

Abstract
Wnt signaling is essential for osteogenesis and also functions as an adipogenic switch, but it is not known if interrupting wnt signaling via knockout of β-catenin from osteoblasts would cause bone marrow adiposity. Here, we determined whether postnatal deletion of β-catenin in preosteoblasts, through conditional cre expression driven by the osterix promoter, causes bone marrow adiposity. Postnatal disruption of β-catenin in the preosteoblasts led to extensive bone marrow adiposity and low bone mass in adult mice. In cultured bone marrow-derived cells isolated from the knockout mice, adipogenic differentiation was dramatically increased, whereas osteogenic differentiation was significantly decreased. As myoblasts, in the absence of wnt/β-catenin signaling, can be reprogrammed into the adipocyte lineage, we sought to determine whether the increased adipogenesis we observed partly resulted from a cell-fate shift of preosteoblasts that had to express osterix, (lineage-committed early osteoblasts), from the osteoblastic to the adipocyte lineage. Using lineage tracing both in vivo and in vitro we demonstrated that the loss of β-catenin from preosteoblasts caused a cell-fate shift of these cells from osteoblasts to adipocytes, a shift that may at least partly contribute to the bone marrow adiposity and low bone mass in the knockout mice. These novel findings indicate that wnt/β-catenin signaling exerts control over the fate of lineage-committed early osteoblasts, with respect to their differentiation into osteoblastic vs. adipocytic populations in bone, and thus offers potential insight into the origin of bone marrow adiposity.
Introduction
Many conditions that can induce bone loss, such as estrogen insufficiency (1), disuse (2), hindlimb unloading (3) and microgravity exposure (4), are accompanied by increased bone marrow adiposity. In patients with osteoporosis, bone marrow adiposity was significantly increased and bone formation rate was inversely related to the adipocytic proportion of the bone marrow (5–7).
Osteoblasts and adipocytes can both arise from the same precursor bone marrow mesenchymal stem cells (BMSCs). Over the past decades, much effort has been devoted to understanding how BMSCs can be allocated into the osteoblastic or adipocytic lineages. Certain crucial factors can direct BMSCs into one lineage and prevent them from differentiating into the other lineage. When adipocyte differentiation signals, such as CCAAT/enhancer binding protein (C/EBP) and the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ) are activated, BMSCs are driven into the adipocytic lineage. Extensive analyses of in vivo and in vitro models have established the basic genetic cascade of adipogenesis. In response to adipocytic inducers, the early transcription factors C/EBPβ, C/EBPδ and PPARγ1 are rapidly activated or induced in BMSCs and initiate the adipogenic cascade, which includes increased expression of two critical transcription factors responsible for adipogenesis, C/EBPα and PPARγ2, followed by upregulation of downstream genes that are characteristic of mature adipocytes, including FABP4, Glut4, and adiponectin, (8–10).
In contrast, entry of BMSCs into the osteoblastic lineage depends upon the activation of Runx2 and wnt/β-catenin signaling. During the process of bone formation, activation of wnt/β-catenin signaling represses differentiation of BMSCs into the adipocytic lineage (11). Thus, in mouse BMSCs, activation of wnt/β-catenin signaling suppresses glycogen synthase kinase 3(GSK-3) and directs multipotential cells into the osteoblastic lineage at the expense of the adipogenic lineage (12). Inactivation of wnt/β-catenin signaling by silencing β-catenin with RNAi promoted adipogenesis in human bone marrow-derived mesenchymal stem cells (13). Similar results obtained with cell lines have also shown that activation of wnt signaling inhibits adipogenic differentiation. Overexpression of wnt10b in bipotential ST2 cells suppresses appearance of the adipocytic phenotype but induces osteoblastogenic gene expression, and this process is mediated by suppression of C/EBPα and PPARγ (14). Stabilization of β-catenin or overexpression of wnt1 in 3T3-L1 preadipocytes inhibits their ability to differentiate into mature adipocytes by reducing the expression of C/EBPα and PPARγ (15), whereas disruption of β-catenin in the C2C12 cell line increases the expression of PPARγ (16). Thus, wnt signaling may play an important role in controlling not only osteogenesis but also adipogenesis in bone and bone marrow. Evidence for this mechanism in vivo is lacking, as altered bone marrow adiposity has not been reported in mice with knockout of β-catenin, a key component of the wnt pathway, either in early cells of the osteoblastic lineage or in mature osteoblasts. In fact, removal in vivo of β-catenin from early cells of the osteoblastic lineage by either Prx1-cre or Dermo1-cre or even with Osx-cre results in an early arrest of osteoblast differentiation and no bone formation (17–20). On the other hand, deletion of β-catenin within mature osteoblasts by either Col1α1-cre or osteocalcin-cre or even in osteocytes by DMP1-cre causes exclusively an increase in osteoclast differentiation and low bone mass with normal bone formation (21–23). Moreover, no increase in bone marrow adiposity was reported in mice with knockout of wnt10b, a typical activator of wnt signaling, although the knockout mice exhibit low bone mineral density (24,25).
In this study, we induced postnatal deletion of β-catenin in early (osterix-expressing) cells of the osteoblastic lineage and observed both a striking reduction in bone mass and a remarkable increase in bone marrow adiposity. Using a global double fluorescence cre reporter mouse, we observed that osterix-expressing preosteoblasts became adipocytic cells, with strong expression of the adipocytic marker FABP4 and positive staining for Oil-Red O. These novel findings provide direct evidence that knockout of β-catenin in early cells of the osteoblastic lineage causes a dramatic change in cell fate of the preosteoblasts from osteoblasts to adipocytes, a shift that may at least partly contribute to the phenotype of increased bone marrow adiposity and low bone mass observed in the knockout mice.
Materials And Methods
Animals
The β-cateninf/f (β-catf/f) (26) and mT/mG double fluorescence reporter mice (27) (both from Jackson Labs), and osterix-cre (osx-cre) transgenic mice (20,26,27) (from McMahon lab) used in the present study were backcrossed to the C57/B6 background for more than 6 generations. The cre expression in the osx-cre mice can be suppressed by administration of doxycycline (Dox) to the pregnant mice and their progeny. Treatment with Dox was accomplished by adding it (1.5 mg/ml) to the drinking water. Animals were maintained in facilities operated by the Center for Comparative Research of the Massachusetts General Hospital, and all animal experimental procedures were approved by the institution’s Subcommittee on Research Animal Care.
Histology
For histological analysis, dissected tibiae and femurs were fixed with 10% formalin for 24 hrs and decalcified with 18% EDTA for 14 days. Paraffin sections from the decalcified bone and plastic sections from un-decalcified bone were prepared by standard histological procedures.
Isolation of BMSCs
After anesthetization, mice at eight weeks of age were sacrificed to dissect tibiae and femurs, and then BMSCs were isolated by flushing the bone marrow with α-MEM medium. After centrifuging for 10 min at 800 rpm, BMSCs were resuspended in α-MEM medium supplemented with 10% fetal bovine serum (FBS), and then filtered with a 70 μm pre-sterilized cell strainer. For cell culture, the total BMSCs per mouse were suspended in 36 ml culture medium and seeded into 6 or 12-well tissue culture plates (1 or 2 ml/well).
In vitro differentiation and adenoviral-cre infection of BMSCs
The isolated BMSCs were cultured in 6 or 12-well plates with α-MEM medium containing 10% FBS for 48h, and then infected with adenoviral-cre or adenoviral-GFP (2×107 pfu adenovirus per well) for 48h, followed by continuing culture with either adipogenic medium containing 10% FBS, 3-isobutyl-1-methylxanthine (IBMX, 0.5 μM), dexamethasone (1 μM) and insulin (10 ng/ml), or osteogenic medium supplemented with 10% FBS, β-glycerophosphate (10 mM), and ascorbic acid (50 μg/ml). Culture medium was replaced every 2 days.
Oil red O stain and quantification
After culture, cells were washed twice with PBS and then fixed with 10% formalin for 20 min. After fixation, cells were washed three times with ddH2O followed by 60% isopropanol for 5 min. After removal of isopropanol, cells were allowed to dry completely and then were incubated with Oil Red O working solution for 20 min followed by rinsing 5 times with ddH2O. For Oil Red O quantification, 200 μl isopropanol was added into each well for 5 min. The supernatants were taken for measurement of OD value with a spectrophotometer at 500 nm, and Oil Red O content per well was calculated with a standard curve. Six to eight individual samples were analyzed for each group.
Von Kossa stain
After culture, cells were washed twice with PBS and then fixed with 10% formalin for 20 min. After fixation, cells were washed three times with ddH2O and incubated with 5% silver nitrate for 20 min under room temperature in the dark. After washing with ddH2O plates were exposed to bright light for 30 min.
