including as tissue engineering scaffolds, drug delivery vehicles, or as space filling agents. As a result, there is a tremendous amount of work in rapidly gelling materials. Although many naturally-derived materials have been exploited for these purposes,1,2 the difficulties associated with the risk of diseases, host immune response, batch-to-batch variability, and tuning properties have increasingly led to the use of wholly synthetic materials. The properties of synthetic materials can be easily tuned, including the rate of gelation, as well as mechanical strength.3-6 In addition, it is often possible to dial in sophisticated function such as cellular cues and ligands.6 There are two main methods of gelation for synthetic materials. Stimuli triggered gelation in which an external stimulus is applied to the liquid form of the material resulting in gel formation. Stimuli include temperature, pH, light or injection into aqueous environment. The other method is based upon mixing of two liquid components, and gelation in this case is a result of efficient cross-linking reactions between the two components or rapid self-assembly.2 Typically, reactions in which the kinetics and the efficiency of the reaction can be tuned are desirable for these applications (i.e. the gelation time can be controlled). Herein, we describe for the first time, the use of oxime Click chemistry as a way to form hydrogels by mixing two components (Scheme 1).
Scheme 1.
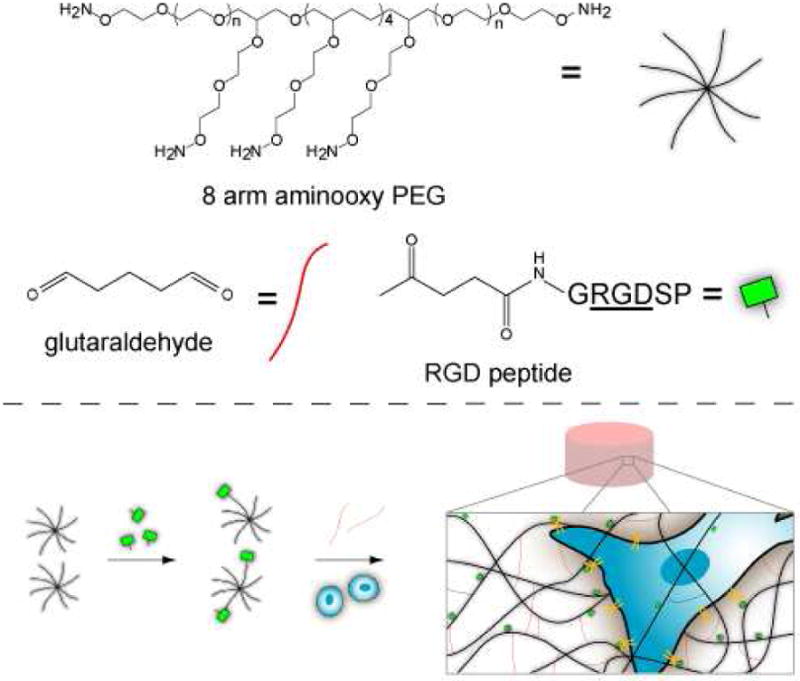
Synthesis and encapsulation of MSCs within RGD-functionalized oxime-cross-linked PEG hydrogels. Note: Glutaraldehyde is generally a mixture of species and thus the structure shown is idealized.
