

Abstract
Purpose
Many tumors exhibit defective cell cycle checkpoint control and increased replicative stress. CHK1 is critically involved in the DNA damage response and maintenance of replication fork stability. We have therefore discovered a novel, potent, highly selective, orally active, ATP competitive CHK1 inhibitor, CCT244747, and present its preclinical pharmacology and therapeutic activity.
Experimental design
Cellular CHK1 activity was assessed using an ELISA assay and cytotoxicity a SRB assay. Biomarker modulation was measured using immunoblotting and cell cycle effects by flow cytometry. Single agent, oral CCT244747 antitumor activity was evaluated in a MYCN-driven transgenic mouse model of neuroblastoma by MRI and in genotoxic combinations in human tumor xenografts by growth delay.
Results
CCT244747 inhibited cellular CHK1 activity (IC50 29-170nM), significantly enhanced the cytotoxicity of several anticancer drugs and abrogated drug-induced S and G2 arrest in multiple tumor cell lines. Biomarkers of CHK1 (pS296 CHK1) activity and cell cycle inactivity (pY15 CDK1) were induced by genotoxics and inhibited by CCT244747 both in vitro and in vivo, producing enhanced DNA damage and apoptosis. Active tumor concentrations of CCT244747 were obtained following oral administration. The antitumor activity of both gemcitabine and irinotecan were significantly enhanced by CCT244747 in several human tumor xenografts, giving concomitant biomarker modulation indicative of CHK1 inhibition. CCT244747 also showed marked antitumor activity as a single agent in a MYCN-driven neuroblastoma.
Conclusion
CCT244747 represents the first structural disclosure of a highly selective, orally active CHK1 inhibitor and warrants further evaluation alone or combined with genotoxic anticancer therapies.
Keywords: CHK1, CCT244747, pharmacology, biomarkers, neuroblastoma
Introduction
The maintenance of genomic integrity is critical for cell survival and proliferation [1]. DNA damage can arise from either intrinsic processes, such as oxidative stress or replication errors, or exogenous sources such as environmental mutagens [1, 2]. Cells respond to genomic stress by activating a number of cell cycle checkpoints as part of the DNA damage response (DDR) to facilitate cell cycle arrest, DNA repair, or apoptosis [3, 4]. The DDR effector kinase CHK1 has been shown to activate the G1 and G2 checkpoints by modulating the expression and function of CDC25 A and C, respectively [5, 6]. In addition, CHK1 has an important role in the S-phase checkpoint where it stabilizes and preserves replication fork complexes following replicative stress, preventing catastrophic replication fork collapse [5, 7]. CHK1 is also involved in homologous recombination repair of DNA through activation of Rad51 [8] and mitosis through direct phosphorylation of Aurora B [9] .
Many tumor cells exhibit an incomplete DDR and harbor defects in genes controlling cell cycle checkpoints and DNA repair [10]. P53 is frequently deregulated in human cancers leading to compromised G1/S arrest and DNA repair [11]. Consequently it has been argued that CHK1 inhibitors may selectively enhance the activity of genotoxic agents in tumors by abrogating the S and G2 checkpoints while normal cells will be rescued due to their competent DDR [5, 12, 13] . Studies using CHK1 RNAi and selective CHK1 inhibitors have provided evidence that this is the case for certain chemotherapeutic agents such as topoisomerase 1 inhibitors and antimetabolites [14-17]. On the basis of these data, a number of CHK1 inhibitors have been developed and are currently undergoing clinical evaluation in combination with specific genotoxic drugs [5, 12, 13, 17].
Accumulating evidence indicates that the activation of a number of oncogenes such as MYC and RAS, can give rise to enhanced replicative stress in tumors due to increased initiation and firing of replication origins in the DNA [1, 18, 19]. In view of the essential role of CHK1 in the preservation of replication fork stability, it has been suggested that CHK1 inhibitors may have single agent activity in defined tumor backgrounds exhibiting high levels of replicative stress [4, 20, 21]. This concept has been consolidated using CHK1 RNAi and several CHK1 inhibitors in tumor types as diverse as AML, C-MYC driven lymphomas, MYCN-regulated neuroblastomas and various solid tumors [17, 20, 22-24]. Consequently CHK1 inhibitors are increasingly attractive potential anticancer agents [5, 12, 13, 25].
In this report we present the preclinical pharmacology, therapeutic activity and pharmacodynamic (PD) biomarker profile of the novel, selective and orally bioavailable CHK1 inhibitor CCT244747 in combination with various genotoxic agents. Moreover we provide clear evidence that CCT244747 has potent single agent antitumor activity in a spontaneous, MYCN-driven transgenic mouse model of neuroblastoma. These data represent the first disclosure, with chemical structure, of an orally active, potent and highly selective CHK1 inhibitor and support the development of this class of compounds as therapeutic agents either alone or in combination with genotoxic anticancer drugs.
Materials and Methods
Drugs and treatments
CCT244747 was synthesized as previously described [26]. Gemcitabine and irinotecan were obtained from Eli Lilly and Pfizer respectively and used as clinical formulations. SN38, the active metabolite of irinotecan, was purchased from LKT laboratories for in vitro studies. All other compounds were purchased from Sigma (Poole, Dorset, UK).
Tissue Culture
The p53 mutant colon tumor cell lines SW620 (p53−/−, KRAS−/−, APC−/−, SMAD4−/− and MAP2K4−/−) and HT29 (p53−/−, APC+/−, PIK3CA +/−, BRAF+/− and SMAD4−/−) , the p53 mutant pancreatic cancer cell line MiaPaCa-2 (p53−/−, KRAS+/−, KDM6A−/−, CDKN2a (p14)−/− and CDKN2A−/−) and the p53 mutant non-small cell lung cancer Calu6 cells (p53−/− and KRAS+/−) were purchased from the American Type Culture Collection (Lot numbers 4487729, 3924081, 57866607 and 58683029, respectively and Sanger Centre data http://www.sanger.ac.uk/genetics/CGP/cosmic). Cells were grown in DMEM containing 10% fetal calf serum and 2mM glutamine under a humidified atmosphere of 5% CO2:air at 37°C. Cells were cultured for <6months before renewal from early passage, frozen stocks. All cell lines were shown to be mycoplasma free using a PCR based assay (VenorGeM, Minerva Biolabs).
In vitro kinase assays
In vitro kinase assays were carried out commercially at 1 and 10μM CCT244747, with ATP concentrations corresponding to the kinase Km against 120-140 human kinases (MRC phosphorylation unit, Dundee, UK). Additional IC50 determinations for FLT3 and CHK2 were carried out using a commercial assay (Z’-Lyte, Invitrogen, UK) or in-house with recombinant human CHK1, on an Ezreader II (Caliper Life Sciences, UK) or CDK1in a DELFIA assay (Perkin-Elmer, Zavenetm, Belgium).
In vitro cytotoxicity, potentiation and G2 checkpoint abrogation assays
All assays were performed as described previously [27]. Cytotoxicity was determined as the growth inhibition 50% (GI50) using a standard 96h incubation (i.e. four cell doublings) and sulphorhodamine B (96h SRB) staining. Potentiation studies were carried out using CCT244747 alone to determine the GI50 of CCT244747 or in combination with a fixed GI50 concentration of the genotoxic to determine the combination GI50 of CCT244747. This approach was validated using gemcitabine in SW620 and shown to be a simple, robust and sensitive assay of CHK1 inhibitor potentiation activity [27, 28] (see Supplementary Fig 1). For contact time studies, cells were treated with a fixed GI50 concentration of gemcitabine combined with different concentrations of CCT244747 and at appropriate times medium was removed and replaced with either fresh medium or medium containing gemcitabine alone and incubated up to 96h. The ability of CCT244747 to enhance drug-induced cell killing was expressed as a potentiation index (PI) equal to the ratio of the GI50 of CCT244747 alone : GI50 for CCT244747 in combination with the genotoxic agent (combination GI50). PI values > 1.0 indicate potentiation of the genotoxic agent by CCT244747. Intracellular inhibition of CHK1 function was determined using a cell-based ELISA assay for G2 checkpoint abrogation (mitosis induction assay, MIA) [27]. The IC50 for G2 checkpoint abrogation (MIA) was determined using nocodazole as a positive control. The activity index (AI) was used as a measure of the compound’s ability to induce mitosis relative to its toxicity (i.e., ratio of MIA IC50 : 96h SRB GI50).
