

Abstract
Pheochromocytomas are rare endocrine tumors that can present insidiously and remain undiagnosed until death or onset of clear manifestations of catecholamine excess. They are often referred to as one of the ‘great mimics’ in medicine. These tumors can no longer be regarded as a uniform disease entity, but rather as a highly heterogeneous group of chromaffin cell neoplasms with different ages of onset, secretory profiles, locations, and potential for malignancy according to underlying genetic mutations. These aspects all have to be considered when the tumor is encountered, thereby enabling optimal management for the patient. Referral to a center of specialized expertise for the disease should be considered wherever possible. This is not only important for surgical management of patients, but also for post-surgical follow up and screening of disease in patients with a hereditary predisposition to the tumor. While preoperative management has changed little over the last 20 years, surgical procedures have evolved so that laparoscopic resection is the standard of care and partial adrenalectomy should be considered in all patients with a hereditary condition. Follow-up testing is essential and should be recommended and ensured on a yearly basis. Managing such patients must now also take into account possible underlying mutations and the appropriate selection of genes for testing according to disease presentation. Patients and family members with identified mutations then require an individualized approach to management. This includes consideration of distinct patterns of biochemical test results during screening and the appropriate choice of imaging studies for tumor localization according to the mutation and associated differences in predisposition to adrenal, extra-adrenal and metastatic disease.
Keywords: pheochromocytoma, paraganglioma, management, clinical presentation, diagnosis, treatment, follow up, genetic testing
Introduction – general aspects
Clinical presentation
Pheochromocytomas are rare tumors usually characterized by secretion of catecholamines and associated signs and symptoms of catecholamine excess. This secretion can arise in a sudden burst leading to paroxysmal symptoms. The classical symptom triad consists of palpitations, headaches and sweating lasting from only minutes to hours and occurring periodically on different occasions [Manger et al., 1996]. Other symptoms, especially in an acute attack, include pallor, nausea and panic attacks, which may last for several minutes and resolve completely.
Apart from the above classic presentation, pheochromocytoma can also present with nonspecific symptoms such as flushing, nausea, tiredness or weight loss. Abdominal pain and constipation or chest pain mimicking myocardial infarction as in the case of inverted takotsubo cardiomyopathy can be caused by sudden catecholamine release [Prejbisz et al., 2011]. A subtle sign may be new onset of diabetes, particularly in the young non-obese patient [La Batide-Alanore et al., 2003]. Due to the diverse clinical manifestations, pheochromocytoma is therefore often referred as one of the great mimics in medicine. The first step in management of pheochromocytoma is to think of this rare disease and to then make the diagnosis [Manger, 2006].
Hypertension and incidentaloma
Pheochromocytoma is a rare cause of hypertension, but important because it is a usually curable cause of high blood pressure. The prevalence of the tumor among 4429 patients investigated for possible secondary hypertension has been reported at 0.3% [Anderson et al., 1994]. This is still higher than revealed in large autopsy studies, where the prevalence ranged from 0.05 to 0.09% in 44,680 and 15,984 deceased individuals respectively [Minno et al., 1954] [Schlegel, 1960] [McNeil et al., 2000]. Thus, pheochromocytoma should be considered in patients with hypertension, but generally only when secondary causes of high blood pressure are being considered or when there are other symptoms or signs of catecholamine excess that can alert the physician to the tumor. Prevalence of pheochromocytoma is much higher, at 4.2 to 6.5%, in patients screened for the tumor due to an incidentally discovered adrenal mass [Mantero et al., 2000] [Mansmann et al., 2004]. All patients with adrenal masses should therefore be screened for pheochromocytoma, irrespective of the presence of hypertension and symptoms of catecholamine excess.
Diagnosis and localization
Diagnosis
As now widely recommended [Pacak et al., 2007c], initial screening of pheochromocytoma should always include either or both measurements of urinary or plasma metanephrines, these metabolites comprising normetanephrine and metanephrine, the respective degradation products of the catecholamines, noradrenaline (NA) and adrenaline (A). Measurements of plasma free metanephrines have been shown by numerous independent studies to provide diagnostic sensitivity exceeding 96% with specificity between 85 to 100% [Pacak et al., 2007c]. To minimize false-positive results blood samples should be drawn under stress free conditions in the supine position. Urinary fractionated metanephrines provide an alternative approach with similar diagnostic sensitivity [Brain et al., 2006]. Diagnostic specificity at cut-offs for optimal sensitivity have, however, been reported as low as 45% for patients tested because of signs and symptoms [Lenders et al., 2002], this representing a limitation of urinary measurements. Urinary fractionated metanephrines are also commonly measured after a deconjugation step and therefore reflect different metabolites from the free metanephrines in plasma. Nevertheless the urinary tests are commonly available and their high diagnostic sensitivity provides justification for use during initial screening, albeit with likelihood to rule out subsequent false-positive results.
Metanephrines in plasma or urine can be measured by three different methods. Measurement by liquid chromatography with electrochemical detection (LC-EC), although a well established method with high accuracy and precision, is a technically demanding procedure. Immunoassays, which are easily established and commonly used in European laboratories for hormone measurements, have disadvantages of calibration, accuracy and precision problems [Singh et al., 2007] [Pillai et al., 2010] [Mullins et al., 2011]. In US and many other laboratories, liquid chromatography with tandem mass spectrometry (LC-MS/MS) has fast become the preferred method for determination of plasma and urinary metanephrines. This high-throughput method provides a precise, rapid, and specific method for biogenic amine measurements, but does require a large capital outlay and significant technical expertise [Lagerstedt et al., 2004].
Localization of pheochromocytoma
Once a pheochromocytoma is confirmed biochemically, the next step is to locate the tumor [Figure 1]. If a pheochromocytoma is located extra-adrenally it is defined as a paraganglioma. Localization is usually achieved by either computed tomography (CT) or magnetic resonance imaging (MRI) of the abdomen, with a sensitivity of 90-100% and a specificity of 70-80% [Maurea et al., 1993] [Francis et al., 1996] [Maurea et al., 1996] [Lenders et al., 2005]. However, specificity of MRI or CT imaging has been reported by some investigators as low as 50% [Ilias et al., 2004]. Unsatisfactory results, for example, may also be obtained in patients with SDHB and SDHD mutations in whom paragangliomas can be located at extra-abdominal locations. For this reason, algorithms that include whole body MRI as part of the combination of conventional and functional imaging have been proposed for disease localization and confirmation.
Figure 1.
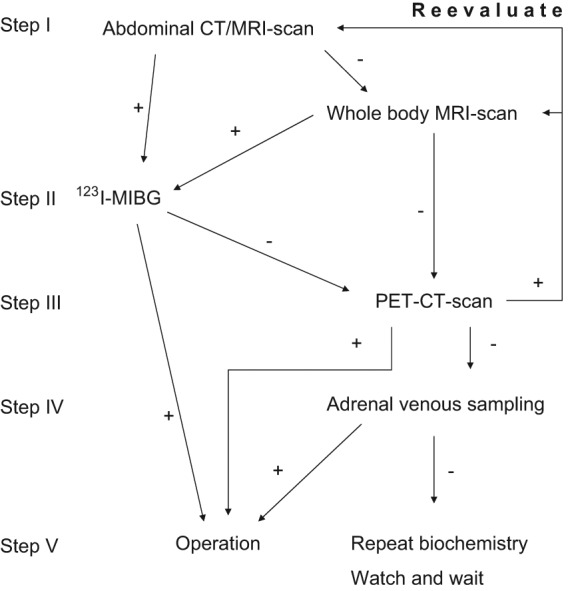
Algorithm for localization and confirmation of pheochromocytoma and paraganglioma
Imaging by [sup]123[/sup]I-MIBG (metaiodobenzylguanidine) scintigraphy provides a widely used secondary imaging modality [Shapiro et al., 1985] [Pacak et al., 2001a]. This test has a high specificity and therefore should be considered in all patients with findings of adrenal tumors by conventional imaging. If the mass shows up on MIBG, the diagnosis is confirmed justifying surgical resection [Bravo, 1994] [Shapiro et al., 1995] [Sisson et al., 1999b] [Miskulin et al., 2003]. MIBG can also point to additional lesions not indicated by conventional imaging in patients with metastatic disease or multifocal disease associated with mutations of disease-susceptibility genes. However, MIBG has lower sensitivity for detection of extra-adrenal tumors than adrenal tumors [Bhaita et al., 2008]. MIBG is also nevertheless not as sensitive for detection of metastases compared to some other functional techniques [Timmers et al., 2009].
If available, positron emission tomography, ideally combined with CT (PET/CT), and utilizing a range of labelled ligands provides an alternative functional imaging approach with improved resolution over scintigraphic methods. Such methods are particularly useful when MIBG scintigraphy, CT or MRI fail to locate the tumor. Radiotracers which have proven useful include [sup]18[/sup]F-fluorodopamine [Pacak et al., 2001b], especially in the diagnosis of metastatic disease [Ilias et al., 2003], [sup]18[/sup]F-fluorodopa [Hoegerle et al., 2002], [sup]11[/sup]C-hydroxyephedrine [Trampal et al., 2004] [Pacak et al., 2004], and [sup]68[/sup]Ga-DOTATOC (tetraazacyclododecanetetraaceticacid-tyr3-octreotide) [Kroiss et al., 2011]. Accumulating evidence suggests that [sup]18[/sup]F-fluorodeoxyglucose (FDG) is particularly valuable for detecting metastases in malignant pheochromocytoma and paraganglioma [Timmers et al., 2007b] [Timmers et al., 2009]. Adrenal venous sampling can be useful for localization of the tumor in selected cases when imaging results remain equivocal [Darr et al., 2011].
Considerations for management of hereditary syndromes
Advances in genetics
Classically, three syndromes are associated with pheochromocytoma, namely von Hippel Lindau (VHL) syndrome, multiple endocrine neoplasia type 2 (MEN2) and neurofibromatosis type 1 (NF1) [Neumann et al., 1993] [Bryant et al., 2003]. After germline mutations of succinate dehydrogenase (SDH) subunit genes were identified as responsible for familial paraganglioma, it became apparent that up to 30% of all pheochromocytomas were due to genetic mutations [Neumann et al., 2002] [Burnichon et al., 2011].