Calcium content quantification
Cultured cells were washed twice with PBS and incubated with 400 μl 0.6N HCl per well on the shaker (200 rpm) overnight at room temperature. The content was collected into 1.5ml tubes and centrifuged for 2 min at 10,000rpm to obtain the supernatants. 20 μl of each supernatant was taken for measurement of OD value with a spectrophotometer at 620 nm and calcium content in each well was determined as described previously (28).
Quantitative Real-Time PCR (qRT-PCR)
Total RNA for qRT-PCR was extracted from cultured cells using Trizol reagents (Invitrogen) according to the manufacturer’s protocol. For qRT-PCR, cDNA was prepared with random primer using the SuperScript First-Strand Synthesis System (Invitrogen) and analyzed with the SYBR GreenMaster Mix (QIAGEN) in the thermal cycler with two sets of primers specific for each targeted gene. Relative expression was calculated for each gene by the 2-ΔΔCT method with β-actin for normalization. Primers used for qRT-PCR were: Osterix (F) AGTGCACGAGCCAGCGACAA and (R) CCAGCGCTTGGAGATCTTAGA; Runx2 (F) GCACAGACAGAAGCTTGATGA and (R) TGCCTGGGATCTGTAATCTGA; ALP (F) TCTCCAGACCCTGCAACCTC and (R) CATCCTGAGCAGACCTGGTC; Col1α1 (F) CGAGTCACACCGGAACTTGG and (R) GCAGGCAGGGCCAATGTCTA; Osteocalcin (F) CTCTGTCTCTCTGACCTCACAG and (R) CAGGTCCTAAATAGTGATACCG; Rb1 (F) CTCACGCTGCCCAGGAGACCT and (R) AGGAGGCCTGGTGGAGGCATAC; Zfp467 (F) TGGCTCCCCAGAGTGAGCCC and (R) TGTGGGGTACTGGCCTGGCT; C/EBPα (F) ACTTCTACGAGGTGGAGCCGCGG and (R) CCGGGTCGATGTAGGCGCTGA; FABP4 (F) CACCTGGAAGACAGCTCCTCCTCG and (R) ACCGGATGGTGACCAAATCCCCA; Maf (F) CAATTCCGACCTGCCCACCAGT and (R) CATGGGGGTGGAGGACAGCG; PPARγ(F) TCCGTGATGGAAGACCACTCGCA and (R) TGCAGGTTCTACTTTGATCGCACTT; Axin2 (F) ATGTGTGGATACGCTGGACTT and (R) TTCTTGATGCCATCTCGTATG; Lef1 (F) TCTCAAGGACAGCAAAGCTC and (R) CACTTGAGGCTTCATGCACAT; Tcf1 (F) ACATGAAGGAGATGAGAGCCA and (R) CTTCTTCTTTCCGTAGTTATC; β-catenin (F) TGCCACCACCACAGCTCCTT and (R) GGAACATGGCAGCTCGGACCC, and β-actin (F) CGTGCGTGACATCAAAGAGAA and (R) GCTCGTTGCCAATAGTGATGA.
Cellular immunofluorescence
Isolated BMSCs were seeded in 8-well slide chambers and cultured with the adipogenic medium for up to 21 days, and then the cells were fixed with 10% formalin for 15 min, followed by 0.25% Triton for 5 min. After rinsing with ddH2O, cells were processed for Oil Red O staining, followed by incubation with rabbit anti-FABP4 antibody (Abcam, 1:500 dilution) and chicken anti-GFP antibody (Abcam, 1:500 dilution) for 45 min. After rinsing 3 times with PBS, cells were incubated with fluorescence-conjugated secondary antibodies (goat anti-rabbit-Alexa Fluor 647 (far red) and goat anti-chicken-Alexa Fluor 488 (green), (both secondary antibodies were obtained from Invitrogen and used at 1:500 dilution) for 30 min followed by rinsing 5 times with PBS. Slides were mounted with a mount medium containing DAPI and examined under fluorescence and confocal microscopes.
Tibial immunofluorescence and quantitative assessment of the GFP+ adipocytes
Five-months old tibiae were dissected from mT/mG; osx-cre and mT/mG; osx-cre;β-catf/f mice treated with Dox until 2-month old. The dissected tibiae were fixed with formalin/sucrose (10%) in PBS for 2 hours and decalcified with 18% EDTA at cold room for a week, and then processed for frozen sections. 8 μm sections were incubated with rabbit anti-FABP4 antibody (1:500 dilution) and chicken anti-GFP antibody (1:500 dilution) for 45 min. After rinsing with PBS, sections were incubated with fluorescence-conjugated goat anti-rabbit-Alexa Fluor 647 antibody (1:500 dilution) and goat anti-chicken-Alexa Fluor 488 antibody (1:500 dilution) for 30 min, followed by rinsing with PBS. Slides were mounted with a mount medium containing DAPI and examined using confocal microscopes. To determine the number of osterix-derived adipocytes, we counted the total number of cells, the cells positive for FABP4 and the cells positive for both FABP4 and GFP in the metaphyseal trabecular region up to 50 microns away from the growth plate; this was the region with the most dramatic increase in fat in the mutant mice. Cells were counted blindly on at least three cross-sections for each animal. The FABP4-positive cells with GFP surrounding less than 50% of the red FABP4 fluorescence were classified as not certainly GFP positive. These cells, representing roughly 10% of the FABP4+ cells in the control mice and 20% of the FABP4+ cells in the mutant mice, were classified as GFP-negative for purposes of calculations. This classification was conservatively designed to minimize the number of fat cells certainly derived from osterix-expressing cells.
Microcomputed Tomography (μCT)
μCT analysis of the distal femur was performed using a desktop microtomographic image system, as described previously (29).
Statistics
Students’ t test was used to compare two test groups and differences were considered significant for p < 0.05.
Results
Loss of β-catenin in early osteoblasts causes increased bone marrow adiposity
In the course of performing tibial bone histology of adult mice with conditional knockout of β-catenin driven by the osx-cre promoter, we unexpectedly observed a dramatic change in the bone marrow fat content. Thus, in osx-cre;β-catf/f mice treated with doxycyline (Dox) until 4-months of age and then sacrificed at 6 months, two months after withdrawal of Dox (to allow cre expression), an extensive increase in marrow fat was observed in the trabecular region of both the primary and secondary ossification centers, which was accompanied by a striking decrease in trabecular bone (Fig. 1A). To further demonstrate whether this phenomenon was caused by inactivation of β-catenin in early cells of the osteoblastic lineage postnatally, osx-cre;β-catf/f mice were treated with Dox prenatally and until 2 months of age and then sacrificed at six months of age. As shown in Fig. 1B, the bone marrow space in the trabecular region of these mice was mostly occupied by adipose tissue, and the loss of bone mass was even more evident in both trabecular and cortical bones. These observations suggested that β-catenin signaling in early (osx-expressing) cells of the osteoblast lineage is important not only for bone acquisition but also for suppression of fat formation in bone. Notably, this postnatal knockout of β-catenin in the growing mice caused a dramatic decrease in trabecular bone mass, as shown by μCT analysis (Fig. 1C) and von Kossa staining of plastic sections from un-decalcified tibia (Fig. 1D). We also observed a dramatic increase in osteoclast-mediated bone resorption and decreased osteoblast-mediated bone formation in both tibiae and vertebrae of the conditional knockout mice. A detailed histomorphometirc analysis of the bone phenotype in this postnatal knockout model will be presented in a separate publication.
Fig. 1. Progressively increased bone marrow fat in adult mice following postnatal knockout of β-catenin through the osterix promoter.

(A–B) H&E staining of paraffin sections from proximal tibiae of 6-month old control (β-catf/f) and knockout (osx-cre;β-catf/f) mice treated with doxycycline (Dox, 1.5 mg/ml supplemented in drinking water) prenatally and until 4 months of age (A) or until 2 months of age (B). Treatment with Dox suppresses cre expression and thus prevents disruption of β-catenin. The earlier disruption of β-catenin by osx-cre at 2 months (B), led to more marrow adipose tissue and more bone loss in the knockout mice at 6 months. (C) μCT analysis of distal femurs and (D) von Kossa staining of plastic sections of proximal tibiae from 12-wk old mice treated with Dox prenatally and until 7 weeks of age.