Click chemistries are increasingly utilized to form hydrogels.7 The most commonly employed systems are the reaction of thiols with activated disulfides, maleimides, acrylates, or vinyl sulfones.7 These Michael addition reactions occur in aqueous buffer and have been widely used to covalently link proteins and integrin binding peptides to polymer matrices, as well as to crosslink star PEG with enzymatically degradable linkers.8,9 Thiol-ene click has been utilized by combining multi-armed thiolated PEG, alkene- and acrylate-functionalized small molecules, and photoinitiators.10 Norborene functionalized PEG has been cross-linked with an enzymatically degradable linker via photoinitiated thiol-ene allowing for spatial and temporal control of the synthesis of an enzymatically degradable hydrogel.11 While thiol chemistry has been extensively used for hydrogel formation, it is sensitive to oxygen resulting in disulfides; in addition, native cysteine and amine residues can compete with the desired thiol during hydrogel formation. Huisgen cycloaddition has also been employed in the synthesis of PEG gels using a degradable peptide as the cross-linker.12 However, for cell encapsulation this approach could be problematic due to the use of the copper catalyst for cross-linking. Copper-free azide-alkyne click reactions have been recently utilized to circumvent this difficulty.13
Oxime Click chemistry, the reaction between an aminooxy group and an aldehyde or ketone, is ideal for hydrogel formation. The reaction is fast, orthogonal to functionalities found in biomolecules and cells, the byproduct is water, and a catalyst is not required.14 Moreover, the reaction partners are stable compared to thiols. As a result, oxime chemistry has been used to modify surfaces with proteins, peptides, and DNA.15 The reaction has also been employed to prepare protein-polymer conjugates,16 modify cell surfaces,17 and even label tissues in vivo.18 For hydrogel materials, this approach like other click chemistries, should allow for covalent incorporation of signaling molecules, particularly since proteins and peptides can be easily modified with ketones, oxoamide, or aminooxy groups. Thus, we explored the use of eight-armed aminooxy PEG (AO-PEG) and glutaraldehyde as a novel approach to hydrogels for cell incorporation (Scheme 1).
PEG was targeted as the scaffold material because it is bioinert and biocompatible,19-21 and multi-armed PEGs have been widely used to form hydrogels.6,21 AO-PEG was synthesized by Mitsunobu reaction of N-hydroxyphthalimide with 8-arm hydroxyl terminated PEG star, followed by reduction with hydrazine (see supporting information, Scheme S1).22 Glutaraldehyde was utilized as a readily available crosslinker for the oxime hydrogels. The molecule is reported to exist as a mixture of species ranging from oligomers/polymers to cyclic hemiacetals depending on the conditions.23 Because of the variety of ways this molecule can possibly react, a model system was utilized to confirm that oxime bond formation would occur. Glutaraldehyde was mixed with O-hydroxylamine tetra(ethylene glycol) in deuterated water and studied by 1H and 13C NMR spectroscopy. Since the small molecule aminooxy compound could not cause cross-linking, yet had the same chemical structure as the end group of the polymer, it provided for straightforward analysis of the system. Upon mixing of the two substrates, the oxime peaks (syn and anti) where observed in the 1H NMR spectrum at 7.6 and 6.9 ppm, respectively (see supporting Information Figure S4a); the aldehyde peak observed in the starting glutaraldehyde solution at 9.7 ppm was no longer visible. The presence of the oxime species was confirmed by 13C NMR spectroscopy, where the peak at 154 ppm, corresponding to the oxime carbon was seen (see supporting Information Figure S4b). The results demonstrate that upon addition of glutaraldehyde to a hydroxyl amine, reaction by oxime bond formation occurs. Although we can not rule out that the other species may be present in the glutaraldehyde solution that react by different mechanisms, this model study strongly suggests that the predominant species is likely to be oxime bond formation during hydrogel formation.
Rheology was used to assess the stiffness and gelation kinetics of oxime hydrogels. A series of gels were formed with different AO-PEG weight percent with an r = 1.0 (moles of aldehyde/moles of aminooxy) or different r ratios and an AO-PEG concentration of 3 weight percent (wt%) (Figure 1). The AO-PEG/glutaraldehyde system was able to produce hydrogels with a wide range of mechanical properties, from 258 Pa (3% PEG, r ratio = 0.7) to 4196 Pa (7% PEG, r ratio = 1.0). The results of the rheology demonstrate that this system is able to generate materials with a broad range of mechanical properties similar to soft tissue. This is important because mechanical properties have been shown to play a key role in determining cell fate.
Figure 1.

Mechanical characterization of AO-PEG/glutaraldehyde hydrogels. Storage and elastic modulus can be modified by adjusting PEG polymer percentage and/or the r ratio. (A-B) Altering polymer percentage while keeping the crosslinking ratio constant (r = 1.0) can significantly change the mechanical properties of the hydrogel. (C-D) Likewise, changing the crosslinking ratio while keeping the polymer percentage constant (PEG: 3%) can drastically change the storage/loss modulus.