Western Blotting
Immunoblotting was carried out using standard techniques [27]. Briefly, cells or tumor homogenates were lysed in ice cold lysis buffer and protein concentrations determined. Aliquots (50μg) of protein were denatured in Laemmli loading buffer and separated on precast 10 or 16% TRIS-Glycine gels (Novex-Invitrogen). Proteins were transferred to PVDF membranes, which were blocked and probed with 1° antibodies to pS296, pS317, pS345 and total CHK1; pY15 and total CDK1 and cleaved PARP (CST); pS139 H2AX and total H2AX (Upstate) and GAPDH (Chemicon) and detected using appropriate HRPO labelled secondary antibodies. Proteins were visualized using enhanced chemiluminescence (Pierce, Thermo-Fisher) on Hyperfilm (GE Healthcare). Protein bands were quantified by densitometry using ImageQuant 5 software.
Cell Cycle Analysis
Cell cycle distribution was assessed using propidium iodide (PI) or bromodeoxyuridine (BrdU) and PI staining as previously described [27].
Pharmacokinetic and Efficacy Studies
Compound tolerability and pharmacokinetic studies were carried out in BALB/c mice (Charles River). Human tumor xenografts were established in CRTac:Ncr-Fox1(nu) athymic mice and treated as previously described [27]. Hemizygotic animals transgenic for TH-MYCN and predisposed to the spontaneous generation of aggressive neuroblastomas through over expression of human MYCN were bred and utilized for in vivo studies as previously described [29, 30]. Briefly, hemizygous TH-MYCN animals carried a single copy of the transgene construct consisting of a human MYCN cDNA expressed under the control of the rat tyrosine hydroxylase promoter and flanked on the 3′ end by a rabbit beta-globin enhancer element. Tumors commonly arose in the peri-adrenal and abdominal paraspinal sympathetic ganglia. Mice were monitored for tumor development and assigned on a rolling trial basis to CCT244747 or vehicle treatment when significant loco-regional tumor was detected by palpation (at least 8 × 8 × 8 mm size at approximately 65 days of age).
For oral administration, CCT244747 was given in 5% DMSO, 20% Tween 20, 65% PEG400, 10% water at 0.01ml/g at the doses indicated. Mice bearing MYCN neuroblastoma tumors were administered CCT244747 as a single agent, bolus dose (100mg/kg p.o.) on seven consecutive days. Neuroblastoma tumor size was assessed by MRI and final tumor weights were measured at necropsy 24h after the last dose. In some animals, anti-tumor activity was determined by measuring tumor volume using MRI prior to and following treatment. This was performed on a 7T Bruker horizontal bore micro imaging system (Bruker Instruments, Ettlingen, Germany) using a 3 cm birdcage coil. Anatomical T2-weighted coronal images were acquired from twenty contiguous 1 mm thick slices through the mouse abdomen, from which tumor volumes were determined using segmentation from regions of interest drawn on each tumor-containing slice.
For combination studies, CCT244747 was administered p.o. to mice bearing established HT29 tumors 24h and 48h after cytotoxic drug administration. In the first study, gemcitabine was administered on days 0, 7 and 14 at 100mg/kg i.v. and CCT244747 at 75mg/kg p.o. on days 1, 2, 8, 9, 15 and 16. In a second study, irinotecan was administered on days 0, 4 and 8 at 25mg/kg i.p. and CCT244747 at 150mg/kg p.o. on days 1, 2, 5, 6, 9, 10. In the Calu6 xenograft model, gemcitabine was administered on days 0, 4 and 8 at 100mg/kg i.v. and CCT244747 at 75mg/kg p.o. on days 1, 2, 5, 6, 9 and 10. In the SW620 model, gemcitabine was administered on days 0, 4 and 8 at 100mg/kg i.v. and CCT244747 at 75mg/kg p.o. on days 1, 2, 5, 6, 9 and 10. The SN38 and gemcitabine doses employed were the highest, minimally active concentrations in each tumor model to facilitate further potentiation with CCT244747. Doses of CCT244747 were the highest non-toxic concentrations achievable on each schedule. Initial treatment groups consisted of 6 mice and animals were inspected daily and tumor size and mouse body weight measured every 2 or 3 days. Tumor volume was determined from two orthogonal measurements and growth delay was calculated from individual growth curves at 300% tumor volume, a value that gave linear re-growth and through which all tumors grew. All mice were treated in accordance with local and national animal welfare guidelines [31].
Pharmacokinetic analyses
Drugs were extracted from plasma and tissue homogenates using methanol containing internal standard. CCT244747 concentrations were determined by LC/MS/MS with an Agilent Infinity 1290 binary pump and a 6410 triple quadrupole mass spectrometer. Pharmacokinetic parameters were calculated using non-compartmental analysis on Pharsight WinNonLin software version 5.2. Protein binding was carried out using ultra filtration (10μM CCT244747 × 1h at 37°C). In-vitro metabolism studies of 10 μM compound were performed using male CD1 mouse microsomes (Tebu-bio, Peterborough, U.K.) for 30 minutes at 37°C with 1 mg/ml protein in the presence of NADPH and UDPGA with LC/MS/MS analysis as above.
Statistics
Statistical significance (*, P<0.05; **, P<0.01; *** P<0.001) was determined using an unpaired, one-tailed, t-test or one-way ANOVA and either Tukey’s or Dunnett’s test as appropriate, with GraphPad Prism 5 software.
Results
Chemical structure and in vitro kinase activity of CCT244747
The chemical structure of CCT244747 is shown in Figure 1A and a model of this compound bound into the ATP pocket of human CHK1 in Supplementary Figure 2. This model was derived using constrained scaffold docking of CCT244747 into the SAR-020106 x-ray crystal structure (PDB 2ym8) [32] . It suggests that CCT244747 can retain key interactions in the ATP-binding site through hydrogen-bonding to Glu85 and Cys87 in the hinge region, the hydrogen-bonding of the nitrile to Lys38 and the dimethylamine situated in the ribose pocket, as noted for other 2-aminopyrazine-5-carbonitrile CHK1 inhibitors [32]
Figure 1.
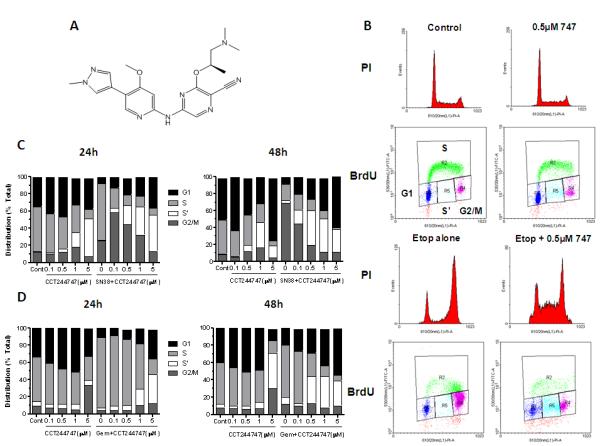
Structure and effects of CCT244747 on drug induced cell cycle arrest in human colon cancer cell lines. A, Structure of CCT244747. B, Effects of CCT244747 exposure (0.5μM × 23h) on etoposide induced (25μM × 1h) cell cycle arrest in HT29 tumor cells measured 23h following cytotoxic treatment. Histograms: cell cycle distribution was assessed by propidium iodide DNA staining; Dot blots: DNA synthesis and distribution were measured by BrdU incorporation and DNA staining with PI. Cell cycle distribution (G1, S, S’ and G2/M) was quantified using BrdU staining as shown and described in Materials and Methods. C, Quantification of the effects of different concentrations of CCT244747 on SN38 (20nM) induced cell cycle arrest in HT29 cells measured following 24h and 48h exposure. D, Comparable data for the effects of different concentrations of CCT244747 on gemcitabine (10nM) induced cell cycle arrest in SW620 cells following 24 and 48h treatments. Similar results were obtained in repeat experiments.