Deletion of β-catenin in cultured BMSCs
The increased bone marrow adiposity caused by postnatal knockout of β-catenin in early cells of the osteoblast lineage is consistent with the possibility that BMSCs may preferentially differentiate into adipocytic lineages instead of osteoblastic lineages when wnt/β-catenin signaling is disrupted in such cells. To test this possibility, we developed an in vitro system to disrupt β-catenin in BMSCs using adenoviral infection. We first determined the infection efficiency of adenoviral cre (ade-cre) and adenoviral GFP (ade-GFP, as control) in BMSCs isolated from control β-catf/f mice that additionally carry a double fluorescence reporter gene (mT/mG) (27). As expected (Fig. 2A), the infection of BMSCs with the ade-cre led to removal of sequences encoding the membrane tomato (mT, red fluorescence) to allow selective expression of the membrane GFP (mG), which presents a distinctive pattern of cellular GFP expression from that generated by the control infection with ade-GFP (cytoplasmic GFP) (Fig. 2A). We next measured levels of mRNAs encoding β-catenin using qRT-PCR. Dramatically decreased expression of β-catenin was observed in the cre-infected cells (Fig. 2A), indicating that the ade-cre effectively disrupted the β-catenin gene in the infected BMSCs.
Fig. 2. Deletion of β-catenin causes increased adipogenesis and decreased osteogenesis.
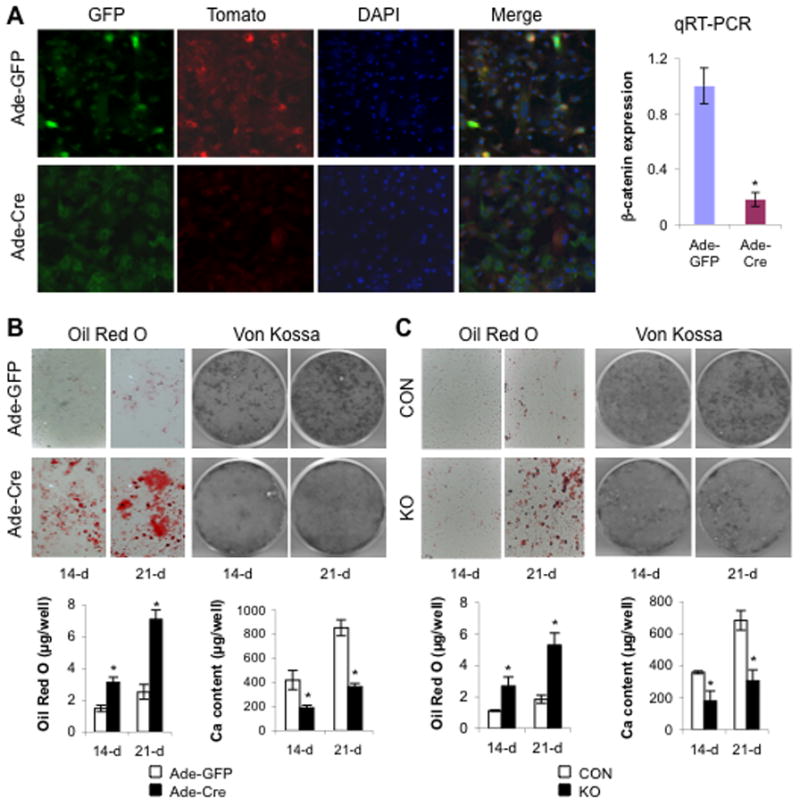
(A) Confocal images of BMSCs isolated from femurs and tibiae of (mT/mG);β-cat(f/f) mice at 8 weeks of age and infected with ade-GFP (upper panel) or ade-Cre (lower panel) at 10 days. Bar graph shows qRT-PCR analysis of mRNAs encoding β-catenin. (B) Representative images of Oil Red O and Von Kossa staining and quantification of Oil Red O and extracellular calcium content measurement (bar graphs) from infected BMSCs cultured in adipogenic or osteogenic medium for the indicated time. (C) Representative images of Oil Red O and Von Kossa staining and quantification of Oil Red O and extracellular calcium content measurement (bar graphs) from BMSCs isolated from 8-wk old femurs and tibiae of β-catf/f (CON) and osx-cre;β-catf/f (KO) mice treated with Dox until sacrifice and cultured in adipogenic or osteogenic medium for the indicated time. Data are plotted as mean ± SEM. Similar results were obtained from four independent experiments. *p < 0.05 vs control cells cultured for the same time.
Removal of β-catenin in BMSCs increases adipogenesis and decreases osteogenesis
Highly effective removal of β-catenin by ade-cre enabled us to assess the requirement for β-catenin in adipogenesis and osteogenesis of BMSCs. BMSCs were isolated from β-catf/f mice and then infected with ade-cre or control adenovirus. When exposed to adipogenic medium, the BMSCs infected with ade-cre exhibited accelerated and increased adipogenic differentiation, as shown by many more cells with accumulation of lipid droplets (visualized by Oil Red-O staining) in the BMSCs infected with ade-cre than with the ade-GFP control (Fig. 2B). This accelerated adipogenesis was evident at 14 days after infection and was even more dramatic at 21 days (Fig. 2B). In contrast, when the infected cells were cultured in osteogenic medium, mineralization, a measure of osteoblastic differentiation in vitro, was dramatically attenuated in the BMSCs infected with ade-cre, as assessed by von Kossa stain and direct measurement of calcium content (Fig. 2B). These results indicate that wnt/β-catenin signaling in bone marrow-derived mesenchymal stem cells is important for directing these cells to differentiate into osteogenic versus adipogenic lineages.
Removal of β-catenin increases transcription of adipogenic genes in BMSCs
As expected, levels of mRNAs encoding both β-catenin and axin2, a wnt target gene, were strikingly reduced in the cre-infected BMSCs cultured in either adipogenic or osteogenic medium, indicating that cre infection effectively disrupted β-catenin gene expression in both culture conditions (Fig. 3A). To address the molecular mechanisms whereby bone marrow-derived cells deprived of β-catenin in vitro increase differentiation to adipocytes, we next examined, by qRT-PCR, expression levels of several crucial genes involved in adipogenesis in the cultured BMSCs after adenoviral infection. When cultured in adipogenic medium for 14 days, levels of mRNAs encoding PPARγ, the vital transcription factor for adipogenic differentiation, were increased by about 2 fold in the BMSCs infected with ade-cre compared to control infection. This increased expression was further augmented to more than 4 fold at 21 days in adipogenic conditions (Fig. 3B). A similar but less extensive, increase in expression of C/EBPα, another key transcription factor for adipogenic differentiation, was also observed in the cultured BMSCs infected with ade-cre (Fig. 3B). As expected, levels of mRNAs encoding FABP4, a marker for mature adipocytes and a target gene of both PPARγ and C/EBPα, was increased by more than 6 fold at 14 days and by up to 26 fold at 21 days in the cre infected BMSCs compared to the control-infected cells cultured for the same intervals (Fig. 3B). Interestingly, we observed no significant effect of deletion of β-catenin in the cultured BMSCs on levels of mRNAs encoding Zfp467 (Zfp), Rb1 or Maf; three genes recently identified as key regulators for adipogenesis (30–32). In addition, a similar increase, although to a much lesser extent, in expression of PPARγ, C/EBPα and FABP4 was also observed in the cre-infected BMSCs cultured under osteogenic conditions (Fig. 3B). Taken together, these results are consistent with the possibility that the increased adipogenesis caused by deletion of β-catenin in bone marrow-derived mesenchymal stem cells was attributable to the increased expression of PPARγ and C/EBPα, two essential transcription factors for adipogenesis.
Fig. 3. Deletion of β-catenin in BMSCs increases adipogenic but suppresses osteogenic gene expression.