Mutations of all four SDH subunits are now identified as causes of hereditary paragangliomas: These include SDHA [Burnichon et al., 2010] [Korpershoek et al., 2011], SDHB [Astuti et al., 2001], SDHC [Niemann et al., 2000] and SDHD [Baysal et al., 2000]. Together with other mutations such as of the succinate dehydrogenase assembly factor 2 (SDHAF2) [Kunst et al., 2011] [Korpershoek et al., 2011], the transmembrane protein 127 (TMEM127) [Qin et al., 2010] and the MAX-gene [Comino-Mendez et al., 2011], there are now 10 known genes in which mutations predispose to pheochromocytoma and paraganglioma.
Screening for pheochromocytoma in mutation carriers
With the exception of mutations associated with low disease penetrance (e.g., NF1), all patients identified with a genetic mutation require periodic screening for pheochromocytoma and must be tracked and traced lifelong on a usually recommended yearly basis. Surveillance for pheochromocytoma and paraganglioma should be individualized according to the underlying genetic mutation. In particular, biochemical screening should account for the distinct mutation-dependent profiles of catecholamine production, best assessed by differences in plasma normetanephrine, metanephrine and methoxytyramine [Eisenhofer et al., 2011b].
Pheochromocytomas in VHL tumors are characterized by a solely noradrenergic biochemical phenotype so that, screening should be directed to measurements of normetanephrine. Adrenal tumors are common and the tumor is bilateral in 40% of the cases [Neumann et al., 2002], but extra-adrenal occurrence is possible and should always be considered in VHL syndrome. In contrast tumors in patients with MEN2 and NF1 also produce epinephrine, so that management of these patients should be directed to identification of increases in both metanephrine and normetanephrine. Since tumors are invariably intra-adrenal [Lairmore et al., 1993] imaging studies should focus mainly on the adrenals, with MIBG-scanning a particularly useful functional imaging modality due to highly active uptake of the agent by these tumors.
Tumors due to SDHB and SDHD mutations are somewhat similar to those due to VHL mutations in that they lack significant epinephrine production, so are not characterized by increases in metanephrine [Timmers et al., 2007a] [Eisenhofer et al., 2010]. However, these tumors often produce dopamine (D). Thus, in addition to measurement of normetanephrine, management of these patients can be improved by extending biochemical testing with measurements of dopamine and its metabolite methoxytyramine.
Extraadrenal tumors predominate in patients with SDHB or SDHD mutations, so imaging studies should be extensive, best achieved by whole body MRI. Head and neck paragangliomas, which are particularly common in SDHD mutation carriers, should always be considered and are best screened for using magnetic resonance angiography [Neumann et al., 2002] [Neumann et al., 2004]. Periodic screening with whole body MRI has been advocated in patients with SDHB mutations since tumors are particularly primitive and abdominal paragangliomas have been reported that do not produce any significant increases in normetanephrine or methoxytyramine [Timmers et al., 2008]. Tumors in SDHB mutation carriers also have a high potential for malignancy, demanding particular close and careful management of these patients [Amar et al., 2005b] [Brouwers et al., 2006]. Where malignancy is suspected, PET scanning with [sup]18[/sup]F-FDG is the preferred technique [Zelinka et al., 2008].
Screening for genetic mutations
Due to the high prevalence of germline mutations in pheochromocytoma and paragangliomas it is generally recommended that possible mutations be considered in all patients with the tumors. The extent of testing remains highly debatable, but it is now clear that management of this testing should always take into account clinical features that can point to a particular mutation [Grossman et al., 2006] [Pacak et al., 2007b].
Of primary importance for individualized genetic testing is to first check for any clinical stigmata suggestive of a particular syndrome or whether there may be any relatives with a history of or suggestive of the tumors or a particular syndrome. Testing then should be directed to those particular genes. In the absence of a family history or characteristic clinical signs, a younger age at presentation carries a higher likelihood of underlying genetic defect [Neumann et al., 2002] [Amar et al., 2005b]. Generally, VHL, SDHB and SDHD mutations present at a younger age compared with tumors in patients with NF1 and MEN2 [Eisenhofer et al., 2011a]. Bilateral adrenal tumors or multifocal extra-adrenal tumors carry a particularly high risk of germ line mutations that should always be explored.
Apart from the above factors, other considerations that can be used to point to mutations in the management of patients include locations of the tumor, catecholamine biochemical profiles and presence of metastatic disease [Table 1]. Mutations of RET, TMEM127 and MAX genes are rarely associated with extra-adrenal primary tumors [Yao et al., 2010] [Comino-Mendez et al., 2011], the presence of which dictates emphasis of genetic testing on mutations involving genes for succinate dehydrogenase subunits, which more often give rise to extra-adrenal than adrenal tumors. Patients with SDHB and SDHD mutations in particular often have tumors that produce methoxytyramine, usually with elevations of normetanephrine, but no or relatively insignificant elevations of metanephrine. Thus, in patients with these catecholamine biochemical profiles mutations of SDHB and SDHD genes should be considered. These mutations need not be considered for patients with tumors that produce metanephrine. Similarly, VHL mutations do not lead to tumors that produce epinephrine or metanephrine, so that this gene need not be considered in patients with tumors associated with increases in metanephrine. In contrast, pheochromocytomas in MEN 2 always produce increases in metanephrine, so that RET mutations should be considered in patients with these tumors.
Table 1.
Main features of hereditary mutations associated with pheochromocytoma, paraganglioma, and sporadic disease
| Onset | Adrenal | Bilateral | Extra-adrenal | Malignancy | Type | ||
|---|---|---|---|---|---|---|---|
| Sporadic | 70% | 40-50 | 90% | 10% | 10% | 10% | |
| Hereditary | 30% | ||||||
| Gene | Chromosome | ||||||
| SDHB | 1p36.13 | 30 | 28% | multifocal | 59% abdominal thoracic |
60% | NA/D |
| SDHD | 11q23 | 30 | 53% | multifocal | 79% head&neck |
17% | NA/D |
| VHL | 3p25-26 | 30 | 88% | 40% | 12% | 4% | NA |
| NF1 | 17q11.2 | 40 | 84% | 10% | 6% | 11% | NA/A |
| RET | 10q11.2 | 40 | 100% | 38% | rare | 4% | NA/A |
| SDHA | 5p15 | n.a. | n.a. | n.a. | n.a. | n.a. | Unknown |
| SDHAF2 | 11q12.2 | 30 | n.a. | n.a. | extraadrenal | n.a. | Unknown |
| SDHC | 1q23.3 | n.a. | rare | rare | rare | rare | Unknown |
| TMEM127 | 2q11.2 | n.a. | adrenal | bilateral | n.a. | 5% | Unknown |
| MAX-gene | 14q23 | n.a. | n.a. | n.a. | n.a. | n.a. | Unknown |
Immunohistochemical staining for SDHB in resected tumor samples can also provide useful information to guide genetic testing, which for SDHB or SDHD mutations is only indicated in SDHB negative tumors [Van Nederveen et al., 2009]. SDHB gene mutations should also be strongly suspected in patients with metastatic disease, but not those where disease is associated with large increases in metanephrine.
Complications and potential pitfalls in the treatment of the discovered pheochromocytoma
Preoperative considerations
Once a pheochromocytoma has been diagnosed and localized the next step is preparation for surgery. This ideally should be an interdisciplinary task involving endocrinologists, radiologists, nuclear medicine physicians, anaesthesiologists and surgeons but may also involve oncologists, cardiologists and clinical chemists. The presence of potential metastatic disease should always be considered and ruled out, since any indication of this is likely to impact treatment options beyond surgery. Patient management related to extent and choice of functional imaging to check for additional tumors or metastases should take into account stratification of risk for malignancy. Patients with large or extra-adrenal tumors, tumors associated with elevated plasma concentrations of methoxytyramine or due to SDHB mutations all have increased likelihood of metastatic disease [Tischler, 2008] [Eisenhofer, 2011c]. Risk increases when these factors associate in any combination and can be used to justify more extensive functional imaging to rule out metastases than might otherwise be considered. With no evidence of malignant disease, surgery is justified.
Due to the effects of circulating catecholamines on the cardiovascular system and possible preexisting cardiomyopathy in a patient with pheochromocytoma [Schurmeyer et al., 1997], cardiovascular function should be evaluated prior to surgery. This may include an electrocardiogram, ambulatory blood pressure monitoring to assess blood pressure variations, supine and standing blood pressure measurements to evaluate for postural hypotension, and echocardiography to determine heart dimensions and function.
Cardiac arrhythmias due to sudden catecholamine release during surgery and cardiac ischemia may occur [Quezado et al., 1992], and preexisting coronary artery disease may be aggravated by sudden vasospasms due to catecholamine release [Mannelli, 2006]. Coronary vasospasms may mimick cardiac ischemia [Liao et al., 2000], and direct catecholamine action may lead to pulmonary oedema [Tauzin-Fin et al., 1999]. Also, cerebrovascular accidents are a severe complication of hypertensive crisis in patients with pheochromocytoma.
Although some have suggested otherwise [Lentschener et al., 2011], it remains widely recommended that appropriate medications should always be employed prior to surgery to block the effects of circulating catecholamine in all patients with catecholamine-producing tumors [Russell et al., 1998] [Kinney et al., 2000]. There are no official guidelines on the types of blocking drugs, with both alpha-adrenergic blockers and calcium channel blockers representing the main medications chosen for the purpose [Tokioka et al., 1988] [Boutros et al., 1990] [Young, 1997] [Ulchaker et al., 1999] [Van Der Horst-Schrivers et al., 2006] [Pacak, 2007d].
Both slow-release nifedipine, a dihydropyridine calcium-channel blockers and diltiazem, a non-dihydropyridine calcium-channel blocker [Tokioka et al., 1988], have been described to control hypertension in pheochromocytoma. Immediate-release nifedipine, however, must be avoided because of potentially life-threatening cardiac accidents associated with this formulation. Otherwise calcium channel blockers can safely be used also in the cardiovascular patient, mitigating coronary vasospasms due to sudden noradrenaline release [Bravo et al., 2003]. Nifedipine is given at a dose of 30-60 mg once daily, doses up to 120 mg per day have also been described [Proye et al., 1989].