In vitro kinase profiling showed that CCT244747 was a potent and highly selective inhibitor of recombinant human CHK1 (Supplementary Table 1) with an IC50 of 8nM. There was 75-fold selectivity against FLT3 (IC50 600nM) and more than 1,000-fold selectivity against the functionally important kinases CHK2 and CDK1 (IC50 > 10,000 nM). Kinase profiling at 1μM CCT244747 showed some activity (>50% inhibition) against 8 kinases from a panel of 140, including IRAK 1, TrKA, RSK, AMPK, NUAK1,AMPK, Aurora B and MAP4K with marked inhibition of CHK1. At 10μM CCT244747 inhibited by ≥80% only 9 out of a total of 121 kinases (including 5 from the 8 listed above). These data confirm that CCT244747 is a potent (1μM) and selective (10μM) inhibitor of CHK1 in vitro.
CCT244747 overcomes genotoxic-induced S and G2 cell cycle arrest
CHK1 has been implicated in stabilizing replication forks and maintaining G2 checkpoint integrity. As a part of our studies we therefore explored the capacity of the CHK1 inhibitor CCT244747 to abrogate drug-induced cell cycle checkpoints. The ability of CCT244747 to abolish a G2 arrest induced by etoposide in HT29 colon carcinoma cells was initially investigated as this forms the basis of the cellular CHK1 assay (see later). Figure 1B shows that there were minimal effects of CCT244747 alone on cell cycle distribution at 0.5μM × 24h. In contrast, acute etoposide exposure (25μM × 1h) caused a marked G2/M arrest at 24h (70.5% in G2/M). The addition of 0.5μM CCT244747 for 23h following acute etoposide treatment caused a substantial loss of G2/M cells (30%) with a corresponding increase in G1 (9.6%). There was a marked increase in cells trapped in S-phase but not actively incorporating BrdU (S’ population increased from 1.2 to 17.7%). These results confirm that CCT244747 can abolish an etoposide-induced G2/M checkpoint. Other cell cycle studies were carried out using SN38 and CCT244747 in HT29 cells or gemcitabine and CCT244747 in SW620 cells (Figure 1C and D, respectively). CCT244747 alone had minimal effects on the cell cycle of HT29 cells up to a concentration of 0.5μM × 24h, with a progressive loss of S and increase in S’-phase cells possibly indicative of replicative crisis and increased cell killing of S-phase cells at higher concentrations (Figure 1C). SN38 alone (20nM × 24h) induced a marked S-phase and G2/M arrest. The addition of CCT244747 reduced this S-phase arrest by 42.3% at 0.1μM with a further 33.8% of cells accumulating in G2/M. Increasing concentrations of CCT244747 caused progressive loss of the G2/M population and a marked increase in S’-phase cells from 5 to 42.1%, potentially indicative of a cytotoxic event in S-phase. Similar results were obtained at 48h. In contrast, CCT244747 had minimal effects alone up to 1μM × 24h in the SW620 colon carcinoma cell line (Figure 1D). Gemcitabine (10nM × 24h) induced a marked S-phase arrest and the addition of CCT244747 overcame this arrest with a 28.8% reduction in S-phase, a small increase in G1 (4.4%) but a marked increase in S’-phase (19%). At 48h similar cell cycle effects were observed but the S’-phase population was more extensively induced by the combination, consistent with a delayed cytotoxic event. These observations provide clear evidence that CCT244747 can abolish both SN38 and gemcitabine-induced S and G2/M-phase cell cycle arrests, although with subtle differences in the kinetics.
Measurement of cellular CHK1 inhibition and potentiation of anticancer drug cytotoxicity by CCT244747
An ELISA-based cellular assay was used to measure functional CHK1 inhibition [27] giving IC50s ranging from 29nM to 170nM for cellular G2 checkpoint abrogation (MIA, mitosis induction assay) in the four cell lines employed (Table 1). The corresponding GI50 for this compound was between 0.33 and 3μM. The potent inhibition of cellular CHK1 function versus low cellular cytotoxicity resulted in activity indices (AI; ratio of G2 checkpoint abrogation IC50 : SRB GI50) of between 3.5 and 23 consistent with good target selectivity. Table 1 summarizes these data and the ability of CCT244747 to enhance the cytotoxicity of numerous anticancer treatments in several different human tumor cell lines. With the exception of CDDP in HT29, CCT244747 significantly enhanced the cytotoxicity of all the treatments tested in all four cell lines, with more marked potentiation occurring with S-phase active drugs such as the topoisomerase I inhibitor SN38 (the active metabolite of irinotecan), the antimetabolite gemcitabine (Supplementary Figure 1) and the thymidylate synthase inhibitor 5-fluoro-2′-deoxyuridine (5FdU). Interestingly, there was minimal selective potentiation by CCT244747 of SN38 or gemcitabine cytotoxicity in a HPV16E6 transfected A549 or p53 recombinant knockout HCT116 cell line relative to the wild type p53 controls (data not shown). On the basis of the results shown here and other published data on active genotoxic combinations with CHK1 inhibitors [14-17, 27] we evaluated the ability of CCT244747 to enhance gemcitabine and irinotecan antitumor activity in vivo.
Table 1.
Summary of cellular CHK1 cytotoxicity and therapeutic activity of CCT244747 in combination with various genotoxic anticancer agents in human tumor cell lines.
| Cells | MIA (μM) | GI50 (μM) | Genotoxic | Combination GI50 (μM) | Potentiation Index (PI) |
|---|---|---|---|---|---|
| HT29 | 0.029±0.0031 (n=4) | 0.62±0.23 (n=3) | SN38 | 0.32±0.022 (n=4) | 1.9±0.14*** (n=4) |
| Gem | 0.074±0.014 (n=4) | 8.5±1.6** (n=4) | |||
| Etop | 0.36±0.13 (n=4) | 1.9±0.63* (n=4) | |||
| CDDP | 0.34±0.067 (n=3) | 1.8±0.32 (n=3) | |||
| 5FU | 0.29±0.029 (n=3) | 2.2±0.2** (n=3) | |||
| 5FdU | 0.065±0.0078 (n=3) | 9.6±1.2** (n=3) | |||
| IR −1h | 0.20±0.021 (n=4) | 3.9±0.32* (n=4) | |||
| IR +1h | 0.22±0.059 (n=4) | 3.0±0.67* (n=4) | |||
| SW620 | 0.17±0.0058 (n=3) | 3.0±1.2 (n=4) | SN38 | 1.2±0.24 (n=3) | 2.6±0.6* (n=3) |
| Gem | 0.26±0.05 (n=5) | 12.2±2.7*** (n=5) | |||
| Etop | 0.68±0.25 (n=3) | 4.9±2.2* (n=3) | |||
| CDDP | 0.73±0.16 (n=3) | 4.2±0.87* (n=3) | |||
| 5FU | 0.91±0.20 (n=3) | 4.9±0.8* (n=3) | |||
| 5FdU | 0.068±0.019 (n=3) | 47±14* (n=3) | |||
| IR −1h | 0.71±0.19 (n=4) | 4.5±1.2* (n=4) | |||
| IR +1h | 0.66±0.18 (n=4) | 4.9±1.6** (n=4) | |||
| MiaPaCa-2 | 0.044±0.010 (n=3) | 1.0±0.29 (n=3) | SN38 | 0.21±0.037 (n=5) | 5.0±0.82*** (n=5) |
| Gem | 0.064±0.023 (n=5) | 16±6.3** (n=5) | |||
| Calu6 | 0.094±0.066 (n=3) | 0.33±0.051 (n=3) | SN38 | 0.24±0.040 (n=3) | 1.4±0.24* (n=3) |
| Gem | 0.061±0.010 (n=3) | 5.6±0.96*** (n=3) |
G2 checkpoint abrogation (MIA, IC50), cytotoxicity (SRB GI50) and activity index (AI) values were determined as described in Material and Methods. Potentiation index (PI) is the ratio of SRB GI50 / Combination GI50, where combination GI50 is the concentration of CCT244747 that causes 50% inhibition of cell growth in combination with a fixed (GI50 ) concentration of the cytotoxic agent . PI values > 1 indicate potentiation of the cytotoxic agent. Values are mean±SD of n independent determinations. Abbreviations: 5FdU , 5-Fluoro-2′-deoxyyuridine and IR , ionizing radiation . Statistical significance: P<0.05;
P<0.01;
P<0.001
CCT244747 inhibits CHK1 phosphorylation and cell cycle biomarkers in vitro
To ensure that target inhibition has occurred in cells it is important to monitor appropriate biomarker readouts such as the phosphorylation status of kinase specific substrates. Figure 2A shows that SN38 induced S296 CHK1 autophosphorylation in HT29 cells after 24h and this was inhibited by combination with CCT244747 at ≥0.05μM with complete loss of signal at ≥0.5μM. Phosphorylation on S317 and S345 was markedly increased by SN38 exposure and these increases were reversed at ≥0.1μM. However, neither phosphorylation was completely abolished by up to 5μM CCT244747. Phosphorylation of the cell cycle marker pY15 CDK1 was induced by SN38 treatment and this was substantially inhibited by combination with ≥0.5μM CCT244747. The inhibition of CHK1 autophosphorylation and CDK1 phosphorylation coincided with the induction of γH2AX (pS139 H2AX) and PARP cleavage, markers of DNA damage and apoptosis, respectively. Similar results were obtained with gemcitabine and CCT244747 combinations in SW620 cells (Figure 2B), although SW620 cells may be slightly less sensitive than HT29 to CHK1 inhibition. Additional studies confirmed that a non-cytotoxic concentration of 0.3μM CCT244747 was able to inhibit CHK1 activity in combination with either SN38 or gemcitabine and this was associated with an increase in DNA damage and apoptosis (Supplementary Figure 3). Consequently, these results clearly demonstrate that CCT244747 can inhibit SN38 and gemcitabine-induced CHK1 activity in tumor cells and this correlates with abrogation of cell cycle arrest, induction of DNA damage and apoptosis.