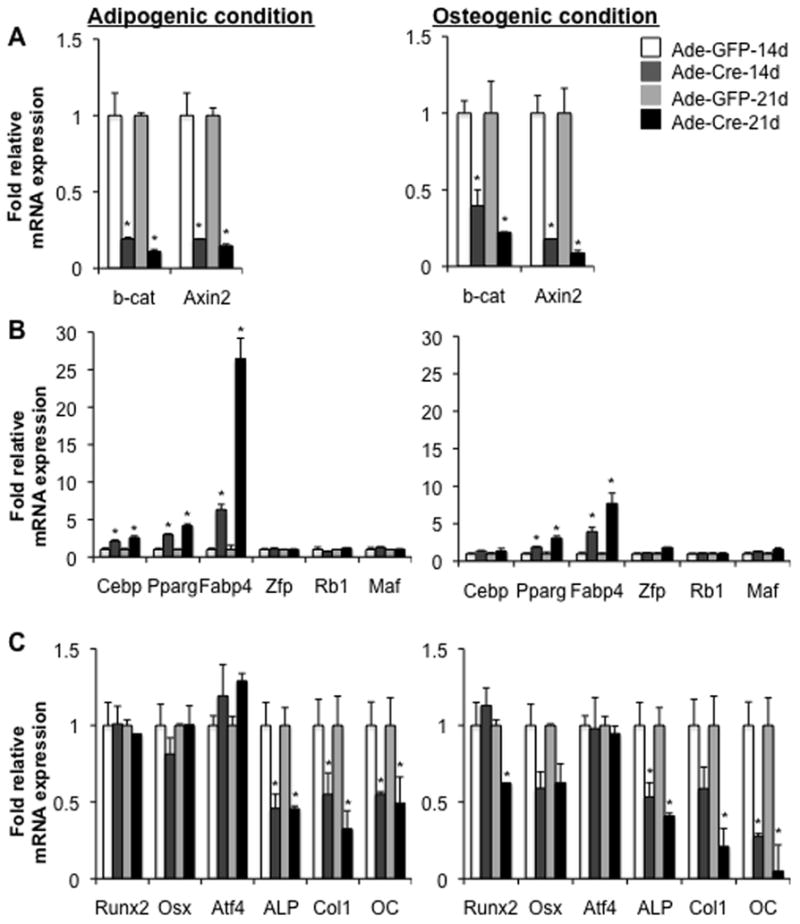
(A–C) qRT-PCR analysis of mRNAs encoding β-catenin (β-cat) and axin2 (A), adipogenic genes (B) and osteogenic transcripts (C) in BMSCs isolated from 8-wk old femurs of β-catf/f mice, infected with adenoviral-Cre (Ade-cre) or with control adenovirus (Ade-GFP), and then cultured under either adipogenic or osteogenic medium for the indicated times. Data are plotted as mean ± SEM of fold expression of each mRNA relative to the expression level observed in the control infected cells cultured at the same intervals. *p < 0.05 vs control infected cells cultured for the same time. Similar results were obtained from four independent experiments.
Deletion of β-catenin in BMSCs leads to decreased expression of osteogenic genes
To explore the molecular mechanism underlying the reduced osteoblastic differentiation, as determined by mineralization, of the cultured BMSCs following deletion of β-catenin, we next examined the effect of β-catenin knockout on osteogenic gene expression in the cultured BMSCs. Analysis by qRT-PCR showed a modest reduction in mRNAs encoding Runx2 and osterix, two essential osteogenic transcription factors in vivo, in cre-infected cells under osteogenic but not adipogenic conditions (Fig. 3C). Interestingly, levels of mRNAs encoding alkaline phosphatase (ALP) and collagen-1 (Col1α1), two early markers for osteoblastogenesis, and osteocalcin (OC), a later marker for osteoblast differentiation, were significantly reduced in the knockout cells cultured under either osteogenic or adipogenic conditions (Fig. 3C). These data suggest that deletion of β-catenin in the cultured BMSCs suppressed osteoblastic differentiation in vitro.
Decreased osteogenic differentiation and increased adipogenesis of BMSCs isolated from osx-cre;β-catf/f mice
We then sought to determine whether deletion of β-catenin in cells that had ever expressed osterix might affect adipogenesis and osteogenesis of cultured BMSCs. For this purpose, BMSCs were isolated from osx-cre;β-catf/f (KO) or β-catf/f (control) mice that had been treated with Dox until sacrifice to continuously suppress cre expression in vivo. BMSCs then were cultured under adipogenic or osteogenic conditions in the absence of Dox. We did not observe any significant changes in bone mass or bone marrow fat in knockout mice continuously treated with Dox until sacrifice (data not shown). To determine whether β-catenin disruption through the osx-cre occurs in the BMSCs isolated from KO mice and then cultured under either adipocytic or osteogenic conditions, we first examined levels of mRNAs encoding β-catenin, axin2 and lef1. As expected, mRNAs encoding β-catenin, axin2 and lef1 in the BMSCs isolated from KO mice all were significantly reduced, relative to those from control (CON) mice, and this was especially evident when cells were cultured under osteogenic conditions (Fig. 4A). This suggests that a large fraction of the isolated BMSCs had initially occupied, or subsequently traversed, the population(s) of preosteoblasts that express osterix, with resultant ablation of β-catenin under both culture conditions. We next assessed adipogenesis of the BMSCs from KO mice. Many more cells from KO than control mice differentiated into adipocytic cells, and they accumulated more lipid droplets in cytoplasm, as demonstrated by Oil Red-O staining and quantification (Fig. 2C). Consistent with the increase in fat cells, levels of mRNAs encoding FABP4, a marker of mature adipocytes, were dramatically elevated in the cultured BMSCs isolated from KO versus control mice (Fig. 5B). We further examined mRNAs encoding genes essential for adipogenesis to determine the molecular mechanism underlying the increased adipogenesis of the BMSCs derived from KO mice. The increased expression of PPARγ and C/EBPα but not of Zfp, Rb1 or Maf, in the cultured BMSCs isolated from KO mice was very similar to that observed previously in the cultured BMSCs that had been isolated from β-catf/f control mice and then infected with adenoviral-cre in vitro (Fig. 3B and 4B).
Fig. 4. Loss of β-catenin through the osterix-cre causes increased adipogenic and reduced osteogenic gene expression.
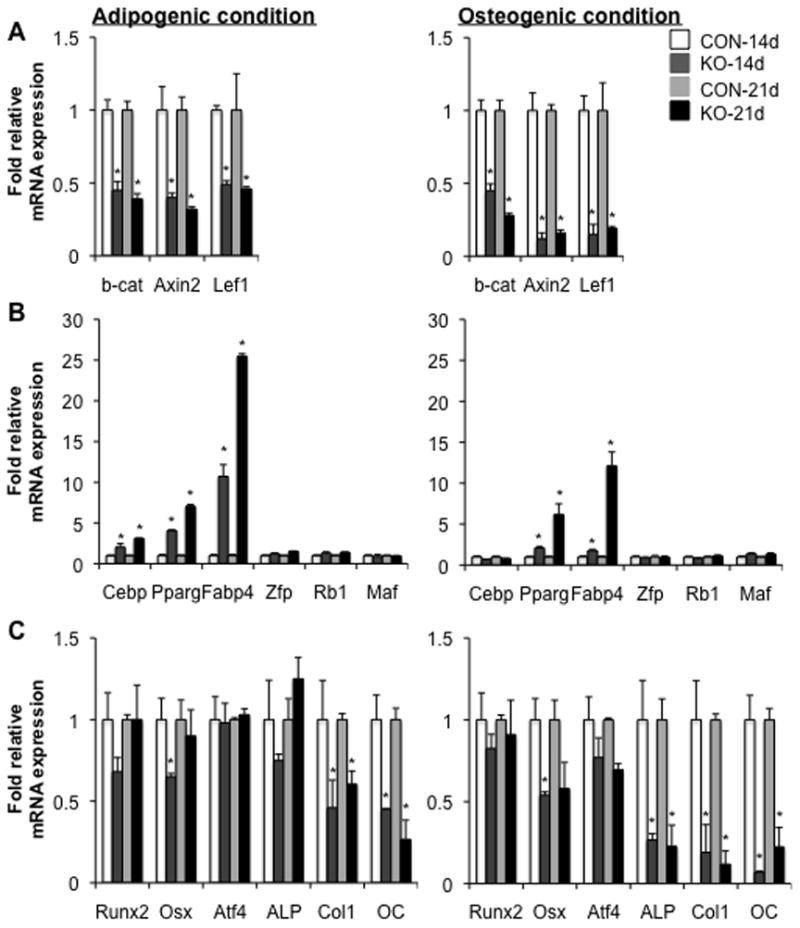
(A–C) qRT-PCR analysis of mRNAs encoding wnt target genes (A), adipogenic genes (B) and osteogenic transcripts (C) in BMSCs isolated from 8-wk old tibiae and femurs of β-catf/f (CON) and osx-cre;β-catf/f (KO) mice treated with Dox until sacrifice, and cultured under either adipogenic or osteogenic condition for 14 or 21 days, as indicated. Data are plotted as mean ± SEM. *p < 0.05 vs CON-derived cells cultured for the same time. Results were obtained from four independent experiments (4 mice per genotype).
Fig 5. Tracing the preosteoblasts that had to express osterix with the double fluorescence reporter model.