The ability to tune the rate of gelation is critical for Click chemistry-induced gelation, where crosslinking is initiated immediately following crosslinker addition. Rapid gelation does not readily allow for the homogeneous incorporation of cells, yet if gelation is too slow, it might not be able to be used as an injectable hydrogel in vivo. Since oxime bond formation is acid catalyzed we investigated the rate of gelation over a range of pH values. Gelation kinetics were characterized by plate to plate rheometry measuring the storage modulus in situ after mixing hydrogel components and incubating at 37 °C (Figure 2A). The final pH of the hydrogel precursor solution was modified by dissolving the polymer in solutions with different pHs. AO-PEG solutions at 3 wt% (r = 1) at pH 6.5 gelled within 5 minutes. When the pH was raised to 7.2, gelation occurred in 30 min. At a pH of 8.0 hydrogel formation was considerably slowed, and the gel was only partially crosslinked at 30 minutes. The data demonstrate that by controlling the pH, the rate of gelation can be tuned. This result is particularly useful for wound/disease models that require a minimally invasive surgery and biomaterials that form hydrogels.
Figure 2.

Hydrogel gelation kinetics can be altered by adjusting the pH of the solution. (A) Making the solution more acidic can increase the rate of gelation whereas a more basic solution slows down the gelation kinetics for 3 wt% AO-PEG (r = 1). Increasing the storage modulus by adjusting the PEG percentage (PEG = 3, 5, 7% from left to right) while keeping the crosslinking ratio constant at r = 1 (open circles) decreases the water content (B) and increases the swell ratios (C). Increasing the storage modulus by changing the r ratio (r = 0.7, 0.8, 1.0 from left to right) while keeping the PEG percentage constant at 3 wt% (closed triangles) decreases the water content (B) and decreases the swelling ratio (C). Data is displayed as the average and standard deviation of three independent experiments.
The amount of water absorbed by the gel and the amount that it is able to swell are important properties of materials for biomedical applications. The water content and swelling ratio of different hydrogel formulations were quantified for the different weight percent gels as well as different r ratios. The water content was quantified by dividing the water weight of the gel by total weight. Increasing the r ratio from 0.7 to 1.0 of 3% PEG hydrogels resulted in a decrease in the water content from 99.26% +/- 0.08 to 98.49% +/- 0.14 (Figure 2B). The r ratio also had an effect on the swelling ratio of the gel. The swelling ratio was calculated by dividing swollen gel weight by non-swollen gel weight. As the r ratio increased to 1.0 (moles of aldehyde = moles of aminooxy) for the 3 wt% gels, the swelling ratio decreased from 1.76 +/- 0.07 to 1.09 +/- 0.02 (Figure 2C) indicating that the more tightly the network is crosslinked, the lower water content and swelling ratio. Varying the polymer percentage (3 wt%, 5 wt%, and 7 wt%) with a constant crosslinking ratio (r =1) resulted in decreasing water content from 98.49% +/- 0.14 to 95.78% +/-0.25 as the weight percent increased (Figure 2B). The swelling ratio increased with increasing weight percent from 1.09 +/- 0.02 to 1.45 +/- 0.17 (Figure 2C). Thus, by changing the amount of AO-PEG and the r ratio it is possible to tune to the amount of water absorbed and the swelling ratio.