Figure 2.
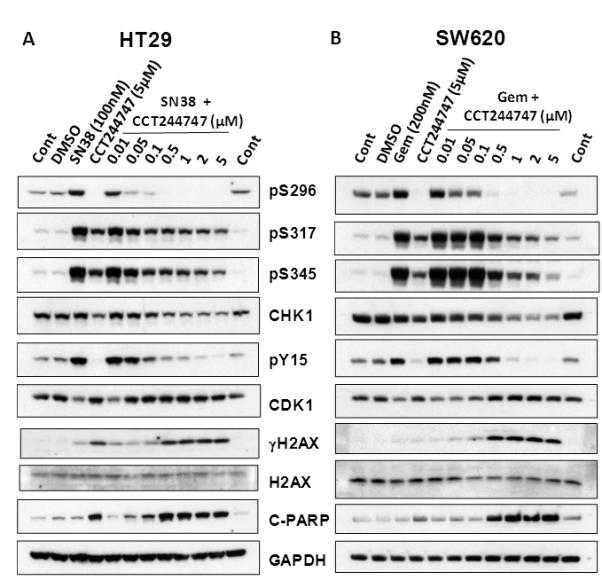
Characterization of the effects of CCT244747 on drug-induced CHK1 and cell cycle biomarker changes in HT29 and SW620 colon cancer cell lines. A, HT29 cells were treated with SN38 (100nM) or CCT244747 alone or with SN38 in combination with increasing concentrations of CCT244747 for 24h. B, SW620 cells were treated with either gemcitabine (200nM) or CCT2447447 alone or gemcitabine in combination with increasing concentrations of CCT244747 for 24h. Cells were pre-treated with CCT244747 1h prior to cytotoxic exposure. Protein expression was assessed by western blotting (50μg sample per lane) as described in Materials and Methods. CHK1 autophosphorylation on S296 and phosphorylation on S317 and S345 were used as biomarkers of CHK1 activity, pY15CDK1 as a biomarker of cell cycle inhibition with pS139 (γ) H2AX and cleaved-PARP (C-PARP) as biomarkers of DNA damage and apoptosis, respectively. GAPDH was used as a loading control. Similar results were obtained in repeat experiments with both HT29 and SW620 tumor cell lines.
Schedule dependency of CHK1 inhibitor enhanced gemcitabine cytotoxicity in vitro
In order to investigate the mechanism of action of CHK1 inhibition and optimize in vivo combinations, a series of experiments was undertaken to determine the most efficacious scheduling of two structurally distinct CHK1 inhibitors, SAR-020106 [27] and CCT244747, on gemcitabine cytotoxicity in SW620 cells. This cell line and the drug combinations were selected due to their large potentiation indices (see Table 1 and [27]). Supplementary Figure 4A shows that SAR-020106 caused maximum potentiation of gemcitabine cytotoxicity following ≥48h continuous genotoxic exposure. Exposures <24h resulted in minimal enhancement of gemcitabine activity. These data suggest that SAR-020106 could be administered 24h following gemcitabine and still maximally enhance gemcitabine cytotoxicity. It is possible that this schedule dependency was the result of synchronization of these cells following initial genotoxic drug exposure and that CHK1 inhibitor activity was maximized in the second cell cycle (24-48h following initial drug exposure). In order to test this hypothesis, SW620 cells were synchronized in M-phase by nocodazole treatment and subsequently released and exposed to gemcitabine in combination with SAR-020106 prior to S-phase entry, for various times to maximize activity in the first cell cycle (Supplementary Figure 4B). The results in Supplementary Figure 4C show that cell synchronization had minimal effects on the ability of SAR-020106 to enhance gemcitabine cytotoxicity. Supplementary Figure 4D shows more detailed scheduling studies with gemcitabine and CCT244747 which confirm the requirement for continuous CHK1 inhibition for 24-48h following genotoxic administration for maximal enhancement of gemcitabine cytotoxicity in SW620 cells. Furthermore the degree of potentiation was similar following either continuous or 24h delayed CCT244747 exposure, justifying a delayed CHK1 inhibitor treatment schedule in vivo. These data are consistent with the notion that a lethal event is induced in the second cell cycle following gemcitabine treatment and that prolonged CHK1 inhibitor contact (48h) will be required for maximum activity in vivo.
Pharmacokinetics of CCT244747 in mice and human tumor xenografts
The pharmacokinetic properties of CCT244747 alone were determined in BALB/c mice to ensure that adequate exposure could be achieved for CHK1 inhibition in vivo (Figure 3A). Following 10mg/kg i.v. administration, CCT244747 reached a peak plasma concentration of 3.62μM at 5 min with a relatively short half-life of 0.67h giving an AUC0-∞ of 3.93μMh. By contrast, the peak plasma concentration was only 0.98μM following 10mg/kg oral administration, but the half-life was longer at 0.92h giving an AUC0-∞ of 2.43μMh. Oral bioavailability was therefore 62% and clearance 6.2l/h/kg. Mouse plasma protein binding was 75.6% and microsomal metabolism was 45%. Figure 3B shows that there was a dose-dependent increase in the plasma concentration of CCT244747 up to 150mg/kg p.o. at 12 and 24h in tumor bearing mice treated with gemcitabine (100mg/kg i.v.). Corresponding HT29 tumor concentrations were also dose-dependent over a similar dose range and there was clear evidence of enhanced tumor drug uptake relative to plasma with tumor:plasma ratios 47±21 fold higher than plasma at 12h following 100mg/kg p.o. (mean±SD, n=3). There was no apparent effect of gemcitabine (100mg/kg iv) on CCT244747 pharmacokinetics in mice (data not shown). Figures 3C & D show that gemcitabine induced S296 CHK1 autophosphorylation in HT29 xenografts in vivo. Combination with CCT244747 significantly decreased this phosphorylation (P<0.05). By comparison, the gemcitabine-induced increase in S317 CHK1 and Y15 CDK1 phosphorylation was less markedly inhibited by CCT244747 in keeping with the in vitro observations (see Figure 2). Perhaps more importantly, these studies indicated that potentially active tumor concentrations of CCT244747 could be achieved for at least 24h following oral administration in vivo and this was associated with a significant decrease in CHK1 activity.