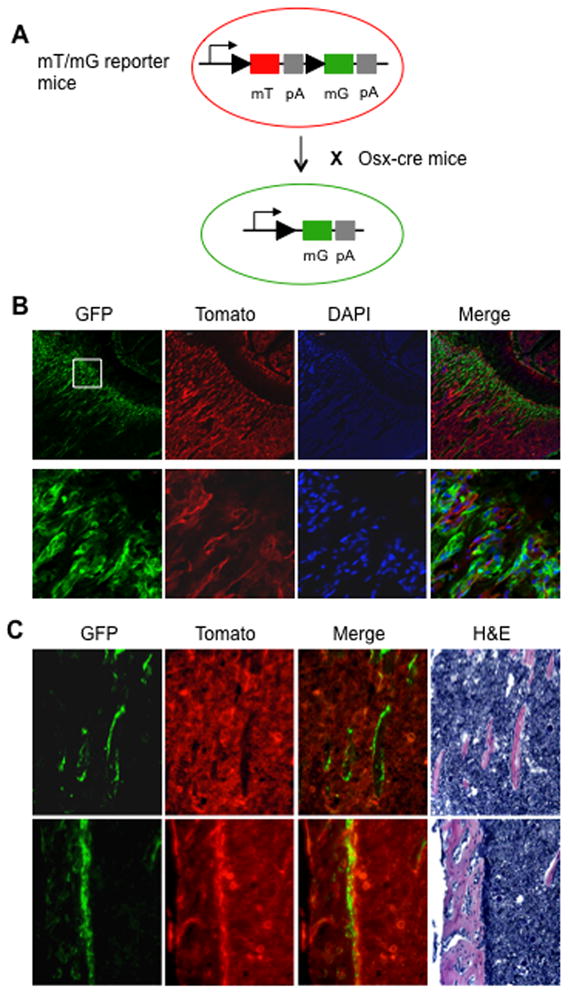
(A) Double fluorescence mT/mG reporter mice were mated to osx-cre mice. (B) Representative images by confocal microscopy of frozen sections from 4-wk old proximal tibia of mT/mG; osx-cre mice without administration of doxycycline; the lower row of images are higher-power views of the area delimited by the white box indicating the primary spongiosa. (C) Representative images first by fluorescence microscopy and then histology of same frozen sections from the same proximal tibia shown above; the upper row of images showing the secondary spongiosa and the lower row of images showing the cortical bone. All the images clearly demonstrate that the GFP-positive cells are restricted on bone but not in the marrow area.
We next evaluated osteoblastogenesis of the cultured BMSCs isolated from KO mice. As expected, mineralized nodules and calcium content were reduced in the BMSCs cultured from KO mice compared to that observed in the BMSCs from control mice, indicating that osteoblastogenesis of BMSCs was attenuated by deletion of β-catenin in the cells that had to express osterix (Fig. 2C). We further analyzed mRNAs encoding transcription factors essential for osteogenesis and markers for osteoblastogenesis. Expression levels of osterix and Runx2 were modestly reduced, whereas expression of ALP, Col1α1 and OC was dramatically attenuated, in the cultured BMSCs isolated from KO mice (Fig. 4C).
Deletion of β-catenin causes cell fate shift of preosteoblasts to adipocytes
Finally, we determined whether the increased adipogenesis of BMSCs isolated from KO mice was attributed to a cell fate change of preosteoblasts from osteoblasts to adipocytes. To test this hypothesis, we cultured BMSCs isolated from both osx-cre; mT/mG and osx-cre;β-catf/f; mT/mG Dox-treated mice under adipogenic conditions, and then performed cellular immune fluorescence and Oil Red-O staining to identify the adipocytic cells. Cells that had expressed osx-cre, and presumably thereby undergone deletion of β-catenin, would also be expected to express GFP (green) and not the (cre-sensitive) tomato marker. Thus, by using the mT/mG double fluorescence reporter model, we could trace the preosteoblasts that had ever expressed osterix and in which β-catenin had been knocked out (Fig. 5). Using this lineage tracing we observed no bone marrow cells that expressed GFP, further confirming the previous finding that osterix is not expressed in the bone marrow cells including uncommitted bone marrow mesenchymal cells (Fig. 5) (33). We observed that Oil Red O and anti-FABP4 staining showed an identical pattern, indicating that the cells positive for FABP4 represent adipocytic cells. Strikingly, 21 days after culture under adipogenic conditions, a number of bone marrow-derived cells from osx-cre;β-catf/f; mT/mG mice had differentiated into FABP4-positive cells that also expressed GFP at the same time. Such FABP4-positive cells co-expressing GFP comprised approximately 40% of total FABP4-positive cells (Fig. 6 and 7). However, no FABP4-positive cells that expressed GFP at the same time were observed in the bone marrow-derived cells from control osx-cre; mT/mG mice (Fig. 6 and 7), and notably, the total number of FABP4-positive cells was much higher in the cultured cells from KO mice than that observed in control cells (Fig. 6A bar graph). These novel findings indicate that loss of wnt/β-catenin signaling determines reprogramming of osterix-expressing marrow preosteoblasts from osteoblasts to adipocytes.
Fig. 6. Loss of β-catenin causes cell fate shift of preosteoblasts.
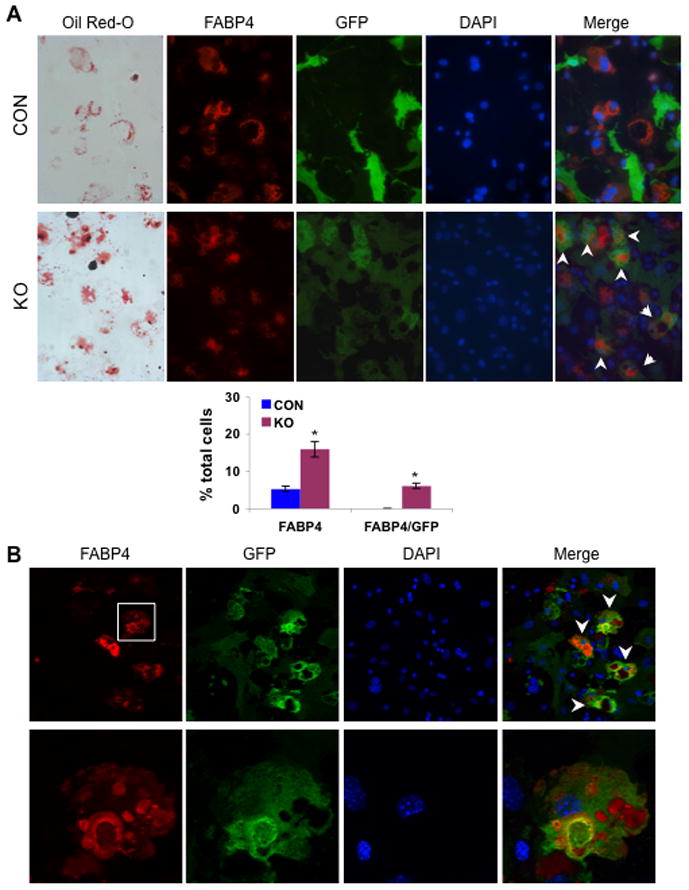
Bone marrow derived cells were isolated from 8-wk old femurs and tibiae of mT/mG; osx-cre (CON) and mT/mG; osx-cre;β-catf/f (KO) mice treated with Dox until sacrifice, cultured under adipogenic condition for 21 days, and analyzed by Oil Red O staining and cellular immunofluorescent detection for FABP4 (red) and GFP (green). (A) Fluorescent microscope images show adipocytic cells that expressed both FABP4 and GFP at the same time (indicated by white arrows) in the cultured BMSCs exclusively from KO mice but not from CON mice at all, and bar graph quantitatively demonstrates a dramatic increase in number of FABP4 positive cells in the KO-derived culture. Data are plotted as mean ± SEM. *p < 0.05 vs CON-derived cells. (B) Representative confocal images of the cultured cells from KO mice (the lower row of images indicating magnified views of the cells delimited by the white box) show the adipocytic cells that strongly express both FABP4 and GFP at the same time (as indicated by arrowheads).
Fig. 7. Confocal microscopy of adipocytic cells positive for FABP4 and either GFP or Tomato.