Stem cell encapsulation is a key area of research in the fields of drug delivery, tissue engineering, and regenerative medicine.24 Of the available stem cell lineages, mesenchymal stem cells (MSCs) are particular interesting because they are multipotent and offer an autologous treatment approach.25,26 MSCs have been differentiated in vitro and in vivo into osteogenic, chondrogenic, and adipogenic lineages, for example, making MSCs an attractive treatment option for degenerative disease and tissue/organ repair.25,27 For the therapeutic effect of stem cells to be realized it is necessary that the cells be delivered efficiently to the desired location and effectively encapsulated such that the cells stay at the site of delivery.24,28 Two main approaches exist for stem cell delivery/encapsulation: 1) seeding cells onto preformed scaffolds and 2) encapsulating cells during scaffold formation. The former strategy offers access to a wide range of materials and engineering approaches; however, nutrient diffusion and uniform cellular distribution in larger scale constructs can be problematic. The latter approach is advantageous because the cells and scaffold precursors can be mixed prior to scaffold formation allowing for uniform distribution of cells and synthesis of large areas of cell-laden material. This also allows the material and cellular cargo to be injectable and delivered directly to the site of interest.
Encapsulation of MSCs within oxime cross-linked hydrogels functionalized with a RGD adhesion peptide was performed to determine if cells could survive the gelation process, to investigate the potential to use these gels as a three-dimmensional gel matrix, and the stability of the oxime bond. Ketone-modified RGD was synthesized following standard solid phase peptide synthesis with the ketone added to the N-terminus using Fmoc-5-aminolevulinic acid (see supporting information for details). AO-PEG was modified with RGD (PEG-RGD) to achieve a 100 μM final concentration of RGD in the hydrogel. Mouse MSCs (5,000 cells/μL of gel) in complete medium were added to PEG-RGD (in PBS buffer pH = 7.4). This mixture was then added to the cross-linker, glutaraldehyde, to result in a 3.0 wt% AO-PEG gel with an r ratio of 0.7. Thus, the hydrogel was able to form in the presense of cells and serum, indicating that it is amenable for rapid hydrogel formation in the presence of a variety of different biological functional groups and living cells.
Live/dead staining of the encapsulated cells at days 1, 4, and 7 post-gelation showed high viability (Figure 3A-C). Metabolic activity of the encapsulated cells was assessed by MTT assay (Figure 3H). The relative absorbance by MTT doubled from 1 to 7 days indicating that the cells were metabolicaly active and proliferating. Glutaraldehyde is known as a cell fixative, yet the cells not only survived the crosslinking procedure, they proliferated. We hypothesize that the ability to encapsulate living cells may be due in part to the reactivity of aminooxy groups to form stable oxime bonds in aqueous solution compared to the unstable imines formed by amine groups.29 It is well known that oxime bond formation occurs in the presence of amines. In fact, this reaction has been widely used for site specific modification of proteins29 and to specifically label aldehyde moieties on living cells and tissues.18 Thus, with the reaction conditions used, it is likely that the glutaraldehyde preferentially reacts with the aminooxy groups of the polymer, leaving the MSCs unharmed. Exclusive reaction with more reactive amines on proteins, leaving other less reactive amines untouched has been reported for glutaraldehyde.30
Figure 3.
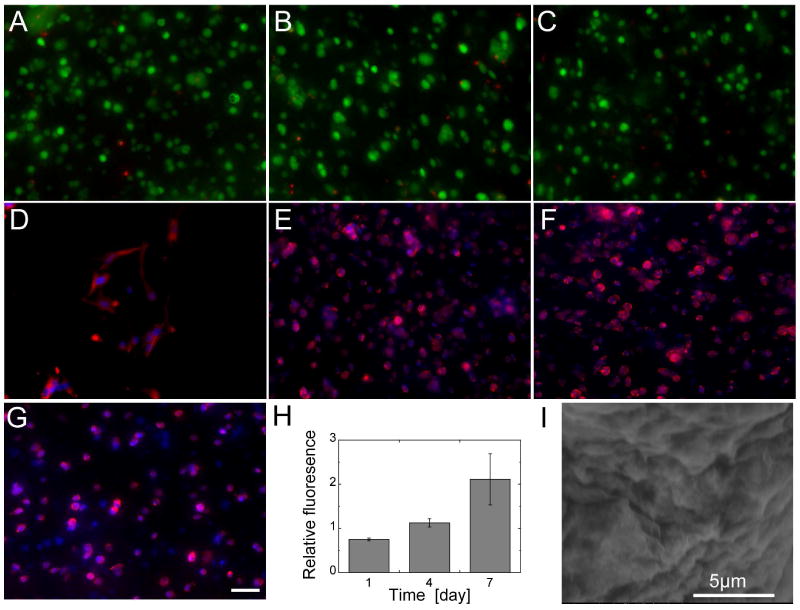
Live/Dead staining of the encapsulated mouse MSCs shows good viability at (A) day 1, (B) day 4, (C) and day 7. (D) Cells seeded on top of the hydrogel spread after one day, whereas cells encapsulated inside did not spread at (E) day 1, (F) day 4, (G) and day 7. (H) MTT assay shows an increased reduction over time, which indicates that cells are proliferating inside the hydrogel. (I) SEM image of hydrogel structure.