Most usually pre-operative blockade of catecholamine action employs the irreversible non-selective alpha-adenergic receptor, phenoxybenzamine. The drug is titrated according to patient needs. The starting dose is 10 mg twice a day, with stepwise increases of 10 to 20 mg every 2-3 days until the final dose of 1 mg/kg of body weight is reached. This dose is given in three divided doses over the day and is normally reached within 10-14 days, while the patient can be followed on an outpatient basis [Witteles et al., 2000]. Blood pressure monitoring should be ensured two to three times a day, but even more important is measurement of standing blood pressure to gauge eventual orthostatic hypotension. Clinical signs of the optimal dose are a stuffy nose and slight dizziness due to postural hypotension.
Alternatively to phenoxybenzamine, the selective and reversible alpha1-blocker doxazosin may be used [Bravo et al., 2003]. Doxazosin is started at a dose of 1 mg and increased thereafter to 32 mg once a day. In contrast to phenoxybenzamine, strong catecholamine bursts can displace doxazosin from its receptor binding site and thus reduce efficacy. The advantage on the other hand is thought to be a reduced hypotensive response after surgery but this issue still remains unresolved [Kinney et al., 2002] [Kocak et al., 2002] [Prys-Roberts et al., 2002]. In our outpatient setting, we normally use phenoxybenzamine for the preoperative treatment. Exceptions from this routine are the operation of head and neck paraganglioma especially if this tumor does not produce catecholamines or only dopamine [Mannelli, 2006].
In order to reduce tachycardic reflex responses, a low dose beta-blockade can be initiated in patients with evidence of tachycardia before surgery. Cardioselective beta1-blockers such as metoprolol at 50 to 100 mg, bisoprolol at 5-10 mg or atenolol at 25-50 mg once daily [Prys-Roberts, 2000] are recommended. Intraoperative tachycardias may be controlled by esmolol [Gray, 1988]. Although labetalol has been used in the pharmacological treatment of pheochromocytoma [Van Stratum et al., 1983] [Yabe et al., 1987], its use is controversial [Reach et al., 1980]. Briggs et al. reported about a hypotensive reaction in a patient in postural position and a reactive hypertensive crisis in supine position after labetalol, cautioning against the use of this drug in pheochromocytoma [Briggs et al., 1978].
Alpha-methyl-para-tyrosine (metyrosine) has been used for additional preoperative treatment of pheochromocytoma [Perry et al., 1990], but today is primarily relegated to patients in whom disease is extensive and accompanied by large increases in catecholamines. Metyrosine blocks catecholamine biosynthesis by inhibiting the conversion of tyrosine to L-dopa [Sjoerdsma et al., 1965]. Usually metyrosine is started with an oral dose of 250 mg 3 to 4 times per day and gradually increased to a total dose of 1,5 to 4,0 g/d [Pacak et al., 2007a].
Acute hypertensive attacks can be managed with the short acting alpha-blocker phentolamine as an infusion of e.g. 100 mg in 500 ml of 5% dextrose in water with 1mg/min [Pacak et al., 2007a]. Other drugs in these emergency cases proven useful are vasodilating agents such as nitroprusside and nitroglycerin [Prys-Roberts, 2000] [Kinney et al., 2002] [Prys-Roberts et al., 2002] as well as urapidil in Europe [Tauzin-Fin et al., 2004]. Interestingly good results in treating hypertension in patients with pheochromocytoma have been achieved with magnesium sulphate and therefore magnesium may be tried as a second line therapy for blood pressure control [James, 1989]. The action of magnesium is thought to be an inhibition of catecholamine release from the normal adrenal gland and from adrenergic nerve terminals [Douglas et al., 1963] [Von Euler et al., 1973].
Anaesthesia – aspects
Premedication with a benzodiazepine can be employed in patients with pheochromocytoma and no premedication has been shown to be superior to others [Kinney et al., 2002]. For operation usually general anaesthesia is chosen. For induction, thiopental has been used with success [Desmonts et al., 1984] and seems to decrease plasma catecholamine levels [Joyce et al., 1983]. Also propofol and etomidate have been used with etomidate providing more cardiovascular stability [Hull, 1986]. Causing indirect catecholamine release, ketamine, morphine or meperidine, as well as droperidol [Sumikawa et al., 1977] and ephedrine are not recommended [Kinney et al., 2002].
For muscle relaxation, vecuronium [Gencarelli et al., 1981], pancuronium [Desmonts et al., 1984] and atracurium [Prys-Roberts, 2000] have been used, with atracurium possibly causing catecholamine release via histamine. As this also applies for tubocurarine and curare, and gallamine causing vagolytic effects, all those drugs should be avoided in patients with pheochromocytoma. Pancuronium has been used with success in the past but has been associated with severe hypertension [Jones et al., 1981]. Because of its cardiac side effects, succinylcholine should be avoided [Stoner et al., 1968]. Also, succinycholine may result in muscle fasciculation with ensuing mechanical stimulation of the tumor, and potentially life threatening catecholamine bursts [Desmonts et al., 1984].
Regarding maintenance of anesthesia with volatile agents, sevoflurane [Kinney et al., 2002], isoflurane [Kinney et al., 2000] and enflurane [Janeczko et al., 1977] can safely be used. Desflurane causes sympathetic activation but has also been administered [Lippmann et al., 1994].
Operation - aspects
Today, pheochromocytomas of the abdomen normally can be operated on laparoscopically [Janetschek et al., 1998] [Gill, 2001]. Single access retroperitoneoscopic adrenalectomy (SARA) may be an elegant alternative [Walz et al., 2010]. The laparoscopic approach, together with adequate preoperative management, reduces perioperative mortality to 2.4% [Plouin et al., 2001]. It also reduces morbidity, hospital stay and costs [Fernandez-Cruz et al., 1996] [Sprung et al., 2000], and is feasible and safe in solitary, bilateral, multiple, and recurrent pheochromocytoma [Walz et al., 2002] [Jaroszewski et al., 2003].
Partial adrenalectomy has been recommended in hereditary phechromocytoma to avoid adrenal insufficiency and lifelong hormone replacement therapy [Neumann et al., 1999] [Walther et al., 2000]. However partial adrenalectomy may predispose patients to an increased risk of recurrent disease. When performed in hereditary disease [Brauckhoff et al., 2004], recurrence rate has been described in the range between 21% and 60% [Lee et al., 1996] [Inabnet et al., 2000]. Furthermore, partial adrenalectomy may not always ensure cortisol independency postoperatively [Yip et al., 2004]. Therefore the benefits and risks of partial and total adrenalectomy must be discussed with the patient in each single case.
Open surgery may be indicated, when tumors are multiple or very large [Vargas et al., 1997] but even tumors sized more than 9 cm may safely be removed laparascopically depending on the experience of the surgeon [Pacak et al., 2007a]. Nevertheless, it is now becoming increasingly clear that risk of malignant disease increases with tumor size [Eisenhofer, 2011c]. Minimizing risk of spillage associated with laparoscopic surgery may therefore be important to consider with large tumors or for those associated with increased risk of malignancy due to other established risk factors (i.e., SDHB mutations, extra-adrenal locations). Difficult extra-adrenal operative sites include tumors located in the bladder, the chest or the head and neck [Manger, 2006].
Since head and neck paragangliomas rarely secrete catecholamines these tumors mostly do not require preoperative treatment. The main risks are related to operative difficulties. Complications include intraoperative nerve lesions, especially of the recurrent laryngeal nerve with subsequent hoarseness and difficulties of speaking and breathing. Other cranial nerves may be injured causing impairment of swallowing, hearing or facial muscle paralysis. In some cases, loss of blood pressure control due to baroreflex trauma may occur [Mannelli, 2006].
Postoperative considerations
Main challenges of the immediate postoperative period are blood pressure instability and heart rate control, as well as symptomatic hypoglycaemia, necessitating close patient monitoring for at least 24 h after operation [Mannelli, 2006]. Loss of peripheral vasoconstriction after operation may lead to hypovolemic hypotension, despite preoperative loading with normal saline. The amount of fluid required may be as large as 6-7 liters within the first two days after operation. Also, the effect of irreversible preoperative alpha-blockade may persist for more than 36 hours postoperatively [Bergman et al., 1978]. Under these circumstances hypotension may be nor- and adrenaline resistant, and vasopressin may be needed for blood pressure control [Augoustides et al., 2004].
Hypoglycaemia may occur because of rebound hyperinsulinemia [Isles et al., 1983] [Ostenson et al., 1989], and may be aggravated by prolonged postoperative alpha-blockade. Also, overt hypoglycaemic reactions may be masked due to impaired gluconeogenesis and glucogenolysis after beta2-receptor blockade [Kinney et al., 2002]. Therefore, close monitoring of blood glucose levels is mandatory, and prompt dextrose-infusions should be initiated when necessary [Meeke et al., 1985]. If both hypotension and hypoglycaemia occur in a patient after bilateral partial or complete adrenalectomy [Costello et al., 1988], suspicions about hypocortisolism and its most severe form, Addisonian crises should be raised, and plasma and urinary cortisol and plasma adrenocorticotropic hormone (ACTH) levels measured. If confirmed immediate steroid replacement should be started [Pacak et al., 2007a].
Incomplete tumor removal, coexisting essential hypertension, volume overload, autonomic instability, postoperative pain or accidental ligation of the renal artery may result in postoperative hypertension [Engelman, 1977] [Young, 1997] [Pacak et al., 2007a]. If hypertension persists the underlying cause should be determined and therapy instituted accordingly.
Follow-up
To ascertain that resection was complete and successful, biochemical testing should be undertaken dependent on the patient’s recovery within 2-6 weeks after surgery. As recurrence rates of up to 14% have been reported in pheochromocytoma and of up to 30% in extra-adrenal disease [Amar et al., 2005a], lifelong follow up is recommended in all patients [Scott et al., 1984] [Jaroszewski et al., 2003]. Also, tumors may recur beginning from one year for up to 16 years after initial diagnosis and operation [Plouin et al., 1997]. Therefore all patients with histologically proven pheochromocytoma or paraganglioma should be referred to and followed in specialized centers to ensure appropriate follow-up measurements [Mannelli, 2006]. Yearly follow up is also indicated in patients who are not willing to be or cannot be operated on for reasons such as previous adrenalectomy, putting them at increased risk for lifelong hormone replacement therapy, or because of an unacceptably high patient morbidity. In patients with sporadic pheochromocytoma follow up should be 10 years at least. Patients with either extra-adrenal disease or familial pheochromocytoma should be screened indefinitely in an individualized manner on a yearly basis [Lenders et al., 2005]. Follow up visits every 6 months are indicated in patients with metastatic pheochromocytoma.