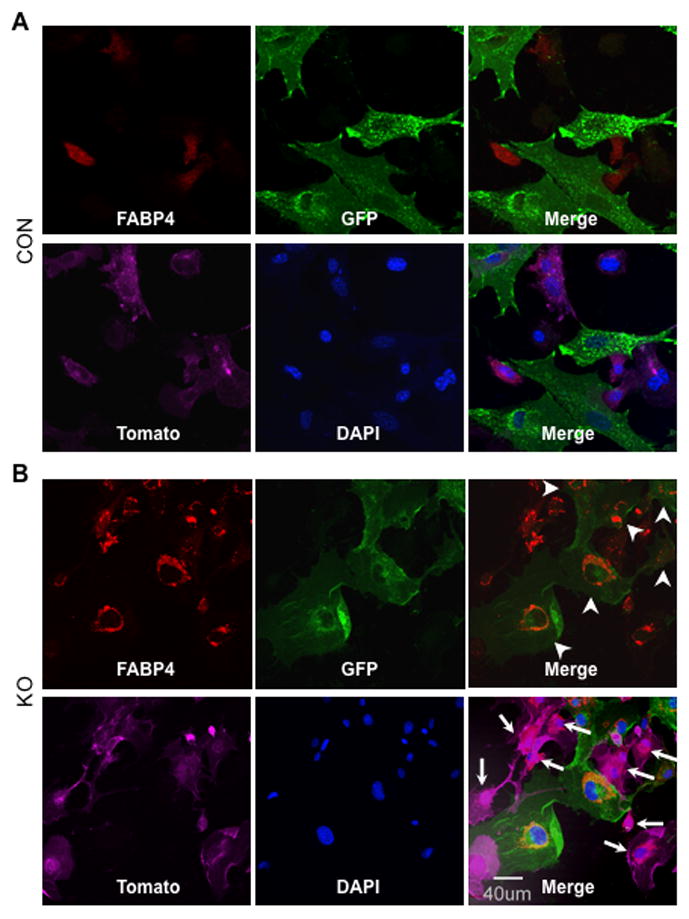
Bone marrow derived cells were isolated from 8-wk old femurs and tibiae of mT/mG; osx-cre (CON) and mT/mG; osx-cre;β-catf/f (KO) mice treated with Dox until sacrifice, and cultured under adipogenic condition for 21 days. The cultured cells underwent immunofluorescent staining only for FABP4 and then were analyzed by confocal microscopy for FABP4 (red), GFP (green), tomato (pink) and DAPI (blue). (A) The confocal images show that in CON-derived BMSCs the FABP4-positive cells only express tomato and none express GFP. (B) The confocal images of the cultured cells from KO mice demonstrate FABP4-positive cells that also express either tomato (arrows) or GFP (arrowheads), but not both, at the same time.
In vivo lineage tracing demonstrates the preosteoblast-derived adipocytes in the KO bone marrow
To further investigate whether removal of β-catenin causes a cell fate shift of the preosteoblasts from osteoblasts to adipocytes, we performed an in vivo lineage tracing study using the mT/mG reporter mice. As expected, five-month old KO mice treated with Dox until two months of age exhibited a dramatic increase in bone marrow fat as revealed by Oil Red O staining of frozen sections derived from the proximal tibia (Fig. 8D). The increased adiposity in the KO tibiae was also indicated by a significant increase in mRNAs encoding adipogenic genes in marrow cells from the mice (Fig. 8E). We also observed a dramatic increase in the number of FABP4-positive cells in the KO bone marrow in vivo (Fig. 8A–C and Fig. S1). More strikingly, and consistent with the findings of the in vitro lineage tracing experiments, a large number of FABP4-positive cells that co-express GFP were visible in the bone marrow in KO mice but few were seen in CON mice (Fig. 8A–C and Fig. S1 and S2). Some of the GFP-positive cells in the KO bone marrow did not express FABP4, while a large fraction of the FABP4 positive cells did not express GFP (Fig. 8A–C, and Fig. S1 and S2). These findings observed from the in vivo lineage tracing further indicate that loss of β-catenin can lead to the cell fate shift of preosteoblasts from osteoblasts to adipocytes, likely through trans- and/or de-differentiation mechanisms.
Fig. 8. In vivo lineage tracing and assessment of adiposity.
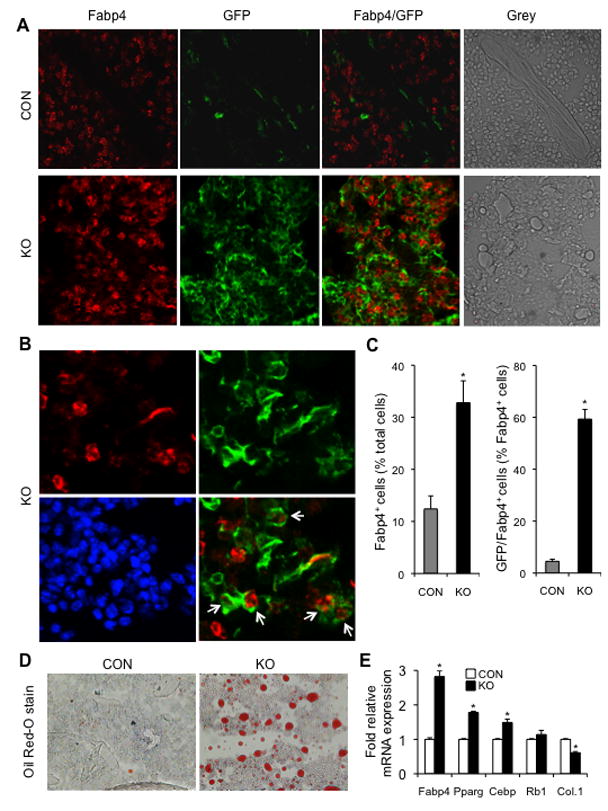
(A) Confocal microscopy of 5-month old proximal tibiae from mT/mG; osx-cre (CON) and mT/mG; osx-cre;β-catf/f (KO) mice treated with Dox until 2-month old. “Grey” denotes the images without fluorescence. Many GFP-positive cells and cells double positive for GFP and FABP4 in the bone marrow were observed in KO but few in CON mice, and FABP4-positive cells were dramatically increased in KO mice. (B) Representative images from the KO confocal microscopy show some FABP4 positive cells that co-expressed GFP (as indicated by white arrows in the merged image of FABP4/GFP; Red: FABP4, Green: GFP and blue: DAPI stain for nucleus). (C) Bar graphs represent a quantitative assessment of FABP4 positive cells and the cells positive for both FABP4 and GFP in the metaphyseal trabecular region of the proximal tibia. Data are expressed as mean ± SEM. *p < 0.01 vs CON mice. We counted blindly the cells positive for both GFP and FABP4 from a total of approximately 1500 or 2100 FABP4 positive cells derived from four CON or four KO mice, respectively. (D) Representative micrographs of Oil Red O stain from the frozen sections of the proximal tibiae described in A and B. (E) qRT-PCR analysis of mRNAs encoding adipogenic genes; total RNA was isolated from one tibia of the same mice described in A and B. There were four mice in each group and data are plotted as mean ± SEM; *p < 0.05 vs CON mice.
Discussion
Osterix is a zinc-finger domain-containing transcription factor that is required for the differentiation of preosteoblasts into functional osteoblasts, and is specifically expressed in osteoblast-lineage cells and at lower levels in prehypertrophic chondrocytes (34). Osterix is not expressed in bone marrow cells (see Fig. 5) or in uncommitted bone marrow mesenchymal cells (33). In osterix-null mice, which die at birth, a full cartilage skeleton is formed but the embryos completely lack bone formation (34). We deleted β-catenin and traced the deleted cells, using the osterix-cre transgenic, which uses osterix promoter sequences to express cre in the preosteoblasts (20). The data reported here indicate that wnt/β-catenin signaling plays a crucial role in controlling osteoblast and adipocyte differentiation, not only in multipotent BMSCs but also in lineage-committed preosteoblasts that express (or had once expressed) osterix. Removal of β-catenin from BMSCs in vitro caused these cells to more readily differentiate into the adipocytic lineage, and, more strikingly, deletion of β-catenin in osterix-expressing preosteoblasts using adenoviral-cre in vitro resulted in the cell fate shift of these cells from osteoblasts to adipocytes.