Figure 3.
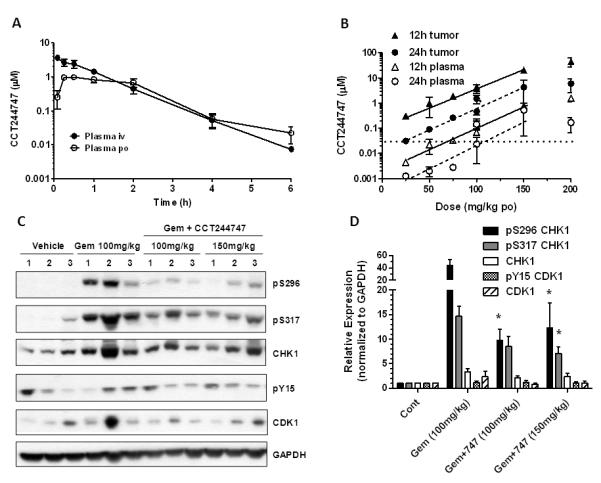
The pharmacokinetic characteristics and pharmacodynamic effects of CCT244747 alone or in combination with gemcitabine in vivo. A, Plasma pharmacokinetics of a single bolus dose of CCT244747 alone (10mg/kg either i.v. or p.o.) in BALB/c mice. B, Concentrations of CCT244747 occurring in mouse plasma and HT29 human tumor xenografts treated with gemcitabine (100mg/kg i.v.) measured 12 and 24h following administration of different doses of CCT244747 p.o. The horizontal dotted line represents the concentration of CCT244747 required for G2 checkpoint abrogation in vitro (MIA, 29nM, see Table 1). C, Biomarker changes induced by gemcitabine treatment alone (100mg/kg i.v.) or in combination with CCT244747 (100 or 150mg/kg p.o) measured in HT29 xenografts 24h following final CCT244747 treatment. Protein expression was characterized by western blotting as described in Materials and Methods and the legend to Figure 2. D, Quantification of the immunoblots shown in C. Values are mean±SE, n=3. Statistical analysis was by one way ANOVA using Dunnett’s correction and * P<0.05 indicates a significant difference from gemcitabine treatment alone.
Antitumor and PD effects of CCT244747 combined with irinotecan and gemcitabine
In all combination antitumor studies each cycle of treatment consisted of a single dose of the genotoxic followed by two doses of CCT244747 24h and 48h later, in keeping with the results of the in vitro scheduling studies. Figure 4 and Supplementary Table 2 summarize the antitumor activity of CCT244747 in combination with irinotecan or gemcitabine in several human tumor xenograft models. Figure 4A shows a minimal effect of CCT244747 alone in HT29 xenografts and a slight but insignificant growth delay of 2.3 days with gemcitabine alone. The addition of CCT244747 to gemcitabine increased the growth delay to 11.4 days giving a statistically significant difference of 9.1 days (P<0.001) in this model with minimal body weight loss (i.e. treatment body weights ≥ initial control body weight, see Supplementary Table 3). Figure 4B shows the effects of gemcitabine and CCT244747 treatment on CHK1 and CDK1 phosphorylation in HT29 xenografts on day 18 of a similar treatment regimen to that used in Figure 4A. Gemcitabine clearly caused induction of both pS296 and pS317 CHK1 and both signals were markedly decreased by combination with CCT244747. Similarly, gemcitabine enhanced the phosphorylation on Y15 CDK1 and this was substantially reduced by CCT244747 treatment. These data demonstrate a relationship between the CCT244747-enhanced antitumor activity of gemcitabine and decreased CHK1 and increased CDK1 activity in tumors. Figure 4C once again shows that CCT244747 alone had minimal activity in HT29 tumors, with a growth delay of only 0.3 days. By contrast, irinotecan alone had significant activity compared with CCT244747 alone, with a growth delay of 6.9 days (P<0.001). Nevertheless, the addition of CCT244747 increased the growth delay to 13.5 days giving a significant increase of 7.1 days (P<0.001) with a nadir of only 1.3% body weight loss on day 10 (see Supplementary Table 4). Figure 4D shows that there was minimal activity associated with CCT244747 alone or gemcitabine alone in Calu6 xenografts. However, the addition of CCT244747 to gemcitabine enhanced the growth delay to 15.9 days giving a significant increase of 8.7 days (P<0.001) with a nadir of body weight loss on day 8 of 5.4% (see Supplementary Table 5). Modest activity of CCT244747 combined with gemcitabine was also confirmed in SW620 xenografts with minimal toxicity (see Supplementary Table 2 and 6). These results demonstrate that oral CCT244747 administration can significantly enhance the antitumor activity of irinotecan and gemcitabine in several human tumor xenograft models and that this activity is associated with CHK1 inhibition.
Figure 4.
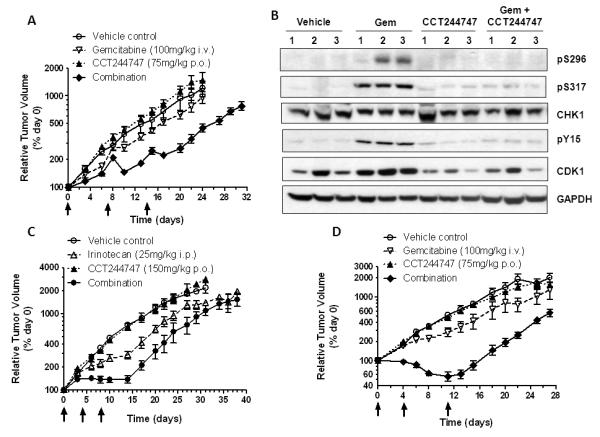
The antitumor and pharmacodynamic effects of CCT244747 combined with irinotecan or gemcitabine in human tumor xenografts. A, Antitumor activity of gemcitabine 100mg/kg i.v. and CCT244747 75mg/kg p.o. alone or in combination in HT29 colon tumor xenografts. B, Biomarker changes occurring 48h (day 18) after the last dose of CCT244747 following drug treatment as shown in A. Protein expression was measured using western blotting as described in Materials and Methods and the legend to Figure 2. C, Efficacy of irinotecan (25mg/kg i.p.) or CCT244747 (150mg/kg p.o.) alone and in combination in SW620 human colon tumor xenografts. D, Efficacy of gemcitabine (100mg/kg i.v.) or CCT244747 (75mg/kg p.o.) alone or combined in Calu6 human lung cancer xenografts. Values are mean±SE for 4-6 animals per point. Tumor size and body weight were assessed as described in Materials and Methods.
CCT244747 has single agent activity in MYCN-driven neuroblastoma
Several studies have indicated that CHK1 inhibitors may have single agent activity in defined tumor types and recent work has identified that MYCN-driven neuroblastoma is exquisitely sensitive to CHK1 inhibition [24]. In view of these observations we evaluated the ability of CCT244747 to inhibit the growth of neuroblastomas which arise spontaneously in a hemizygotic transgenic mouse model characterized by over expression of MYCN (TH-MYCN) [29]. Figure 5A shows that CCT244747 caused significant tumor shrinkage (P<0.001) compared to vehicle treated controls following 7 days of continuous treatment at 100mg/kg p.o.. CCT244747 treatment caused marked reduction in neuroblastoma tumor volume as assessed by MRI (e.g. 79% volume reduction, see Figure 5B) and the treatment was well tolerated with no drug-related morbidity, consistent with previous tolerability studies (Supplementary Figure 5). These data confirm the antitumor activity of CCT244747 as a single agent in a molecularly defined MYCN-driven model of neuroblastoma and warrant further investigation.