The patient with malignant pheochromocytoma and paraganglioma
Although attempts have been made to identify malignant pheochromocytoma by histomorphologic features [Thompson, 2002], proof of malignant disease to date relies on pathological confirmation of tumor metastases at sites where chromaffin tissue normally is absent, including bones, lungs, liver and lymph nodes. There are, however, several factors that can be used to predict future development of malignant disease. The presence of an SDHB mutation represents the single most important predictor for increased risk of malignancy [Gimenez-Roqueplo et al., 2003]. As shown by Eisenhofer [Eisenhofer, 2011c], the high risk of malignancy assocociated with SDHB tumors entirely reflects both the predominant extra-adrenal locations and large size reached by tumors in patients with these mutations. Both extra-adrenal location and large tumor size are independent factors that contribute to increased risk of malignancy. Additionally, elevations of methoxytyramine, the O-methylated metabolite of dopamine, indicate an increased likelihood of metastases [Eisenhofer, 2011c].
Treatment options of malignant disease are limited. Disappointing results with only temporary remissions lasting for up to two years [Eisenhofer et al., 2004] have been obtained with i.v. cyclophosphamide, vincristine and dacarbazine combination chemotherapy [Averbuch et al., 1988]. Future therapy options may include LB1, a small molecule inhibitor of serine/threonine protein phosphatase 2A (PP2A), combined with temzolamide [Martiniova et al., 2011] or sunitinib [Joshua et al., 2009]. Symptomatic medical therapy is directed towards control of blood pressure and largely follows preoperative disease management rules. In addition alpha-methyl-para-tyrosine has been shown effective in case of high levels of plasma catecholamines [Decoulx et al., 1987].
[sup]131[/sup]MIBG-radiotherapy either alone or in combination with chemotherapy has been reported to result in partial tumor remission [Sisson et al., 1999a]. However, progressive disease has also been reported using this treatment modality [Schlumberger et al., 1992]. It is presently unclear, whether higher doses of radiation might improve patient survival [Rose et al., 2003]. The low remission rate observed may at least in part be due to the fact that malignant pheochromocytoma do not readily accumulate [sup]131[/sup]MIBG [Ilias et al., 2003].
Surgery is restricted to debulking strategies as there is no curative approach [Eisenhofer et al., 2004]. Surgery can reduce the total amount of the catecholamine producing mass and the deleterious results of elevated catecholamines on the heart [Mishra et al., 2000]. Furthermore, surgery can remove lesions that are life threatening due to their location [Nonaka et al., 2000]. Finally, surgery can serve as an adjuvant measure prior to radiotherapy or chemotherapy. Nevertheless, prognosis of malignant disease is poor. In a postoperative survival study, 5 year survival rate has been estimated to be 20%, while no patient was alive ten years after surgery [John et al., 1999]. In one single report a patient was alive more than 25 years after initial diagnosis [Yoshida et al., 2001].
Pheochromocytoma in pregnancy - special considerations
With a prevalence of 1 in 50,000, pheochromocytoma is only rarely seen in pregnancy, where it shares two main symptoms with pre-eclampsia, namely spells of hypertension [Mannelli et al., 2002] and headaches. Suspicions of pheochromocytoma should be raised in any pregnant woman with new onset hypertension [Manger, 2009], especially in the first half of pregnancy [Lenders, 2011]. Hypertensive crisis may compromise placental circulation while placental enzymes protect the fetus from direct catecholamine actions [Brunt, 2001] [Pacak et al., 2007a].
Phenoxybenzamine in the pretreatment period before surgery seems to be safe for the fetus [Ahlawat et al., 1999]. In hypertensive crisis, phentolamine can be used with 1 mg/min, as well as sodium nitroprusside as a final backup but only at a low dose and not over a prolonged time period to avoid fetal cyanide poisoning [Molitch, 1992]. Also, control of hypertension may be achieved with magnesium sulphate provided adequate magnesium levels are reached [James et al., 1988]. Care should be taken to avoid hypotension since similar to hypertension, this can compromise placental circulation [Griffen et al., 1969].
Special regards are necessary concerning diagnosis and tumor localization. The clonidine test cannot be performed in pregnant women, but measurement of urinary metanephrines after overnight sampling may be a useful alternative [Sullivan et al., 1975] [Peaston et al., 1996]. Abdominal ultrasound has a low diagnostic sensitivity especially for small tumors. Magnetic resonance tomography with gadolinium enhancement is the imaging procedure of first choice. However confirmation of a mass as pheochromocytoma or paraganglioma by scintigraphy is not feasible, leaving physicians and the mother in doubt of its origin.
Due to its infrequent occurrence, management of pheochromocytoma in pregnancy remains a medical challenge. If unrecognized, the risk of miscarriage, and of both maternal and fetal mortality is high, with reported mean ranges of 40 through 48% and 44 through 56%, respectively [Griffen et al., 1969] [Schenker et al., 1982] [Pacak et al., 2007a], while rapid diagnosis reduces these rates approximately by half [Grodski et al., 2006]. However, despite unquestionable progress in recent years still antenatal diagnosis is made in only 30% to 50% of patients. These are sobering results considering that with antenatal diagnosis maternal mortality rate decreases nearly to zero and mortality of the unborn child to 15-18%, respectively [Harper et al., 1989] [Oishi et al., 1994].
There is general agreement that the tumor should be resected before the third trimenon and also that the method of choice should be laparascopy [Demeure et al., 1998]. The risk of spontaneous abortion is greatest in the first trimenon [Yumi, 2008]. In the third trimenon, cesarean section should be performed prior to tumor removal [Manger et al., 1996]. The issue of whether both operations should be carried out simultaneously is unsettled, unless in case of uncontrolled hypertension, hemorrhage or other emergency situations [Pacak et al., 2007a].
Footnotes
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
The Authors declare that there is no conflict of interest.
References
- Ahlawat S.K., Jain S., Kumari S., Varma S., Sharma B.K. (1999) Pheochromocytoma Associated with Pregnancy: Case Report and Review of the Literature. Obstet Gynecol Surv 54: 728–737 [DOI] [PubMed] [Google Scholar]
- Amar L., Servais A., Gimenez-Roqueplo A.P., Zinzindohoue F., Chatellier G., Plouin P.F. (2005a) Year of Diagnosis, Features at Presentation, and Risk of Recurrence in Patients with Pheochromocytoma or Secreting Paraganglioma. J Clin Endocrinol Metab 90: 2110–2116 [DOI] [PubMed] [Google Scholar]
- Amar L., Bertherat J., Baudin E., Ajzenberg C., Bressac-De Paillerets B., Chabre O., et al. (2005b) Genetic Testing in Pheochromocytoma or Functional Paraganglioma. J Clin Oncol 23: 8812–8818 [DOI] [PubMed] [Google Scholar]
- Anderson G.H., Jr., Blakeman N., Streeten D.H. (1994) The Effect of Age on Prevalence of Secondary Forms of Hypertension in 4429 Consecutively Referred Patients. J Hypertens 12: 609–615 [DOI] [PubMed] [Google Scholar]
- Astuti D., Latif F., Dallol A., Dahia P.L., Douglas F., George E., et al. (2001) Gene Mutations in the Succinate Dehydrogenase Subunit Sdhb Cause Susceptibility to Familial Pheochromocytoma and to Familial Paraganglioma. Am J Hum Genet 69: 49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augoustides J.G., Abrams M., Berkowitz D., Fraker D. (2004) Vasopressin for Hemodynamic Rescue in Catecholamine-Resistant Vasoplegic Shock after Resection of Massive Pheochromocytoma. Anesthesiology 101: 1022–1024 [DOI] [PubMed] [Google Scholar]
- Averbuch S.D., Steakley C.S., Young R.C., Gelmann E.P., Goldstein D.S., Stull R., et al. (1988) Malignant Pheochromocytoma: Effective Treatment with a Combination of Cyclophosphamide, Vincristine, and Dacarbazine. Ann Intern Med 109: 267–273 [DOI] [PubMed] [Google Scholar]