The relationship between osteoblastogenesis and adipogenesis is complex. During development, mesenchymal precursor cells can selectively differentiate into adipocytes, osteoblasts, or other cell types (1), and so it is widely accepted that decreased bone formation is typically accompanied by increased fat formation (6). Many factors including wnt signaling are shown essential for the lineage commitment of mesenchymal precursor cells during development. In vitro, wnt10b supplementation increases osteoblastogenesis and decreases adipogenesis in ST2 cells, a bone marrow-derived stromal cell line, and similar findings were observed when cultured cells were treated with wnt1 (14,25). β-catenin is the key component of the canonical wnt pathway, and it regulates wnt target gene transcription when this pathway is activated by any of a variety of wnt ligands (wnt10b, wnt1, wnt3, etc). Thus, β-catenin is a critical target for exploring the function of the canonical wnt pathway. Stabilization of β-catenin in a bone marrow-derived stromal cell line can promote osteoblastogenesis and inhibit adipogenesis (25), and over-expression of β-catenin in preadipocytes can also inhibit their differentiation into mature adipocytes (15). These in vitro data showed the importance of the canonical wnt pathway for determining both osteoblast and adipocyte differentiation. Interestingly, however, increased bone marrow adiposity has not been reported in mice with deletion of β-catenin either from early progenitors of the osteoblast lineage in fetal life (17–20) or from mature osteoblasts (21–23). Here we observed that blocking the canonical wnt pathway by disrupting β-catenin within early cells of the osteoblast lineage in postnatal mice causes osteopenia and increased bone marrow adiposity. Our data indicate that wnt/β-catenin signaling in early cells of the osteoblast lineage is not only essential for bone formation but also controls (restrains) bone marrow fat formation in adult animals. Our lineage tracing experiment in bone in vivo further indicates that removal of β-catenin in preosteoblasts causes a cell fate shift of these cells from osteoblasts to adipocytes. In vivo lineage tracing shows that inactivation of β-catenin from preosteoblasts through the osterix cre induces many preosteoblast-derived cells to become adipogenic cells in the bone marrow microenvironment, and that normally such preosteoblast-derived adipocytic cells are few in number in the bone marrow microenvironment. This reprogramming of preosteoblasts determined by β-catenin could be explained by two possible mechanisms. First, preosteoblasts, upon removal of β-catenin, may trans-differentiate directly into the adipocytic lineage and proceed to become mature adipocytes. Alternatively, preosteoblasts deficient in β-catenin may first de-differentiate into multipotent progenitor cells and then enter into the adipocytic lineage. It will be important to determine which one or both of these scenarios pertains following osterix-directed deletion of β-catenin. We also observed, in our cultures of BMSCs isolated from knockout mice, that, by comparison with the control cells, there were many more adipocytes that did not express the GFP marker, while such increase in the GFP-negative adipocytes in the KO mice was less prominent in the lineage tracing in vivo. It seems, at least in vitro, that knockout of β-catenin from preosteoblasts may cause an indirect effect on adjacent cells that do not express osterix, stimulating the production of more adipocytes.
We further explored the molecular mechanism(s) underlying the increased adipogenesis and reprogramming of preosteoblasts from osteoblasts to adipocytes, and found that blockage of wnt/β-catenin signaling in preosteoblsts dramatically upregulated expression of two transcription factors determinant for adipogenesis, C/EBPα and PPARγ, but caused only modest effects on two crucial transcription factors for osteoblastogenesis, Runx2 and osterix. Several lines of evidence indicate that wnt/β-catenin signaling regulates expression of C/EBPα and PPARγ. Transient activation of wnt signaling suppresses expression of mRNAs encoding C/EBPα and PPARγ (14). GSK3β, which can block the canonical wnt pathway by degrading β-catenin, directly regulates the activity of C/EBPβ, an early inducible adipogenic transcription factor, by phosphorylation of its Ser-184 and Thr-179 with subsequent activation of transcription of C/EBPα and PPARγ (35). These studies indicated that wnt/β-catenin signaling controls the adipogenic process through directly regulating the activity of early transcription factors for adipogenesis. Moreover, PPARγ may bind to β-catenin and regulate its activity. For example, in mouse fibroblasts activation of PPARγ promotes the degradation of phosphorylated β-catenin, which is GSK3β-dependent (36) and this interaction is mediated by a region within PPARγ that is highly homologous to the β-catenin binding domain in TCF/LEF (37). Furthermore, in multipotent mesenchymal precursor cells the balance between osteogenic and adipogenic transcription factors is also important for the cells to maintain their quiescence and to determine lineage commitment. Osteogenic and adipogenic transcription factors can regulate each other by reciprocal inhibition. Once activated, PPARγ not only suppresses the expression of Runx2 but also interferes with the transcriptional ability of Runx2, and thus inhibits osteoblast differentiation (38,39). In our study, the decreased osteoblastic differentiation of BMSCs upon adenoviral-cre deletion of β-catenin and of osteoblast progenitors following β-catenin removal through osterix-cre expression might be attributed to the decreased transcriptional activity of Runx2 caused by the increased expression of PPARγ and C/EBPα.
In addition to PPARγ and C/EBPα, both retinoblastoma gene (Rb1) (31) and Maf (32) were recently identified as important factors that can regulate mesenchymal cell bifurcation into osteoblasts versus adipocytes. The retinoblastoma protein (pRb), product of the Rb1 gene, can bind to Runx2 and potentiate its ability to promote osteogenic differentiation in vitro (40). pRb also acts with E2F to suppress PPARγ, the master activator of adipogenesis (41). Knockdown of Rb1 promotes PPARγ expression, and its restoration induces downregulation of adipogenic markers. Moreover, its appearance is sufficient to switch lineage specification between osteoblastic commitment and multipotency. The crucial role of Maf in osteoblast and adipocyte lineage commitment is determined by the severe low bone mass phenotype, with increased bone marrow adiposity, seen in Maf knockout mice (32). Maf disturbs adipocyte differentiation by suppressing the interaction of Cebpδ and CREB-binding protein gene (Crebbp) and controls osteoblast differentiation through binding to Runx2 and directly regulating Bglasp1 (encoding osteocalcin). However, in our study, we did not observe any significant changes in the expression of these two genes following removal of β-catenin from either BMSCs or from the preosteoblasts that had to express osterix. Taken together, our data indicate that wnt/β-catenin signaling controls the fate of preosteoblasts, the lineage-committed early osteoblasts, with respect to differentiation into osteoblastic vs. adipocytic populations in bone, and that this correlates with increased expression of PPARγ and C/EBPα but not of Rb1 and Maf. The molecular mechanism underlying the reprogramming of preosteoblasts is very similar to that observed previously in myoblasts (15). This is the first demonstration that loss of β-catenin can actively reprogram preosteoblasts from osteoblasts to adipocytes. Our findings may offer potential insight into the mechanisms underlying increased bone marrow adiposity.
Supplementary Material
Acknowledgments
We thank Andrew P. McMahon (Harvard University) for generously providing osx-cre mice, Mary L. Bouxsein for μCT analysis of bone phenotype and Jie Zhao (MGH-Core Center for Photomedicine) for expert assistance in confocal microscopy. This work was supported by the MGH-Department of Medicine (F.R.B. and J.G.), and by a grant (PO1 DK11794 to H.M.K.) from the National Institutes of Health.
Footnotes
Disclosure
All authors state that they have no conflicts of interest.
Authors’ roles: Study design: JG. Study conduct: LS, ML, NO and JG. Data analysis: LS, NO, FRB, HMK and JG. Drafting manuscript: LS and JG. Revising manuscript: FRB, HMK, NO, and JG.