Figure 5.
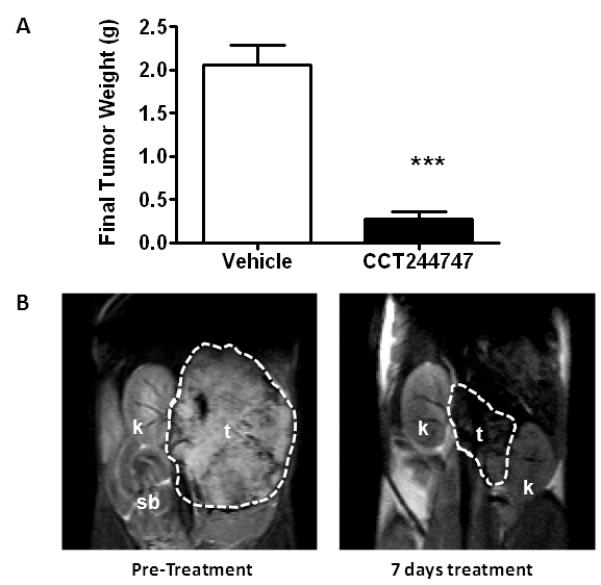
Single agent antitumor activity of CCT244747 in a MYCN-driven transgenic mouse model of neuroblastoma (TH-MYCN). A, Effect of CCT244747 treatment (100mg/kg p.o. × 7 days) on final neuroblastoma tumor weight at necropsy relative to vehicle treated controls. Values are mean±SE for 6 mice. Statistical differences were assessed using a t-test, *** P<0.001. B, T2-weighted MR images of a TH-MYCN neuroblastoma tumor prior to (left panel) and following treatment with CCT244747 (100mg/kg p.o. × 7days). The pre- and post-treatment tumor volumes were calculated as 1960mm3 and 417mm3, respectively, representing a 79% decrease in tumor volume. Tissues are: t, tumor; k, kidney and; sb, small bowel. Tumor size was assessed as described in Materials and Methods.
Discussion
CCT244747 is a novel, potent, highly selective and orally bioavailable CHK1 inhibitor which is a considerable improvement over our previous inhibitor SAR-020106 [27] and represents the first structural disclosure of an orally active CHK1 inhibitor. Kinetic and modelling studies have established that CCT244747 is an ATP competitive inhibitor of recombinant human CHK1. The selectivity profile of CCT244747 shows minimal cross-reactivity with CHK2 and CDK1. Both of these characteristics were deemed desirable as we and others have shown that CHK2 inhibition appears to have limited therapeutic potential [5, 33] and both CHK2 and CDK1 inhibition have been shown to antagonize the ability of CHK1 inhibitors to abrogate checkpoints [34]. Some cross-reactivity with the tyrosine kinase inhibitors FLT3, IRAK1 and RSK1 and 2 at 1μM was noted and correspondingly greater kinase inhibition occurred at 10μM, although this concentration of free drug is unlikely to be achieved or maintained in vivo.
Cellular CHK1 inhibition was measured using an ELISA-based G2 checkpoint abrogation/mitotic induction assay (MIA) while toxicity was determined using a growth delay endpoint (GI50) the ratio of these two values giving a useful therapeutic window of18 and 21-fold in SW620 and HT29 colon carcinoma cells, respectively. CCT244747 enhanced the cytotoxicity of several anticancer drugs and ionizing radiation. The latter was independent of whether the inhibitor was given prior to or after radiation, suggesting a potential effect on either checkpoint abrogation or DNA repair rather than increased radiation damage per se. Similar results have been obtained with other CHK1 inhibitors in vitro [17, 35, 36]. Interestingly, the most marked potentiation by CCT244747 was with gemcitabine or 5FdU, which both target DNA synthesis through inhibition of ribonucleotide reductase or thymidylate synthase respectively, and frequently lead to nucleotide misincorporation [37-39]. This in turn may explain the requirement with gemcitabine for CHK1 inhibitor exposure 24-48h after genotoxic exposure, as the initial misincorporation may undergo subsequent processing in the second cell division giving rise to a lethal event consistent with the increased S’-phase population in the cell cycle results [38, 40, 41]. Several other CHK1 inhibitors also require exposure 24 to 48h following gemcitabine or camptothecin treatment in vitro [15, 17, 41]. However, this may not be a universal observation and could depend on the ability of the different CHK1 inhibitors to deplete total CHK1 protein through S345 CHK1 phosphorylation and ubiquitination with subsequent compromised checkpoint recovery [42]. Other factors could also contribute to the scheduling requirements such as the selectivity profile of the CHK1 inhibitor, the genotoxic agent involved and the cell line under investigation leading to different effects on transcription and DNA repair versus checkpoint abrogation. The role of p53 in enhancing the activity of CHK1 inhibitors in combination with genotoxic drugs is also unclear with some reports suggesting that p53 functional status does not always affect either combinations or single agent CHK1 inhibitor activity in agreement with our observations [20, 43].
We have characterized the pharmacokinetic and pharmacodynamic relationships between drug exposure and the modulation of a number of potentially useful biomarkers for assessing CHK1 inhibition both in vitro and in vivo. Such studies constitute an important component of drug discovery and ensure that target inhibition can be monitored throughout the preclinical and clinical drug evaluation program via a “pharmacological audit trail” [44]. Pharmacokinetic studies established that CCT244747 was concentrated or retained in tumor tissue following oral administration and adequate exposures for CHK1 inhibition could be achieved and maintained using this route. Studies with SN38 and gemcitabine in HT29 and SW620 cells showed that S296 CHK1 autophosphorylation was markedly enhanced together with induction of phosphorylation on S317 and S345. Combination with CCT244747 abolished S296 CHK1 phosphorylation but only partially inhibited the pS317 and pS345 signals, which may require prolonged exposure for complete inhibition (see Figure 4B). Substantial induction of pY15 CDK1 (a cell cycle biomarker) was also observed following genotoxic exposure in vitro and this signal was abolished by CCT244747, consistent with abrogation of a genotoxic-induced cell cycle arrest. The observation that pS139 H2AX and PARP cleavage occurred at concentrations of CCT244747 which abolished pS296 CHK1 and pY15 CDK1 suggest that CHK1 inhibition was associated with DNA damage and enhanced apoptosis. Subsequent in vivo studies confirmed that CCT244747 could inhibit a gemcitabine-induced increase in both pS296 CHK1 and pY15 CDK1 in tumor xenograft tissues using treatment regimens that induced significant antitumor activity. These studies support the use of pS296 CHK1 as a direct readout of CHK1 inhibition and pY15 CDK1 as a biomarker of cell cycle modulation. However, it is likely that several biomarkers will be required to ensure the fidelity of CHK1 inhibition in the clinical setting and these might include down-stream readouts of DNA damage such as RAD51 foci and γH2AX as well as apoptosis markers e.g. cleaved PARP and caspase-3 [12, 27, 29, 34, 36].
Efficacy studies showed that CCT244747 could significantly enhance the growth inhibitory properties of irinotecan and gemcitabine in several human tumor xenograft models. Moreover, the use of CCT244747 alone or in combination was well tolerated with minimal body weight loss. The precise mechanism of this enhanced antitumor activity is still unclear and may involve replication fork collapse, checkpoint abrogation and inhibition of DNA repair [8, 34, 36, 45]. Nevertheless, our studies clearly show that the CHK1 inhibitor needs to be present for at least 24 to 48h following genotoxic drug administration, possibly requiring multiple administrations, which the oral activity of CCT244747 will facilitate.