- Baysal B.E., Ferrell R.E., Willett-Brozick J.E., Lawrence E.C., Myssiorek D., Bosch A., et al. (2000) Mutations in Sdhd, a Mitochondrial Complex II Gene, in Hereditary Paraganglioma. Science 287: 848–851 [DOI] [PubMed] [Google Scholar]
- Bergman S.M., Sears H.F., Javadpour N., Keiser H.R. (1978) Postoperative Management of Patients with Pheochromocytoma. J Urol 120: 109–112 [DOI] [PubMed] [Google Scholar]
- Bhatia K.S., Ismail M.M., Sahdev A., Rockall A.G., Hogarth K., Canizales A., et al. (2008) 123i-Metaiodobenzylguanidine (Mibg) Scintigraphy for the Detection of Adrenal and Extra-Adrenal Phaeochromocytomas: Ct and Mri Correlation. Clin Endocrinol (Oxf) 69: 181–188 [DOI] [PubMed] [Google Scholar]
- Boutros A.R., Bravo E.L., Zanettin G., Straffon R.A. (1990) Perioperative Management of 63 Patients with Pheochromocytoma. Cleve Clin J Med 57: 613–617 [DOI] [PubMed] [Google Scholar]
- Brain K.L., Kay J., Shine B. (2006) Measurement of Urinary Metanephrines to Screen for Pheochromocytoma in an Unselected Hospital Referral Population. Clin Chem 52: 2060–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauckhoff M., Gimm O., Brauckhoff K., Dralle H. (2004) Repeat Adrenocortical-Sparing Adrenalectomy for Recurrent Hereditary Pheochromocytoma. Surg Today 34: 251–255 [DOI] [PubMed] [Google Scholar]
- Bravo E.L. (1994) Evolving Concepts in the Pathophysiology, Diagnosis, and Treatment of Pheochromocytoma. Endocr Rev 15: 356–368 [DOI] [PubMed] [Google Scholar]
- Bravo E.L., Tagle R. (2003) Pheochromocytoma: State-of-the-Art and Future Prospects. Endocr Rev 24: 539–553 [DOI] [PubMed] [Google Scholar]
- Briggs R.S., Birtwell A.J., Pohl J.E. (1978) Hypertensive Response to Labetalol in Phaeochromocytoma. Lancet 1: 1045–1046 [DOI] [PubMed] [Google Scholar]
- Brouwers F.M., Eisenhofer G., Tao J.J., Kant J.A., Adams K.T., Linehan W.M., et al. (2006) High Frequency of Sdhb Germline Mutations in Patients with Malignant Catecholamine-Producing Paragangliomas: Implications for Genetic Testing. J Clin Endocrinol Metab 91: 4505–4509 [DOI] [PubMed] [Google Scholar]
- Brunt L.M. (2001) Phaeochromocytoma in Pregnancy. Br J Surg 88: 481–483 [DOI] [PubMed] [Google Scholar]
- Bryant J., Farmer J., Kessler L.J., Townsend R.R., Nathanson K.L. (2003) Pheochromocytoma: The Expanding Genetic Differential Diagnosis. J Natl Cancer Inst 95: 1196–1204 [DOI] [PubMed] [Google Scholar]
- Burnichon N., Briere J.J., Libe R., Vescovo L., Riviere J., Tissier F., et al. (2010) Sdha Is a Tumor Suppressor Gene Causing Paraganglioma. Hum Mol Genet 19: 3011–3020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnichon N., Vescovo L., Amar L., Libe R., De Reynies A., Venisse A., et al. (2011) Integrative Genomic Analysis Reveals Somatic Mutations in Pheochromocytoma and Paraganglioma. Hum Mol Genet 20: 3974–3985 [DOI] [PubMed] [Google Scholar]
- Comino-Mendez I., Gracia-Aznarez F.J., Schiavi F., Landa I., Leandro-Garcia L.J., Leton R., et al. (2011) Exome Sequencing Identifies Max Mutations as a Cause of Hereditary Pheochromocytoma. Nat Genet 43: 663–667 [DOI] [PubMed] [Google Scholar]
- Costello G.T., Moorthy S.S., Vane D.W., Dierdorf S.F. (1988) Hypoglycemia Following Bilateral Adrenalectomy for Pheochromocytoma. Crit Care Med 16: 562–563 [DOI] [PubMed] [Google Scholar]
- Darr R., Eisenhofer G., Kotzerke J., Zophel K., Stroszczynski C., Deinum J., et al. (2011) Is There Still a Place for Adrenal Venous Sampling in the Diagnostic Localization of Pheochromocytoma? Endocrine 40: 75–79 [DOI] [PubMed] [Google Scholar]
- Decoulx M., Wemeau J.L., Racadot-Leroy N., Grimbert I., Proye C., Plane C. (1987) [Alpha-Methyl-Paratyrosine in the Treatment of Malignant Pheochromocytoma]. Rev Med Interne 8: 383–388 [DOI] [PubMed] [Google Scholar]
- Demeure M.J., Carlsen B., Traul D., Budney C., Lalande B., Lipinski A., et al. (1998) Laparoscopic Removal of a Right Adrenal Pheochromocytoma in a Pregnant Woman. J Laparoendosc Adv Surg Tech A 8: 315–319 [DOI] [PubMed] [Google Scholar]
- Desmonts J.M., Marty J. (1984) Anaesthetic Management of Patients with Phaeochromocytoma. Br J Anaesth 56: 781–789 [DOI] [PubMed] [Google Scholar]
- Douglas W.W., Rubin R.P. (1963) The Mechanism of Catecholamine Release from the Adrenal Medulla and the Role of Calcium in Stimulus-Secretion Coupling. J Physiol 167: 288–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G., Bornstein S.R., Brouwers F.M., Cheung N.K., Dahia P.L., De Krijger R.R., et al. (2004) Malignant Pheochromocytoma: Current Status and Initiatives for Future Progress. Endocr Relat Cancer 11: 423–436 [DOI] [PubMed] [Google Scholar]
- Eisenhofer G., Pacak K., Huynh T.T., Qin N., Bratslavsky G., Linehan W.M., et al. (2010) Catecholamine Metabolomic and Secretory Phenotypes in Phaeochromocytoma. Endocr Relat Cancer 18: 97–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G., Timmers H.J., Lenders J.W., Bornstein S.R., Tiebel O., Mannelli M., et al. (2011a) Age at Diagnosis of Pheochromocytoma Differs According to Catecholamine Phenotype and Tumor Location. J Clin Endocrinol Metab 96: 375–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G., Lenders J.W., Timmers H., Mannelli M., Grebe S.K., Hofbauer L.C., et al. (2011b) Measurements of Plasma Methoxytyramine, Normetanephrine, and Metanephrine as Discriminators of Different Hereditary Forms of Pheochromocytoma. Clin Chem 57: 411–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhofer G., Lenders J.W., Siegert G., Bornstein S., Friberg P., Milosevic D., Mannelli M., Linehan W.M., Adams K., Timmers H., Pacak K. (2011c) Plasma Methoxytyramine: A Novel Biomarker of Metastatic Pheochromocytoma and Paraganglioma in Relation to Established Risk Factors of Tumor Size, Location and Sdhb Mutation Status. Eur J Cancer: in press [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelman K. (1977) Phaeochromocytoma. Clin Endocrinol Metab 6: 769–797 [DOI] [PubMed] [Google Scholar]
- Fernandez-Cruz L., Taura P., Saenz A., Benarroch G., Sabater L. (1996) Laparoscopic Approach to Pheochromocytoma: Hemodynamic Changes and Catecholamine Secretion. World J Surg 20: 762-768; discussion 768 [DOI] [PubMed] [Google Scholar]
- Francis I.R., Korobkin M. (1996) Pheochromocytoma. Radiol Clin North Am 34: 1101–1112 [PubMed] [Google Scholar]
- Gencarelli P.J., Roizen M.F., Miller R.D., Joyce J., Hunt T.K., Tyrrell J.B. (1981) Org Nc45 (Norcuron) and Pheochromocytoma: A Report of Three Cases. Anesthesiology 55: 690–693 [DOI] [PubMed] [Google Scholar]
- Gill I.S. (2001) The Case for Laparoscopic Adrenalectomy. J Urol 166: 429–436 [PubMed] [Google Scholar]
- Gimenez-Roqueplo A.P., Favier J., Rustin P., Rieubland C., Crespin M., Nau V., et al. (2003) Mutations in the Sdhb Gene Are Associated with Extra-Adrenal and/or Malignant Phaeochromocytomas. Cancer Res 63: 5615–5621 [PubMed] [Google Scholar]
- Gray R.J. (1988) Managing Critically Ill Patients with Esmolol. An Ultra Short-Acting Beta-Adrenergic Blocker. Chest 93: 398–403 [DOI] [PubMed] [Google Scholar]
- Griffen W.O., Jr., Dilts P.V., Jr., Roddick J.W., Jr. (1969) Non-Obstetric Surgery During Pregnancy. Curr Probl Surg: 1–56 [PubMed] [Google Scholar]
- Grodski S., Jung C., Kertes P., Davies M., Banting S. (2006) Phaeochromocytoma in Pregnancy. Intern Med J 36: 604–606 [DOI] [PubMed] [Google Scholar]
- Grossman A., Pacak K., Sawka A., Lenders J.W., Harlander D., Peaston R.T., et al. (2006) Biochemical Diagnosis and Localization of Pheochromocytoma: Can We Reach a Consensus? Ann N Y Acad Sci 1073: 332–347 [DOI] [PubMed] [Google Scholar]
- Harper M.A., Murnaghan G.A., Kennedy L., Hadden D.R., Atkinson A.B. (1989) Phaeochromocytoma in Pregnancy. Five Cases and a Review of the Literature. Br J Obstet Gynaecol 96: 594–606 [DOI] [PubMed] [Google Scholar]
- Hoegerle S., Nitzsche E., Altehoefer C., Ghanem N., Manz T., Brink I., et al. (2002) Pheochromocytomas: Detection with 18f Dopa Whole Body Pet–Initial Results. Radiology 222: 507–512 [DOI] [PubMed] [Google Scholar]
- Hull C.J. (1986) Phaeochromocytoma. Diagnosis, Preoperative Preparation and Anaesthetic Management. Br J Anaesth 58: 1453–1468 [DOI] [PubMed] [Google Scholar]
- Ilias I., Yu J., Carrasquillo J.A., Chen C.C., Eisenhofer G., Whatley M., et al. (2003) Superiority of 6-[18f]-Fluorodopamine Positron Emission Tomography Versus [131i]-Metaiodobenzylguanidine Scintigraphy in the Localization of Metastatic Pheochromocytoma. J Clin Endocrinol Metab 88: 4083–4087 [DOI] [PubMed] [Google Scholar]