References
- 1.Nuttall ME, Gimble JM. Is there a therapeutic opportunity to either prevent or treat osteopenic disorders by inhibiting marrow adipogenesis? Bone. 2000;27(2):177–84. doi: 10.1016/s8756-3282(00)00317-3. [DOI] [PubMed] [Google Scholar]
- 2.Minaire P, Neunier P, Edouard C, Bernard J, Courpron P, Bourret J. Quantitative histological data on disuse osteoporosis: comparison with biological data. Calcif Tissue Res. 1974;17(1):57–73. doi: 10.1007/BF02547214. [DOI] [PubMed] [Google Scholar]
- 3.Ahdjoudj S, Lasmoles F, Holy X, Zerath E, Marie PJ. Transforming growth factor beta2 inhibits adipocyte differentiation induced by skeletal unloading in rat bone marrow stroma. J Bone Miner Res. 2002;17(4):668–77. doi: 10.1359/jbmr.2002.17.4.668. [DOI] [PubMed] [Google Scholar]
- 4.Wronski TJ, Morey-Holton E, Jee WS. Skeletal alterations in rats during space flight. Adv Space Res. 1981;1(14):135–40. doi: 10.1016/0273-1177(81)90254-4. [DOI] [PubMed] [Google Scholar]
- 5.Meunier P, Aaron J, Edouard C, Vignon G. Osteoporosis and the replacement of cell populations of the marrow by adipose tissue. A quantitative study of 84 iliac bone biopsies. Clin Orthop Relat Res. 1971;80:147–54. doi: 10.1097/00003086-197110000-00021. [DOI] [PubMed] [Google Scholar]
- 6.Kajkenova O, Lecka-Czernik B, Gubrij I, Hauser SP, Takahashi K, Parfitt AM, Jilka RL, Manolagas SC, Lipschitz DA. Increased adipogenesis and myelopoiesis in the bone marrow of SAMP6, a murine model of defective osteoblastogenesis and low turnover osteopenia. J Bone Miner Res. 1997;12(11):1772–9. doi: 10.1359/jbmr.1997.12.11.1772. [DOI] [PubMed] [Google Scholar]
- 7.Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M. Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology. 2001;2(3):165–71. doi: 10.1023/a:1011513223894. [DOI] [PubMed] [Google Scholar]
- 8.Rosen ED, Spiegelman BM. Molecular regulation of adipogenesis. Annu Rev Cell Dev Biol. 2000;16:145–71. doi: 10.1146/annurev.cellbio.16.1.145. [DOI] [PubMed] [Google Scholar]
- 9.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14(11):1293–307. [PubMed] [Google Scholar]
- 10.Darlington GJ, Ross SE, MacDougald OA. The role of C/EBP genes in adipocyte differentiation. J Biol Chem. 1998;273(46):30057–60. doi: 10.1074/jbc.273.46.30057. [DOI] [PubMed] [Google Scholar]
- 11.Krishnan V, Bryant HU, Macdougald OA. Regulation of bone mass by Wnt signaling. J Clin Invest. 2006;116(5):1202–9. doi: 10.1172/JCI28551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gambardella A, Nagaraju CK, O’Shea PJ, Mohanty ST, Kottam L, Pilling J, Sullivan M, Djerbi M, Koopmann W, Croucher PI, Bellantuono I. Glycogen synthase kinase-3alpha/beta inhibition promotes in vivo amplification of endogenous mesenchymal progenitors with osteogenic and adipogenic potential and their differentiation to the osteogenic lineage. J Bone Miner Res. 2011;26(4):811–21. doi: 10.1002/jbmr.266. [DOI] [PubMed] [Google Scholar]
- 13.Guo W, Flanagan J, Jasuja R, Kirkland J, Jiang L, Bhasin S. The effects of myostatin on adipogenic differentiation of human bone marrow-derived mesenchymal stem cells are mediated through cross-communication between Smad3 and Wnt/beta-catenin signaling pathways. J Biol Chem. 2008;283(14):9136–45. doi: 10.1074/jbc.M708968200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kang S, Bennett CN, Gerin I, Rapp LA, Hankenson KD, Macdougald OA. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J Biol Chem. 2007;282(19):14515–24. doi: 10.1074/jbc.M700030200. [DOI] [PubMed] [Google Scholar]
- 15.Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, MacDougald OA. Inhibition of adipogenesis by Wnt signaling. Science. 2000;289(5481):950–3. doi: 10.1126/science.289.5481.950. [DOI] [PubMed] [Google Scholar]
- 16.Chen JR, Lazarenko OP, Wu X, Tong Y, Blackburn ML, Shankar K, Badger TM, Ronis MJ. Obesity reduces bone density associated with activation of PPARgamma and suppression of Wnt/beta-catenin in rapidly growing male rats. PLoS One. 2010;5(10):e13704. doi: 10.1371/journal.pone.0013704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hill TP, Spater D, Taketo MM, Birchmeier W, Hartmann C. Canonical Wnt/beta-catenin signaling prevents osteoblasts from differentiating into chondrocytes. Dev Cell. 2005;8(5):727–38. doi: 10.1016/j.devcel.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 18.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8(5):739–50. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 19.Hu H, Hilton MJ, Tu X, Yu K, Ornitz DM, Long F. Sequential roles of Hedgehog and Wnt signaling in osteoblast development. Development. 2005;132(1):49–60. doi: 10.1242/dev.01564. [DOI] [PubMed] [Google Scholar]
- 20.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133(16):3231–44. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 21.Glass DA, 2nd, Bialek P, Ahn JD, Starbuck M, Patel MS, Clevers H, Taketo MM, Long F, McMahon AP, Lang RA, Karsenty G. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8(5):751–64. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 22.Holmen SL, Zylstra CR, Mukherjee A, Sigler RE, Faugere MC, Bouxsein ML, Deng L, Clemens TL, Williams BO. Essential role of beta-catenin in postnatal bone acquisition. J Biol Chem. 2005;280(22):21162–8. doi: 10.1074/jbc.M501900200. [DOI] [PubMed] [Google Scholar]
- 23.Kramer I, Halleux C, Keller H, Pegurri M, Gooi JH, Weber PB, Feng JQ, Bonewald LF, Kneissel M. Osteocyte Wnt/beta-catenin signaling is required for normal bone homeostasis. Mol Cell Biol. 2010;30(12):3071–85. doi: 10.1128/MCB.01428-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bennett CN, Ouyang H, Ma YL, Zeng Q, Gerin I, Sousa KM, Lane TF, Krishnan V, Hankenson KD, MacDougald OA. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22(12):1924–32. doi: 10.1359/jbmr.070810. [DOI] [PubMed] [Google Scholar]
- 25.Bennett CN, Longo KA, Wright WS, Suva LJ, Lane TF, Hankenson KD, MacDougald OA. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102(9):3324–9. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brault V, Moore R, Kutsch S, Ishibashi M, Rowitch DH, McMahon AP, Sommer L, Boussadia O, Kemler R. Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development. 2001;128(8):1253–64. doi: 10.1242/dev.128.8.1253. [DOI] [PubMed] [Google Scholar]
- 27.Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double-fluorescent Cre reporter mouse. Genesis. 2007;45(9):593–605. doi: 10.1002/dvg.20335. [DOI] [PubMed] [Google Scholar]
- 28.Yang D, Guo J, Divieti P, Bringhurst FR. Parathyroid hormone activates PKC-delta and regulates osteoblastic differentiation via a PLC-independent pathway. Bone. 2006;38(4):485–96. doi: 10.1016/j.bone.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 29.Yang D, Singh R, Divieti P, Guo J, Bouxsein ML, Bringhurst FR. Contributions of parathyroid hormone (PTH)/PTH-related peptide receptor signaling pathways to the anabolic effect of PTH on bone. Bone. 2007;40(6):1453–61. doi: 10.1016/j.bone.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quach JM, Walker EC, Allan E, Solano M, Yokoyama A, Kato S, Sims NA, Gillespie MT, Martin TJ. Zinc finger protein 467 is a novel regulator of osteoblast and adipocyte commitment. J Biol Chem. 2011;286(6):4186–98. doi: 10.1074/jbc.M110.178251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calo E, Quintero-Estades JA, Danielian PS, Nedelcu S, Berman SD, Lees JA. Rb regulates fate choice and lineage commitment in vivo. Nature. 2010;466(7310):1110–4. doi: 10.1038/nature09264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nishikawa K, Nakashima T, Takeda S, Isogai M, Hamada M, Kimura A, Kodama T, Yamaguchi A, Owen MJ, Takahashi S, Takayanagi H. Maf promotes osteoblast differentiation in mice by mediating the age-related switch in mesenchymal cell differentiation. J Clin Invest. 2010;120(10):3455–65. doi: 10.1172/JCI42528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma’ayan A, Enikolopov GN, Frenette PS. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–34. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 35.Tang QQ, Gronborg M, Huang H, Kim JW, Otto TC, Pandey A, Lane MD. Sequential phosphorylation of CCAAT enhancer-binding protein beta by MAPK and glycogen synthase kinase 3beta is required for adipogenesis. Proc Natl Acad Sci U S A. 2005;102(28):9766–71. doi: 10.1073/pnas.0503891102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu J, Farmer SR. Regulating the balance between peroxisome proliferator-activated receptor gamma and beta-catenin signaling during adipogenesis. A glycogen synthase kinase 3beta phosphorylation-defective mutant of beta-catenin inhibits expression of a subset of adipogenic genes. J Biol Chem. 2004;279(43):45020–7. doi: 10.1074/jbc.M407050200. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Wang H, Zuo Y, Farmer SR. Functional interaction between peroxisome proliferator-activated receptor gamma and beta-catenin. Mol Cell Biol. 2006;26(15):5827–37. doi: 10.1128/MCB.00441-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan E, Abu-Amer Y. Activation of peroxisome proliferator-activated receptor-gamma inhibits differentiation of preosteoblasts. J Lab Clin Med. 2003;142(1):29–34. doi: 10.1016/S0022-2143(03)00058-1. [DOI] [PubMed] [Google Scholar]
- 39.Jeon MJ, Kim JA, Kwon SH, Kim SW, Park KS, Park SW, Kim SY, Shin CS. Activation of peroxisome proliferator-activated receptor-gamma inhibits the Runx2-mediated transcription of osteocalcin in osteoblasts. J Biol Chem. 2003;278(26):23270–7. doi: 10.1074/jbc.M211610200. [DOI] [PubMed] [Google Scholar]
- 40.Thomas DM, Carty SA, Piscopo DM, Lee JS, Wang WF, Forrester WC, Hinds PW. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell. 2001;8(2):303–16. doi: 10.1016/s1097-2765(01)00327-6. [DOI] [PubMed] [Google Scholar]
- 41.Fajas L, Landsberg RL, Huss-Garcia Y, Sardet C, Lees JA, Auwerx J. E2Fs regulate adipocyte differentiation. Dev Cell. 2002;3(1):39–49. doi: 10.1016/s1534-5807(02)00190-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.