Several recent publications have indicated that CHK1 inhibitors may have single agent activity in tumors exhibiting marked genomic instability or specific molecular defects [17, 21, 24] Using a genetically defined MYCN-driven murine transgenic model of neuroblastoma in which tumors arise spontaneously, we have clearly demonstrated that CCT244747 has marked single agent, antitumor activity. Importantly this model recapitulates the major clinico-pathological features of high-risk MYCN-amplified neuroblastoma in humans, which is characterized by an extremely poor response to conventional chemotherapeutics [29]. In addition the chronic oral administration of CCT244747 was well tolerated and caused marked regression of this extremely aggressive tumor. These data are consistent with the observations of Cole et al in MYCN-driven neuroblastoma [24] and the reported single-agent activity of CHK1 inhibitors and CHK1 RNAi in C-MYC driven lymphoma cells [23], in AML [22] and several human tumor cell lines [20]. This may be explained by MYC oncogene expression causing increased replicative stress and enhanced replication origin firing with an associated need for elevated CHK1 to avoid catastrophic replication fork collapse [1, 7, 18, 19, 21]. CHK1 inhibition appears to enhance CDK activity with attendant replication fork collapse and increased DNA double-strand breaks giving rise to enhanced tumor cell killing [4, 46]. Inhibition of CHK1 could compromise homologous recombination repair, through indirectly inhibiting RAD51, a CHK1 substrate and this may also be therapeutically advantageous [8]. It is further interesting to note that other G2 checkpoint abrogators such as the WEE1 inhibitor MK1775, also show single agent activity through deregulation of CDKs in tumors [46, 47].
Finally, our data show that CCT244747 is a novel, potent, highly selective and orally active CHK1 inhibitor, which significantly enhances the antitumor activity of gemcitabine and irinotecan in a variety of human tumor xenografts. Biomarker studies indicate that this enhanced antitumor activity is associated with CHK1 inhibition and cell cycle checkpoint abrogation. Moreover, CCT244747 also showed marked antitumor activity as a single agent in MYCN-driven neuroblastoma. This report is the first disclosure of a highly selective, orally active CHK1 inhibitor and supports further development of these compounds.
Supplementary Material
Statement of translational relevance.
CCT244747 is a novel, potent, highly selective and orally active CHK1 inhibitor, which significantly increased the cytotoxicity of several chemotherapeutic agents in vitro. CCT244747 clearly inhibited genotoxic-induced biomarkers for CHK1 (pS296 CHK1) activity and CDK1 (pY15 CDK1) inactivity in vitro and in vivo, resulting in enhanced DNA damage and apoptosis. Scheduling studies showed CCT244747 must be present 24-48h following initial genotoxic exposure for maximum potentiation. Potentially therapeutic concentrations of CCT244747 were readily achieved in tumors following oral administration. Gemcitabine and irinotecan antitumor activity were significantly enhanced by delayed and repeated CCT244747 administration, a requirement greatly facilitated through its oral bioavailability. Moreover, CCT244747 showed marked antitumor activity as an orally administered single agent in a MYCN-driven transgenic model of neuroblastoma. In conclusion, CCT244747 represents the first structural disclosure of a highly selective, orally active CHK1 inhibitor that justifies further evaluation as a single agent and in combination with genotoxic anticancer therapies.
Acknowledgements
The authors gratefully acknowledge the help of Gowri Vijayaraghavan with flow cytometry studies, Nathan Brown with modelling studies and useful discussions with other members of the Cell Cycle Control Team.
Grant support: This work was supported by Cancer Research UK [CUK] grant number C309/A8274, C309/A11566. We acknowledge the support received for the Cancer Research UK and EPSRC Cancer Imaging Centre, in association with the MRC and Department of Health (England) grant number C1060/A10334, The Wellcome Trust, grant number 091763Z/10/Z and also NHS funding to the NIHR Biomedical Research Centre.
Abbreviation List
- TBS
Tris buffered saline
- PI
potentiation index
- GI50
concentration at which cell growth is inhibited by 50%
- AI
Activity index
- MIA
mitosis induction assay
- 5FdU
5-Fluoro-2′-deoxyuridine
- IR
ionizing radiation
- Gem
gemcitabine
- Irin
irinotecan
- SRB
sulphorhodamine B
- HRPO
horse-radish peroxidase
Footnotes
Conflict of interest statement: M I Walton, P D Eve, A Hayes, M R Valenti, A K De Haven Brandon, G Box, A Hallsworth, E L Smith, K J Boxall, M Lainchbury, T P Matthews, Y Jamin, S P Robinson, G W Aherne, L Chesler, S A Eccles, F I Raynaud, I Collins and M D Garrett are current or former employees of The Institute of Cancer Research, which has a commercial interest in CHK1 inhibitors.
JC Reader is an employee of Sareum Ltd which has a commercial interest in CHK1 inhibitors. Sareum Ltd is a wholly owned subsidiary of Sareum Holdings PLC, of which JC Reader is a shareholder.
Both Sareum and The Institute of Cancer Research have been involved in a commercial collaboration with Cancer Research Technology Ltd (CRT) to discover and develop inhibitors of CHK1.
References
- 1.Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11(3):220–8. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- 2.Kaufmann WK. Initiating the uninitiated: Replication of damaged DNA and carcinogenesis. Cell Cycle. 2007;6(12):1460–7. [PubMed] [Google Scholar]
- 3.Meek DW. Tumour suppression by p53: A role for the DNA damage response? Nat Rev Cancer. 2009;9(10):714–23. doi: 10.1038/nrc2716. [DOI] [PubMed] [Google Scholar]
- 4.Sorensen CS, Syljuasen RG. Safeguarding genome integrity: The checkpoint kinases atr, CHK1 and WEE1 restrain cdk activity during normal DNA replication. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2):376–83. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xiao Z, Chen Z, Gunasekera AH, Sowin TJ, Rosenberg SH, Fesik S, et al. CHK1 mediates S and G2 arrests through CDC25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278(24):21767–21773. doi: 10.1074/jbc.M300229200. [DOI] [PubMed] [Google Scholar]
- 7.Feijoo C. Activation of mammalian chk1 during DNA replication arrest: A role for CHK1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154(5):913–924. doi: 10.1083/jcb.200104099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sorensen CS, Hansen LT, Dziegielewski J, Syljuasen RG, Lundin C, Bartek J, et al. The cell-cycle checkpoint kinase CHK1 is required for mamalian homologous recombination repair. Nat Cell Biol. 2005;7(2):195–201. doi: 10.1038/ncb1212. [DOI] [PubMed] [Google Scholar]
- 9.Peddibhotla S, Lam MH, Gonzalez-Rimbau M, Rosen JM. The DNA-damage effector checkpoint kinase 1 is essential for chromosome segregation and cytokinesis. Proc Natl Acad Sci. 2009;106(13):5159–64. doi: 10.1073/pnas.0806671106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanahan D, Weinberg RA. Hallmarks of cancer: The next generation. Cell. 2011;144(5):646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature. 2000;408(6810):307–10. doi: 10.1038/35042675. [DOI] [PubMed] [Google Scholar]
- 12.Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer. 2008;98(3):523–528. doi: 10.1038/sj.bjc.6604208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17(2):88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matthews DJ, Yakes FM, Chen J, Tadano M, Bornheim L, Clary DO, et al. Pharmacological abrogation of S-phase checkpoint enhances the anti-tumor activity of gemcitabine in vivo. Cell Cycle. 2007;6(1):104–10. doi: 10.4161/cc.6.1.3699. [DOI] [PubMed] [Google Scholar]