- Ilias I., Pacak K. (2004) Current Approaches and Recommended Algorithm for the Diagnostic Localization of Pheochromocytoma. J Clin Endocrinol Metab 89: 479–491 [DOI] [PubMed] [Google Scholar]
- Inabnet W.B., Caragliano P., Pertsemlidis D. (2000) Pheochromocytoma: Inherited Associations, Bilaterality, and Cortex Preservation. Surgery 128: 1007-1011;discussion 1011–1002 [DOI] [PubMed] [Google Scholar]
- Isles C.G., Johnson J.K. (1983) Phaeochromocytoma and Diabetes Mellitus: Further Evidence That Alpha 2 Receptors Inhibit Insulin Release in Man. Clin Endocrinol (Oxf) 18: 37–41 [DOI] [PubMed] [Google Scholar]
- James M.F., Huddle K.R., Owen A.D., Van Der Veen B.W. (1988) Use of Magnesium Sulphate in the Anaesthetic Management of Phaeochromocytoma in Pregnancy. Can J Anaesth 35: 178–182 [DOI] [PubMed] [Google Scholar]
- James M.F. (1989) Use of Magnesium Sulphate in the Anaesthetic Management of Phaeochromocytoma: A Review of 17 Anaesthetics. Br J Anaesth 62: 616–623 [DOI] [PubMed] [Google Scholar]
- Janeczko G.F., Ivankovich A.D., Glisson S.N., Heyman H.J., El-Etr A.A., Albrecht R.F. (1977) Enflurane Anesthesia for Surgical Removal of Pheochromocytoma. Anesth Analg 56: 62–67 [DOI] [PubMed] [Google Scholar]
- Janetschek G., Finkenstedt G., Gasser R., Waibel U.G., Peschel R., Bartsch G., et al. (1998) Laparoscopic Surgery for Pheochromocytoma: Adrenalectomy, Partial Resection, Excision of Paragangliomas. J Urol 160: 330–334 [DOI] [PubMed] [Google Scholar]
- Jaroszewski D.E., Tessier D.J., Schlinkert R.T., Grant C.S., Thompson G.B., Van Heerden J.A., et al. (2003) Laparoscopic Adrenalectomy for Pheochromocytoma. Mayo Clin Proc 78: 1501–1504 [DOI] [PubMed] [Google Scholar]
- John H., Ziegler W.H., Hauri D., Jaeger P. (1999) Pheochromocytomas: Can Malignant Potential Be Predicted? Urology 53: 679–683 [DOI] [PubMed] [Google Scholar]
- Jones R.M., Hill A.B. (1981) Severe Hypertension Associated with Pancuronium in a Patient with a Phaeochromocytoma. Can Anaesth Soc J 28: 394–396 [DOI] [PubMed] [Google Scholar]
- Joshua A.M., Ezzat S., Asa S.L., Evans A., Broom R., Freeman M., et al. (2009) Rationale and Evidence for Sunitinib in the Treatment of Malignant Paraganglioma/Pheochromocytoma. J Clin Endocrinol Metab 94: 5–9 [DOI] [PubMed] [Google Scholar]
- Joyce J.T., Roizen M.F., Eger E.I., 2nd (1983) Effect of Thiopental Induction on Sympathetic Activity. Anesthesiology 59: 19–22 [DOI] [PubMed] [Google Scholar]
- Kinney M.A., Warner M.E., Vanheerden J.A., Horlocker T.T., Young W.F., Jr., Schroeder D.R., et al. (2000) Perianesthetic Risks and Outcomes of Pheochromocytoma and Paraganglioma Resection. Anesth Analg 91: 1118–1123 [DOI] [PubMed] [Google Scholar]
- Kinney M.A., Narr B.J., Warner M.A. (2002) Perioperative Management of Pheochromocytoma. J Cardiothorac Vasc Anesth 16: 359–369 [DOI] [PubMed] [Google Scholar]
- Kocak S., Aydintug S., Canakci N. (2002) Alpha Blockade in Preoperative Preparation of Patients with Pheochromocytomas. Int Surg 87: 191–194 [PubMed] [Google Scholar]
- Korpershoek E., Favier J., Gaal J., Burnichon N., Van Gessel B., Oudijk L., et al. (2011) Sdha Immunohistochemistry Detects Germline Sdha Gene Mutations in Apparently Sporadic Paragangliomas and Pheochromocytomas. J Clin Endocrinol Metab 96: E1472–1476 [DOI] [PubMed] [Google Scholar]
- Kroiss A., Putzer D., Uprimny C., Decristoforo C., Gabriel M., Santner W., et al. (2011) Functional Imaging in Phaeochromocytoma and Neuroblastoma with 68ga-Dota-Tyr 3-Octreotide Positron Emission Tomography and 123i-Metaiodobenzylguanidine. Eur J Nucl Med Mol Imaging 38: 865–873 [DOI] [PubMed] [Google Scholar]
- Kunst H.P., Rutten M.H., De Monnink J.P., Hoefsloot L.H., Timmers H.J., Marres H.A., et al. (2011) Sdhaf2 (Pgl2-Sdh5) and Hereditary Head and Neck Paraganglioma. Clin Cancer Res 17: 247–254 [DOI] [PubMed] [Google Scholar]
- La Batide-Alanore A., Chatellier G., Plouin P.F. (2003) Diabetes as a Marker of Pheochromocytoma in Hypertensive Patients. J Hypertens 21: 1703–1707 [DOI] [PubMed] [Google Scholar]
- Lagerstedt S.A., O’kane D.J., Singh R.J. (2004) Measurement of Plasma Free Metanephrine and Normetanephrine by Liquid Chromatography-Tandem Mass Spectrometry for Diagnosis of Pheochromocytoma. Clin Chem 50: 603–611 [DOI] [PubMed] [Google Scholar]
- Lairmore T.C., Ball D.W., Baylin S.B., Wells S.A., Jr. (1993) Management of Pheochromocytomas in Patients with Multiple Endocrine Neoplasia Type 2 Syndromes. Ann Surg 217: 595-601; discussion 601–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.E., Curley S.A., Gagel R.F., Evans D.B., Hickey R.C. (1996) Cortical-Sparing Adrenalectomy for Patients with Bilateral Pheochromocytoma. Surgery 120: 1064-1070; discussion 1070–1061 [DOI] [PubMed] [Google Scholar]
- Lenders J.W., Pacak K., Walther M.M., Linehan W.M., Mannelli M., Friberg P., et al. (2002) Biochemical Diagnosis of Pheochromocytoma: Which Test Is Best? Jama 287: 1427–1434 [DOI] [PubMed] [Google Scholar]
- Lenders J.W., Eisenhofer G., Mannelli M., Pacak K. (2005) Phaeochromocytoma. Lancet 366: 665–675 [DOI] [PubMed] [Google Scholar]
- Lenders J. (2011) Review Topic: Endocrine Disorders in Pregnancy Phaeochromocytoma and Pregnancy:A Deceptive Connection. Eur J Endocrinol: in press [DOI] [PubMed] [Google Scholar]
- Lentschener C., Gaujoux S., Tesniere A., Dousset B. (2011) Point of Controversy: Perioperative Care of Patients Undergoing Pheochromocytoma Removal-Time for a Reappraisal? Eur J Endocrinol 165: 365–373 [DOI] [PubMed] [Google Scholar]
- Liao W.B., Liu C.F., Chiang C.W., Kung C.T., Lee C.W. (2000) Cardiovascular Manifestations of Pheochromocytoma. Am J Emerg Med 18: 622–625 [DOI] [PubMed] [Google Scholar]
- Lippmann M., Ford M., Lee C., Ginsburg R., Foran W., Raum W., et al. (1994) Use of Desflurane During Resection of Phaeochromocytoma. Br J Anaesth 72: 707–709 [DOI] [PubMed] [Google Scholar]
- Manger W.M., Gifford R.W. (1996) Clinical and Experimental Pheochromocytoma. Blackwell Science [Google Scholar]
- Manger W.M. (2006) An Overview of Pheochromocytoma: History, Current Concepts, Vagaries, and Diagnostic Challenges. Ann N Y Acad Sci 1073: 1–20 [DOI] [PubMed] [Google Scholar]
- Manger W.M. (2009) The Protean Manifestations of Pheochromocytoma. Horm Metab Res 41: 658–663 [DOI] [PubMed] [Google Scholar]
- Mannelli M., Bemporad D. (2002) Diagnosis and Management of Pheochromocytoma During Pregnancy. J Endocrinol Invest 25: 567–571 [DOI] [PubMed] [Google Scholar]
- Mannelli M. (2006) Management and Treatment of Pheochromocytomas and Paragangliomas. Ann N Y Acad Sci 1073: 405–416 [DOI] [PubMed] [Google Scholar]
- Mansmann G., Lau J., Balk E., Rothberg M., Miyachi Y., Bornstein S.R. (2004) The Clinically Inapparent Adrenal Mass: Update in Diagnosis and Management. Endocr Rev 25: 309–340 [DOI] [PubMed] [Google Scholar]
- Mantero F., Terzolo M., Arnaldi G., Osella G., Masini A.M., Ali A., et al. (2000) A Survey on Adrenal Incidentaloma in Italy. Study Group on Adrenal Tumors of the Italian Society of Endocrinology. J Clin Endocrinol Metab 85: 637–644 [DOI] [PubMed] [Google Scholar]
- Martiniova L., Lu J., Chiang J., Bernardo M., Lonser R., Zhuang Z., et al. (2011) Pharmacologic Modulation of Serine/Threonine Phosphorylation Highly Sensitizes Pheo in a Mpc Cell and Mouse Model to Conventional Chemotherapy. PLoS One 6: e14678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurea S., Cuocolo A., Reynolds J.C., Tumeh S.S., Begley M.G., Linehan W.M., et al. (1993) Iodine-131-Metaiodobenzylguanidine Scintigraphy in Preoperative and Postoperative Evaluation of Paragangliomas: Comparison with Ct and Mri. J Nucl Med 34: 173–179 [PubMed] [Google Scholar]
- Maurea S., Cuocolo A., Reynolds J.C., Neumann R.D., Salvatore M. (1996) Diagnostic Imaging in Patients with Paragangliomas. Computed Tomography, Magnetic Resonance and Mibg Scintigraphy Comparison. Q J Nucl Med 40: 365–371 [PubMed] [Google Scholar]
- Mcneil A.R., Blok B.H., Koelmeyer T.D., Burke M.P., Hilton J.M. (2000) Phaeochromocytomas Discovered During Coronial Autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med 30: 648–652 [DOI] [PubMed] [Google Scholar]
- Meeke R.I., O’keeffe J.D., Gaffney J.D. (1985) Phaeochromocytoma Removal and Postoperative Hypoglycaemia. Anaesthesia 40: 1093–1096 [DOI] [PubMed] [Google Scholar]