To determine if the cells could remodel the material in the absense of enzymatically degradable crosslinkers, spreading was assessed at 1, 4 and 7-days for cells plated inside the hydrogels. mMSCs (5000 cells/μL) were encapsulated inside RGD (100 μM) functionalized AO-PEG (3 wt%, r = 0.7) hydrogels as described above or on top of the hydrogel. Although cells plated on top of the hydrogel exhibited spread morphology over the 7-days of culture (Figure 3D and supporting information Figure S5), the MSCs encapsulated in an RGD functionalized hydrogel exhibited a rounded morphology over the course of the 7 day experiment (Figure 3E-G). This result indicates the stability and non-degradability of the oxime bond in the presense of cells and aqueous environment since cells require hydrogel degradation/remodeling to be able to spread. This result also suggests that the hydrogels may be useful for specific degradation of the gel, where a degradable linker for a particular enzyme is introduced into the system. These studies are underway. The SEM imaging looked typical for nano-porous PEG gels and did not show any poors larger than 5 μm in size, which is the resolution of the instrument used (Figure 3I and supporting information Figure S6).
Conclusions
In conclusion, a novel methodology has been developed to synthesize biocompatible and biofunctionalized hydrogels that encapsulate stem cells (3-D) or support adherence of the cells (2-D) by oxime bond formation. The mechanical properties, water absorption, and swelling ratio of this system can be tuned by adjusting the weight percent of the aminooxy-PEG and the reactivity ratio of aldehyde to aminooxy. The rate of gelation can easily be tuned by adjusting the pH of the system, within a mild pH range that is still applicable for tissue engineering applications. Facile installation of an integrin binding peptide (RGD) to the matrix was demonstrated. Mouse MSCs were able to adhere to the gels in 2-D and 3-D. MSCs encapsulated within the oxime cross-linked PEG hydrogel were viable and metabolically active for at least seven days demonstrating the biocompatibility of this approach for cell encapsulation. The material was non-degradable up to seven days, and suggesting that application of an enzymatically degradable cross-linker could facilitate tissue-specific cellular infiltration and spreading throughout the material. The ability to tune the properties and gelation rate of this system as well as encapsulate stem cells will allow this methodology to be applied as a functional coating and injectable material for stem cell therapies in research and clinical settings.
Supplementary Material
Acknowledgments
Funding Sources: HDM thanks the National Institutes of Health (R21 CA 137506-01) for funding. GNG and JL thank the NIH Funded Biotechnology Training Grant (T32 GMO67555) for predoctoral fellowships. Juneyoung Lee and Dr. Sina Saxer are thanked for providing the modified RGD.