- 15.Tse AN, Rendahl KG, Sheikh T, Cheema H, Aardalen K, Embry M, et al. Chir-124, a novel potent inhibitor of CHK1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin Cancer Res. 2007;13(2):591–602. doi: 10.1158/1078-0432.CCR-06-1424. [DOI] [PubMed] [Google Scholar]
- 16.Morgan MA, Parsels LA, Parsels JD, Lawrence TS, Maybaum J. The relationship of premature mitosis to cytotoxicity in response to checkpoint abrogation and antimetabolite treatment. Cell Cycle. 2006;5(17):1983–8. doi: 10.4161/cc.5.17.3184. [DOI] [PubMed] [Google Scholar]
- 17.Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: What, where and when? Trends Pharmacol Sci. 2011;32(5):308–16. doi: 10.1016/j.tips.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 18.Kuschak TI, Kuschak BC, Taylor CL, Wright JA, Wiener F, Mai S. C-MYC initiates illegitimate replication of the ribonucleotide reductase R2 gene. Oncogene. 2002;21(6):909–20. doi: 10.1038/sj.onc.1205145. [DOI] [PubMed] [Google Scholar]
- 19.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444(7119):638–42. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 20.Davies KD, Humphries MJ, Sullivan FX, von Carlowitz I, Le Huerou Y, Mohr PJ, et al. Single-agent inhibition of CHK1 is antiproliferative in human cancer cell lines in vitro and inhibits tumor xenograft growth in vivo. Oncol Res. 2011;19(7):349–63. doi: 10.3727/096504011x13079697132961. [DOI] [PubMed] [Google Scholar]
- 21.Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and CHK1 kinases for cancer treatment: A new model for new (and old) drugs. Mol Oncol. 2011;5(4):368–73. doi: 10.1016/j.molonc.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cavelier C, Didier C, Prade N, Mansat-De Mas V, Manenti S, Recher C, et al. Constitutive activation of the DNA damage signaling pathway in acute myeloid leukemia with complex karyotype: Potential importance for checkpoint targeting therapy. Cancer Res. 2009;69(22):8652–8661. doi: 10.1158/0008-5472.CAN-09-0939. [DOI] [PubMed] [Google Scholar]
- 23.Ferrao PT, Bukczynska EP, Johnstone RW, McArthur GA. Efficacy of CHK inhibitors as single agents in MYC-driven lymphoma cells. Oncogene. 2011 doi: 10.1038/onc.2011.358. [DOI] [PubMed] [Google Scholar]
- 24.Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, et al. Rnai screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc Natl Acad Sci. 2011;108(8):3336–3341. doi: 10.1073/pnas.1012351108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lainchbury M, Collins I. Checkpoint kinase inhibitors: A patent review (2009 - 2010) Expert Opin Ther Pat. 2011;21(8):1191–210. doi: 10.1517/13543776.2011.586632. [DOI] [PubMed] [Google Scholar]
- 26.Collins I, Reader JC, Matthews TP, Cheung KM, Proisy N, Williams DH, et al. Pyrazin-2-yl-pyridin-2-yl-amine and pyrazin-2-yl-pyrimidin-4-yl-amine compounds, their preparation, and their use as chk1 kinase inhibitors for treating proliferative diseases. Chem Abs. 2009:398578. [Google Scholar]
- 27.Walton MI, Eve PD, Hayes A, Valenti M, De Haven Brandon A, Box G, et al. The preclinical pharmacology and therapeutic activity of the novel chk1 inhibitor SAR-020106. Mol Cancer Ther. 2010;9(1):89–100. doi: 10.1158/1535-7163.MCT-09-0938. [DOI] [PubMed] [Google Scholar]
- 28.Lin NH, Xia P, Kovar P, Park C, Chen Z, Zhang H, et al. Synthesis and biological evaluation of 3-ethylidene-1,3-dihydro-indol-2-ones as novel checkpoint 1 inhibitors. Bioorg Med Chem Lett. 2006;16(2):421–6. doi: 10.1016/j.bmcl.2005.09.064. [DOI] [PubMed] [Google Scholar]
- 29.Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of mycn causes neuroblastoma in transgenic mice. EMBO J. 1997;16(11):2985–95. doi: 10.1093/emboj/16.11.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, et al. Inhibition of phosphatidylinositol 3-kinase destabilizes MYCN protein and blocks malignant progression in neuroblastoma. Cancer Res. 2006;66(16):8139–46. doi: 10.1158/0008-5472.CAN-05-2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ, et al. Guidelines for the welfare and use of animals in cancer research. Br J Cancer. 2010;102(11):1555–1577. doi: 10.1038/sj.bjc.6605642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reader JC, Matthews TP, Klair S, Cheung KM, Scanlon J, Proisy N, et al. Structure-guided evolution of potent and selective CHK1 inhibitors through scaffold morphing. J Med Chem. 2011;54(24):8328–42. doi: 10.1021/jm2007326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson VE, Walton MI, Eve PD, Boxall KJ, Antoni L, Caldwell JJ, et al. CCT241533 is a potent and selective inhibitor of CHK2 that potentiates the cytotoxicity of PARP inhibitors. Cancer Res. 2011;71(2):463–72. doi: 10.1158/0008-5472.CAN-10-1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guzi TJ, Paruch K, Dwyer MP, Labroli M, Shanahan F, Davis N, et al. Targeting the replication checkpoint using SCH 900776, a potent and functionally selective CHK1 inhibitor identified via high content screening. Mol Cancer Ther. 2011;10(4):591–602. doi: 10.1158/1535-7163.MCT-10-0928. [DOI] [PubMed] [Google Scholar]
- 35.Tao Y, Leteur C, Yang C, Zhang P, Castedo M, Pierre A, et al. Radiosensitization by CHIR-124, a selective CHK1 inhibitor: Effects of p53 and cell cycle checkpoints. Cell Cycle. 2009;8(8):1196–205. doi: 10.4161/cc.8.8.8203. [DOI] [PubMed] [Google Scholar]
- 36.Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the CHK1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res. 2010;70(12):4972–4981. doi: 10.1158/0008-5472.CAN-09-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mini E, Nobili S, Caciagli B, Landini I, Mazzei T. Cellular pharmacology of gemcitabine. Ann Oncol. 2006;17(Suppl 5):v7–12. doi: 10.1093/annonc/mdj941. [DOI] [PubMed] [Google Scholar]
- 38.Flanagan SA, Robinson BW, Krokosky CM, Shewach DS. Mismatched nucleotides as the lesions responsible for radiosensitization with gemcitabine: A new paradigm for antimetabolite radiosensitizers. Mol Cancer Ther. 2007;6(6):1858–68. doi: 10.1158/1535-7163.MCT-07-0068. [DOI] [PubMed] [Google Scholar]
- 39.Liu A, Yoshioka K, Salerno V, Hsieh P. The mismatch repair-mediated cell cycle checkpoint response to fluorodeoxyuridine. J Cell Biochem. 2008;105(1):245–54. doi: 10.1002/jcb.21824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lazzaro F, Giannattasio M, Puddu F, Granata M, Pellicioli A, Plevani P, et al. Checkpoint mechanisms at the intersection between DNA damage and repair. DNA Repair. 2009;8(9):1055–1067. doi: 10.1016/j.dnarep.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 41.Blasina A, Hallin J, Chen E, Arango ME, Kraynov E, Register J, et al. Breaching the DNA damage checkpoint via PF-00477736, a novel small-molecule inhibitor of checkpoint kinase 1. Mol Cancer Ther. 2008;7(8):2394–2404. doi: 10.1158/1535-7163.MCT-07-2391. [DOI] [PubMed] [Google Scholar]
- 42.Calonge TM, O’Connell MJ. Turning off the G2 DNA damage checkpoint. DNA Repair (Amst) 2008;7(2):136–40. doi: 10.1016/j.dnarep.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29(46):6149–6159. doi: 10.1038/onc.2010.343. [DOI] [PubMed] [Google Scholar]
- 44.Workman P. How much gets there and what does it do?: The need for better pharmacokinetic and pharmacodynamic endpoints in contemporary drug discovery and development. Curr Pharm Des. 2003;9(11):891–902. doi: 10.2174/1381612033455279. [DOI] [PubMed] [Google Scholar]
- 45.McNeely S, Conti C, Sheikh T, Patel H, Zabludoff S, Pommier Y, et al. CHK1 inhibition after replicative stress activates a double strand break response mediated by ATM and DNA-dependent protein kinase. Cell Cycle. 2010;9(5):995–1004. doi: 10.4161/cc.9.5.10935. [DOI] [PubMed] [Google Scholar]
- 46.Beck H, Nahse V, Larsen MS, Groth P, Clancy T, Lees M, et al. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. J Cell Biol. 2010;188(5):629–38. doi: 10.1083/jcb.200905059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective WEE1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Mol Cancer Ther. 2012;11(1):174–82. doi: 10.1158/1535-7163.MCT-11-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.