- Minno A.M., Bennett W.A., Kvale W.F. (1954) Pheochromocytoma; a Study of 15 Cases Diagnosed at Autopsy. N Engl J Med 251: 959–965 [DOI] [PubMed] [Google Scholar]
- Mishra A.K., Agarwal G., Kapoor A., Agarwal A., Bhatia E., Mishra S.K. (2000) Catecholamine Cardiomyopathy in Bilateral Malignant Pheochromocytoma: Successful Reversal after Surgery. Int J Cardiol 76: 89–90 [DOI] [PubMed] [Google Scholar]
- Miskulin J., Shulkin B.L., Doherty G.M., Sisson J.C., Burney R.E., Gauger P.G. (2003) Is Preoperative Iodine 123 Meta-Iodobenzylguanidine Scintigraphy Routinely Necessary before Initial Adrenalectomy for Pheochromocytoma? Surgery 134: 918-922; discussion 922–913 [DOI] [PubMed] [Google Scholar]
- Molitch M.E. (1992) Endocrine Emergencies in Pregnancy. Baillieres Clin Endocrinol Metab 6: 167–191 [DOI] [PubMed] [Google Scholar]
- Mullins F., O’shea P., Fitzgerald R., Tormey W. (2011) Enzyme-Linked Immunoassay for Plasma-Free Metanephrines in the Biochemical Diagnosis of Phaeochromocytoma in Adults Is Not Ideal. Clin Chem Lab Med: in press [DOI] [PubMed] [Google Scholar]
- Neumann H.P., Berger D.P., Sigmund G., Blum U., Schmidt D., Parmer R.J., et al. (1993) Pheochromocytomas, Multiple Endocrine Neoplasia Type 2, and Von Hippel-Lindau Disease. N Engl J Med 329: 1531–1538 [DOI] [PubMed] [Google Scholar]
- Neumann H.P., Reincke M., Bender B.U., Elsner R., Janetschek G. (1999) Preserved Adrenocortical Function after Laparoscopic Bilateral Adrenal Sparing Surgery for Hereditary Pheochromocytoma. J Clin Endocrinol Metab 84: 2608–2610 [DOI] [PubMed] [Google Scholar]
- Neumann H.P., Bausch B., Mcwhinney S.R., Bender B.U., Gimm O., Franke G., et al. (2002) Germ-Line Mutations in Nonsyndromic Pheochromocytoma. N Engl J Med 346: 1459–1466 [DOI] [PubMed] [Google Scholar]
- Neumann H.P., Pawlu C., Peczkowska M., Bausch B., Mcwhinney S.R., Muresan M., et al. (2004) Distinct Clinical Features of Paraganglioma Syndromes Associated with Sdhb and Sdhd Gene Mutations. Jama 292: 943–951 [DOI] [PubMed] [Google Scholar]
- Niemann S., Muller U. (2000) Mutations in Sdhc Cause Autosomal Dominant Paraganglioma, Type 3. Nat Genet 26: 268–270 [DOI] [PubMed] [Google Scholar]
- Nonaka K., Makuuchi H., Naruse Y., Kobayashi T., Goto M. (2000) Surgical Excision of Malignant Pheochromocytoma in the Left Atrium. Jpn J Thorac Cardiovasc Surg 48: 126–128 [DOI] [PubMed] [Google Scholar]
- Oishi S., Sato T. (1994) Pheochromocytoma in Pregnancy: A Review of the Japanese Literature. Endocr J 41: 219–225 [DOI] [PubMed] [Google Scholar]
- Ostenson C.G., Cattaneo A.G., Doxey J.C., Efendic S. (1989) Alpha-Adrenoceptors and Insulin Release from Pancreatic Islets of Normal and Diabetic Rats. Am J Physiol 257: E439–443 [DOI] [PubMed] [Google Scholar]
- Pacak K., Linehan W.M., Eisenhofer G., Walther M.M., Goldstein D.S. (2001a) Recent Advances in Genetics, Diagnosis, Localization, and Treatment of Pheochromocytoma. Ann Intern Med 134: 315–329 [DOI] [PubMed] [Google Scholar]
- Pacak K., Eisenhofer G., Carrasquillo J.A., Chen C.C., Li S.T., Goldstein D.S. (2001b) 6-[18f]Fluorodopamine Positron Emission Tomographic (Pet) Scanning for Diagnostic Localization of Pheochromocytoma. Hypertension 38: 6–8 [DOI] [PubMed] [Google Scholar]
- Pacak K., Eisenhofer G., Goldstein D.S. (2004) Functional Imaging of Endocrine Tumors: Role of Positron Emission Tomography. Endocr Rev 25: 568–580 [DOI] [PubMed] [Google Scholar]
- Pacak K., Eisenhofer G., Lenders J.W.M. (2007a) Pheochromocytoma: Diagnosis, Localization, and Treatment. Blackwell Pub [Google Scholar]
- Pacak K., Eisenhofer G., Ahlman H., Bornstein S.R., Gimenez-Roqueplo A.P., Grossman A.B., et al. (2007b) Pheochromocytoma: Recommendations for Clinical Practice from the First International Symposium October 2005 Nat Clin Pract Endocrinol Metab 3: 92–102 [DOI] [PubMed] [Google Scholar]
- Pacak K., Eisenhofer G. (2007c) An Assessment of Biochemical Tests for the Diagnosis of Pheochromocytoma. Nat Clin Pract Endocrinol Metab 3: 744–745 [DOI] [PubMed] [Google Scholar]
- Pacak K. (2007d) Preoperative Management of the Pheochromocytoma Patient. J Clin Endocrinol Metab 92: 4069–4079 [DOI] [PubMed] [Google Scholar]
- Peaston R.T., Lennard T.W., Lai L.C. (1996) Overnight Excretion of Urinary Catecholamines and Metabolites in the Detection of Pheochromocytoma. J Clin Endocrinol Metab 81: 1378–1384 [DOI] [PubMed] [Google Scholar]
- Perry R.R., Keiser H.R., Norton J.A., Wall R.T., Robertson C.N., Travis W., et al. (1990) Surgical Management of Pheochromocytoma with the Use of Metyrosine. Ann Surg 212: 621–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai D., Callen S. (2010) Pilot Quality Assurance Programme for Plasma Metanephrines. Ann Clin Biochem 47: 137–142 [DOI] [PubMed] [Google Scholar]
- Plouin P.F., Chatellier G., Fofol I., Corvol P. (1997) Tumor Recurrence and Hypertension Persistence after Successful Pheochromocytoma Operation. Hypertension 29: 1133–1139 [DOI] [PubMed] [Google Scholar]
- Plouin P.F., Duclos J.M., Soppelsa F., Boublil G., Chatellier G. (2001) Factors Associated with Perioperative Morbidity and Mortality in Patients with Pheochromocytoma: Analysis of 165 Operations at a Single Center. J Clin Endocrinol Metab 86: 1480–1486 [DOI] [PubMed] [Google Scholar]
- Prejbisz A., Lenders J.W., Eisenhofer G., Januszewicz A. (2011) Cardiovascular Manifestations of Phaeochromocytoma. J Hypertens 29: 2049–2060 [DOI] [PubMed] [Google Scholar]
- Proye C., Thevenin D., Cecat P., Petillot P., Carnaille B., Verin P., et al. (1989) Exclusive Use of Calcium Channel Blockers in Preoperative and Intraoperative Control of Pheochromocytomas: Hemodynamics and Free Catecholamine Assays in Ten Consecutive Patients. Surgery 106: 1149–1154 [PubMed] [Google Scholar]
- Prys-Roberts C. (2000) Phaeochromocytoma– Recent Progress in Its Management. Br J Anaesth 85: 44–57 [DOI] [PubMed] [Google Scholar]
- Prys-Roberts C., Farndon J.R. (2002) Efficacy and Safety of Doxazosin for Perioperative Management of Patients with Pheochromocytoma. World J Surg 26: 1037–1042 [DOI] [PubMed] [Google Scholar]
- Qin Y., Yao L., King E.E., Buddavarapu K., Lenci R.E., Chocron E.S., et al. (2010) Germline Mutations in Tmem127 Confer Susceptibility to Pheochromocytoma. Nat Genet 42: 229–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quezado Z.N., Keiser H.R., Parker M.M. (1992) Reversible Myocardial Depression after Massive Catecholamine Release from a Pheochromocytoma. Crit Care Med 20: 549–551 [DOI] [PubMed] [Google Scholar]
- Reach G., Thibonnier M., Chevillard C., Corvol P., Milliez P. (1980) Effect of Labetalol on Blood Pressure and Plasma Catecholamine Concentrations in Patients with Phaeochromocytoma. Br Med J 280: 1300–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose B., Matthay K.K., Price D., Huberty J., Klencke B., Norton J.A., et al. (2003) High-Dose 131i-Metaiodobenzylguanidine Therapy for 12 Patients with Malignant Pheochromocytoma. Cancer 98: 239–248 [DOI] [PubMed] [Google Scholar]
- Russell W.J., Metcalfe I.R., Tonkin A.L., Frewin D.B. (1998) The Preoperative Management of Phaeochromocytoma. Anaesth Intensive Care 26: 196–200 [DOI] [PubMed] [Google Scholar]
- Schenker J.G., Granat M. (1982) Phaeochromocytoma and Pregnancy–an Updated Appraisal. Aust N Z J Obstet Gynaecol 22: 1–10 [DOI] [PubMed] [Google Scholar]
- Schlegel G.C. (1960) [Recklinghausen’s Neurofibromatosis and Pheochromocytoma]. Schweiz Med Wochenschr 90: 31–39 [PubMed] [Google Scholar]
- Schlumberger M., Gicquel C., Lumbroso J., Tenenbaum F., Comoy E., Bosq J., et al. (1992) Malignant Pheochromocytoma: Clinical, Biological, Histologic and Therapeutic Data in a Series of 20 Patients with Distant Metastases. J Endocrinol Invest 15: 631–642 [DOI] [PubMed] [Google Scholar]
- Schurmeyer T.H., Engeroff B., Dralle H., Von Zur Muhlen A. (1997) Cardiological Effects of Catecholamine-Secreting Tumours. Eur J Clin Invest 27: 189–195 [DOI] [PubMed] [Google Scholar]
- Scott H.W., Jr., Halter S.A. (1984) Oncologic Aspects of Pheochromocytoma: The Importance of Follow-Up. Surgery 96: 1061–1066 [PubMed] [Google Scholar]
- Shapiro B., Copp J.E., Sisson J.C., Eyre P.L., Wallis J., Beierwaltes W.H. (1985) Iodine-131 Metaiodobenzylguanidine for the Locating of Suspected Pheochromocytoma: Experience in 400 Cases. J Nucl Med 26: 576–585 [PubMed] [Google Scholar]