Footnotes
Supporting Information. Experimental procedures, model study, additional cell and SEM images. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Christman KL, Lee RJ. J Am Coll Cardiol. 2006;48:907–913. doi: 10.1016/j.jacc.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 2.Wang HB, Zhou J, Liu ZQ, Wang CY. J Cell Mol Med. 2010;14:1044–1055. doi: 10.1111/j.1582-4934.2010.01046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu L, Ding JD. Chem Soc Rev. 2008;37:1473–1481. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 4.Fisher OZ, Khademhosseini A, Langer R, Peppas NA. Acc Chem Res. 2010;43:419–428. doi: 10.1021/ar900226q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eisenbarth E. Adv Eng Mater. 2007;9:1051–1060. [Google Scholar]
- 6.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 7.Nimmo CM, Shoichet MS. Bioconjugate Chem. 2011;22:2199–2209. doi: 10.1021/bc200281k. [DOI] [PubMed] [Google Scholar]
- 8.Liu SQ, Tian QA, Wang L, Hedrick JL, Hui JHP, Yang YY, Ee PLR. Macromol Rapid Comm. 2010;31:1148–1154. doi: 10.1002/marc.200900818. [DOI] [PubMed] [Google Scholar]
- 9.Patterson J, Hubbell JA. Biomaterials. 2010;31:7836–7845. doi: 10.1016/j.biomaterials.2010.06.061. [DOI] [PubMed] [Google Scholar]
- 10.Yang T, Long H, Malkoch M, Gamstedt EK, Berglund L, Hult A. J Polym Sci Polym Chem. 2011;49:4044–4054. [Google Scholar]
- 11.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang JY, Jacobsen MT, Pan HZ, Kopecek J. Macromol Biosci. 2010;10:445–454. doi: 10.1002/mabi.200900295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeForest CA, Anseth KS. Nat Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kalia J, Raines RT. Chem Int Ed. 2008;47:7523–7526. doi: 10.1002/anie.200802651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christman KL, Broyer RM, Schopf E, Kolodziej CM, Chen Y, Maynard HD. Langmuir. 2011;27:1415–1418. doi: 10.1021/la103978x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heredia KL, Tolstyka ZP, Maynard HD. Macromolecules. 2007;40:4772–4779. [Google Scholar]
- 17.Zeng Y, Ramya TNC, Dirksen A, Dawson PE, Paulson JC. Methods. 2009;6:207–209. doi: 10.1038/nmeth.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baskin JM, Dehnert KW, Laughlin ST, Amacher SL, Bertozzi CR. Proc Natl Acad Sci USA. 2010;107:10360–10365. doi: 10.1073/pnas.0912081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mimeault M, Hauke R, Batra SK. Clin Pharmacol Ther. 2007;82:252–264. doi: 10.1038/sj.clpt.6100301. [DOI] [PubMed] [Google Scholar]
- 20.Yang ZG, Ding JD. Macromol Rapid Comm. 2008;29:751–756. [Google Scholar]
- 21.Zhu JM. Biomaterials. 2010;31:4639–4656. doi: 10.1016/j.biomaterials.2010.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christman KL, Schopf E, Broyer RM, Li RC, Chen Y, Maynard HD. J Am Chem Soc. 2009;131:521–527. doi: 10.1021/ja804767j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Migneault I, Dartiguenave C, Bertrand MJ, Waldron KC. BioTechniques. 2004;37:790–802. doi: 10.2144/04375RV01. [DOI] [PubMed] [Google Scholar]
- 24.Drury JL, Mooney DJ. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 25.Maumus M, Guerit D, Toupet K, Jorgensen C, Noel D. Stem Cell Res Ther. 2011;2 doi: 10.1186/scrt55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Motaln H, Schichor C, Lah TT. Cancer. 2010;116:2519–2530. doi: 10.1002/cncr.25056. [DOI] [PubMed] [Google Scholar]
- 27.Deans RJ, Moseley AB. Exp Hematol. 2000;28:875–884. doi: 10.1016/s0301-472x(00)00482-3. [DOI] [PubMed] [Google Scholar]
- 28.Kretlow JD, Klouda L, Mikos AG. Adv Drug Delivery Rev. 2007;59:263–273. doi: 10.1016/j.addr.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 29.Maynard HD, Broyer RM, Kolodziej CM. In: Click Chemistry for Biotechnology and Materials Science. Lahann J, editor. Wiley, Inc.; West Sussex, UK: 2009. pp. 53–68. [Google Scholar]
- 30.Avrameas S, Ternynck T. Immunochemistry. 1969;6:53–66. doi: 10.1016/0019-2791(69)90178-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.