- Shapiro B., Sisson J.C., Shulkin B.L., Gross M.D., Zempel S. (1995) The Current Status of Meta-Iodobenzylguanidine and Related Agents for the Diagnosis of Neuro-Endocrine Tumors. Q J Nucl Med 39: 3–8 [PubMed] [Google Scholar]
- Singh R.J., Eisenhofer G. (2007) High-Throughput, Automated, and Accurate Biochemical Screening for Pheochromocytoma: Are We There Yet? Clin Chem 53: 1565–1567 [DOI] [PubMed] [Google Scholar]
- Sisson J.C., Shapiro B., Shulkin B.L., Urba S., Zempel S., Spaulding S. (1999a) Treatment of Malignant Pheochromocytomas with 131-I Metaiodobenzylguanidine and Chemotherapy. Am J Clin Oncol 22: 364–370 [DOI] [PubMed] [Google Scholar]
- Sisson J.C., Shulkin B.L. (1999b) Nuclear Medicine Imaging of Pheochromocytoma and Neuroblastoma. Q J Nucl Med 43: 217–223 [PubMed] [Google Scholar]
- Sjoerdsma A., Engelman K., Spector S., Udenfriend S. (1965) Inhibition of Catecholamine Synthesis in Man with Alpha-Methyl-Tyrosine, an Inhibitor of Tyrosine Hydroxylase. Lancet 2: 1092–1094 [DOI] [PubMed] [Google Scholar]
- Sprung J., O’hara J.F., Jr., Gill I.S., Abdelmalak B., Sarnaik A., Bravo E.L. (2000) Anesthetic Aspects of Laparoscopic and Open Adrenalectomy for Pheochromocytoma. Urology 55: 339–343 [DOI] [PubMed] [Google Scholar]
- Stoner T.R., Jr., Urbach K.F. (1968) Cardiac Arrhythmias Associated with Succinylcholine in a Patient with Pheochromocytoma. Anesthesiology 29: 1228–1229 [DOI] [PubMed] [Google Scholar]
- Sullivan J.M., Solomon H.S. (1975) The Diagnosis of Pheochromocytoma. Overnight Excretion of Catecholamine Metabolites. Jama 231: 618–619 [PubMed] [Google Scholar]
- Sumikawa K., Amakata Y. (1977) The Pressor Effect of Droperidol on a Patient with Pheochromocytoma. Anesthesiology 46: 359–361 [DOI] [PubMed] [Google Scholar]
- Tauzin-Fin P., Hilbert G., Krol-Houdek M., Gosse P., Maurette P. (1999) Mydriasis and Acute Pulmonary Oedema Complicating Laparoscopic Removal of Phaechromocytoma. Anaesth Intensive Care 27: 646–649 [DOI] [PubMed] [Google Scholar]
- Tauzin-Fin P., Sesay M., Gosse P., Ballanger P. (2004) Effects of Perioperative Alpha1 Block on Haemodynamic Control During Laparoscopic Surgery for Phaeochromocytoma. Br J Anaesth 92: 512–517 [DOI] [PubMed] [Google Scholar]
- Thompson L.D. (2002) Pheochromocytoma of the Adrenal Gland Scaled Score (Pass) to Separate Benign from Malignant Neoplasms: A Clinicopathologic and Immunophenotypic Study of 100 Cases. Am J Surg Pathol 26: 551–566 [DOI] [PubMed] [Google Scholar]
- Timmers H.J., Kozupa A., Eisenhofer G., Raygada M., Adams K.T., Solis D., et al. (2007a) Clinical Presentations, Biochemical Phenotypes, and Genotype-Phenotype Correlations in Patients with Succinate Dehydrogenase Subunit B-Associated Pheochromocytomas and Paragangliomas. J Clin Endocrinol Metab 92: 779–786 [DOI] [PubMed] [Google Scholar]
- Timmers H.J., Kozupa A., Chen C.C., Carrasquillo J.A., Ling A., Eisenhofer G., et al. (2007b) Superiority of Fluorodeoxyglucose Positron Emission Tomography to Other Functional Imaging Techniques in the Evaluation of Metastatic Sdhb-Associated Pheochromocytoma and Paraganglioma. J Clin Oncol 25: 2262–2269 [DOI] [PubMed] [Google Scholar]
- Timmers H.J., Pacak K., Huynh T.T., Abu-Asab M., Tsokos M., Merino M.J., et al. (2008) Biochemically Silent Abdominal Paragangliomas in Patients with Mutations in the Succinate Dehydrogenase Subunit B Gene. J Clin Endocrinol Metab 93: 4826–4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmers H.J., Chen C.C., Carrasquillo J.A., Whatley M., Ling A., Havekes B., et al. (2009) Comparison of 18f-Fluoro-L-Dopa, 18f-Fluoro-Deoxyglucose, and 18f-Fluorodopamine Pet and 123i-Mibg Scintigraphy in the Localization of Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab 94: 4757–4767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischler A.S. (2008) Pheochromocytoma and Extra-Adrenal Paraganglioma: Updates. Arch Pathol Lab Med 132: 1272–1284 [DOI] [PubMed] [Google Scholar]
- Tokioka H., Takahashi T., Kosogabe Y., Ohta Y., Kosaka F. (1988) Use of Diltiazem to Control Circulatory Fluctuations During Resection of a Phaeochromocytoma. Br J Anaesth 60: 582–587 [DOI] [PubMed] [Google Scholar]
- Trampal C., Engler H., Juhlin C., Bergstrom M., Langstrom B. (2004) Pheochromocytomas: Detection with 11c Hydroxyephedrine Pet. Radiology 230: 423–428 [DOI] [PubMed] [Google Scholar]
- Ulchaker J.C., Goldfarb D.A., Bravo E.L., Novick A.C. (1999) Successful Outcomes in Pheochromocytoma Surgery in the Modern Era. J Urol 161: 764–767 [PubMed] [Google Scholar]
- Van Der Horst-Schrivers A.N., Kerstens M.N., Wolffenbuttel B.H. (2006) Preoperative Pharmacological Management of Phaeochromocytoma. Neth J Med 64: 290–295 [PubMed] [Google Scholar]
- Van Nederveen F.H., Gaal J., Favier J., Korpershoek E., Oldenburg R.A., De Bruyn E.M., et al. (2009) An Immunohistochemical Procedure to Detect Patients with Paraganglioma and Phaeochromocytoma with Germline Sdhb, Sdhc, or Sdhd Gene Mutations: A Retrospective and Prospective Analysis. Lancet Oncol 10: 764–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Stratum M., Levarlet M., Lambilliotte J.P., Lignian H., De Rood M. (1983) Use of Labetalol During Anesthesia for Pheochromocytoma Removal. Acta Anaesthesiol Belg 34: 233–240 [PubMed] [Google Scholar]
- Vargas H.I., Kavoussi L.R., Bartlett D.L., Wagner J.R., Venzon D.J., Fraker D.L., et al. (1997) Laparoscopic Adrenalectomy: A New Standard of Care. Urology 49: 673–678 [DOI] [PubMed] [Google Scholar]
- Von Euler U.S., Lishajko F. (1973) Effects of Mg2+ and Ca2+ on Noradrenaline Release and Uptake in Adrenergic Nerve Granules in Differential Media. Acta Physiol Scand 89: 415–422 [DOI] [PubMed] [Google Scholar]
- Walther M.M., Herring J., Choyke P.L., Linehan W.M. (2000) Laparoscopic Partial Adrenalectomy in Patients with Hereditary Forms of Pheochromocytoma. J Urol 164: 14–17 [PubMed] [Google Scholar]
- Walz M.K., Peitgen K., Neumann H.P., Janssen O.E., Philipp T., Mann K. (2002) Endoscopic Treatment of Solitary, Bilateral, Multiple, and Recurrent Pheochromocytomas and Paragangliomas. World J Surg 26: 1005–1012 [DOI] [PubMed] [Google Scholar]
- Walz M.K., Groeben H., Alesina P.F. (2010) Single-Access Retroperitoneoscopic Adrenalectomy (Sara) Versus Conventional Retroperitoneoscopic Adrenalectomy (Cora): A Case-Control Study. World J Surg 34: 1386–1390 [DOI] [PubMed] [Google Scholar]
- Witteles R.M., Kaplan E.L., Roizen M.F. (2000) Safe and Cost-Effective Preoperative Preparation of Patients with Pheochromocytoma. Anesth Analg 91: 302–304 [DOI] [PubMed] [Google Scholar]
- Yabe R., Suenaga K., Niimura S., Itoh N., Tani M., Kunii N., et al. (1987) Treatment of Pheochromocytoma with Dilevalol. J Med 18: 147–152 [PubMed] [Google Scholar]
- Yao L., Schiavi F., Cascon A., Qin Y., Inglada-Perez L., King E.E., et al. (2010) Spectrum and Prevalence of Fp/Tmem127 Gene Mutations in Pheochromocytomas and Paragangliomas. Jama 304: 2611–2619 [DOI] [PubMed] [Google Scholar]
- Yip L., Lee J.E., Shapiro S.E., Waguespack S.G., Sherman S.I., Hoff A.O., et al. (2004) Surgical Management of Hereditary Pheochromocytoma. J Am Coll Surg 198: 525–534; discussion 534–525 [DOI] [PubMed] [Google Scholar]
- Yoshida S., Hatori M., Noshiro T., Kimura N., Kokubun S. (2001) Twenty-Six-Years’ Survival with Multiple Bone Metastasis of Malignant Pheochromocytoma. Arch Orthop Trauma Surg 121: 598–600 [DOI] [PubMed] [Google Scholar]
- Young W.F., Jr. (1997) Pheochromocytoma: Issues in Diagnosis & Treatment. Compr Ther 23: 319–326 [PubMed] [Google Scholar]
- Yumi H. (2008) Guidelines for Diagnosis, Treatment, and Use of Laparoscopy for Surgical Problems During Pregnancy: This Statement Was Reviewed and Approved by the Board of Governors of the Society of American Gastrointestinal and Endoscopic Surgeons (Sages), September 2007. It Was Prepared by the Sages Guidelines Committee. Surg Endosc 22: 849–861 [DOI] [PubMed] [Google Scholar]
- Zelinka T., Timmers H.J., Kozupa A., Chen C.C., Carrasquillo J.A., Reynolds J.C., et al. (2008) Role of Positron Emission Tomography and Bone Scintigraphy in the Evaluation of Bone Involvement in Metastatic Pheochromocytoma and Paraganglioma: Specific Implications for Succinate Dehydrogenase Enzyme Subunit B Gene Mutations. Endocr Relat Cancer 15: 311–323 [DOI] [PubMed] [Google Scholar]