

Abstract
Endocrine release of insulin principally controls glucose homeostasis. Nutrient-induced exocytosis of insulin granules from pancreatic β-cells involves ion channels and mobilization of Ca2+ and cyclic AMP (cAMP) signalling pathways. Whole-animal physiology, islet studies and live-β-cell imaging approaches reveal that ablation of the kinase/phosphatase anchoring protein AKAP150 impairs insulin secretion in mice. Loss of AKAP150 impacts L-type Ca2+ currents, and attenuates cytoplasmic accumulation of Ca2+ and cAMP in β-cells. Yet surprisingly AKAP150 null animals display improved glucose handling and heightened insulin sensitivity in skeletal muscle. More refined analyses of AKAP150 knock-in mice unable to anchor protein kinase A or protein phosphatase 2B uncover an unexpected observation that tethering of phosphatases to a seven-residue sequence of the anchoring protein is the predominant molecular event underlying these metabolic phenotypes. Thus anchored signalling events that facilitate insulin secretion and glucose homeostasis may be set by AKAP150 associated phosphatase activity.
Keywords: A-kinase anchoring protein (AKAP), calcineurin (PP2B), cyclic-AMP-dependent protein kinase (PKA), glucoregulation, glucose-stimulated insulin secretion (GSIS)
Introduction
Reversible phosphorylation of proteins contributes to the regulation of glucose homeostasis and insulin action. Stimulus-secretion coupling of insulin release from β-cells is a multistep process: Ca2+ influx triggers fusion of insulin granules with the cell membrane and this can be modulated by calmodulin, phospholipid or cAMP-dependent protein kinases (Bratanova-Tochkova et al, 2002; Hiriart and Aguilar-Bryan, 2008). Release of insulin can be influenced by several classes of protein phosphatases, including the Ca2+/calmodulin-dependent enzyme PP2B (calcineurin) (Sim et al, 2003). Yet the same cohort of second messenger-regulated enzymes enacts hormone-stimulated changes in glucose uptake and utilization in insulin target tissues. The differential use of these kinase and phosphatase combinations is made possible by anchoring proteins that provide a molecular framework for the compartmentalization of these enzymes. The principle functions of these non-catalytic regulatory proteins are two-fold: to target protein kinases and phosphatases to cellular microenvironments where they have access to particular substrates, and to segregate their binding partners in a manner that prevents the indiscriminate relay of signals (Scott and Pawson, 2009). Prototypic examples of these signal-organizing elements are A-kinase anchoring proteins (AKAPs), a group of multivalent binding proteins classified on the basis of their ability to interact with the cAMP-dependent protein kinase (PKA). It is recognized that PKA phosphorylation contributes to β-cell physiology (Seino and Shibasaki, 2005), though the role of PKA anchoring is less well understood. While peptides that globally disrupt the PKA–AKAP interface negatively impact incretin-stimulated insulin secretion (Lester et al, 1997), the anchoring proteins responsible have not been identified.
One anchoring protein of particular interest is AKAP79/150 (gene name: AKAP5) that targets enzymes including PKA, a guanine nucleotide exchange factor (Epac2), protein kinase C (PKC) and PP2B to the membrane (Hoshi et al, 2005; Nijholt et al, 2008). We hypothesized that the murine orthologue, AKAP150, could be a potential modulator of glucose homeostasis, as previous studies have shown that this anchoring protein is functionally coupled to G protein-coupled receptors, adenylyl cyclases and ion channels (Gao et al, 1997; Fraser et al, 2000; Bauman et al, 2006). In this report, we show that manipulation of the AKAP150 gene has metabolic implications for glucose homeostasis. AKAP150 null mice secrete less insulin from β-cells, yet display improved glucose handling because of increased insulin sensitivity in target tissues. These metabolically advantageous characteristics are retained in AKAP150ΔPIX mice that lack a seven amino-acid sequence responsible for tethering PP2B. Hence anchoring of PP2B is an hitherto unrecognized molecular and metabolic determinant that contributes to glucose homeostasis.
Results
AKAP150 organizes insulin secretion
Membrane depolarization and the concurrent mobilization of Ca2+ and cAMP signalling cascades drive the regulated exocytosis of insulin granules (Hinke et al, 2004; Hiriart and Aguilar-Bryan, 2008; Seino et al, 2011). Although anchored PKA augments GLP-1 mediated insulin secretion (Lester et al, 1997), a question of broader significance is whether AKAPs facilitate nutrient-induced insulin release. Several RII binding proteins were detected in INS-1(832/13) insulinoma cells by overlay assay (Figure 1A). These included AKAP150 and AKAP220 (Figure 1B; Supplementary Figure S1A and B): two channel-associated AKAPs that integrate Ca2+ and cAMP signals (Gao et al, 1997; Yang et al, 2008). A gene-silencing screen evaluated the role of each AKAP in hormone secretion from INS-1 cells. Co-transfected human growth hormone (hGH) served as a marker of exocytosis (Supplementary Figure S1C–J). Under basal conditions, insulin secretion was unaltered from AKAP150-depleted cells (Figure 1C and D; n=6). Blunting effects on insulin secretion were measured upon administration of 20 mM glucose and in response to GLP-1 or forskolin (Figure 1D and E). In contrast, gene silencing of AKAP220 had no measurable effect on basal or stimulated hormone release from INS-1 cells (Supplementary Figure S1F–J).
Figure 1.

Loss of AKAP150 supresses insulin secretion in vitro and in vivo. (A) Detection of AKAPs in INS-1(832/13) lysate by digoxigenin-labelled RII overlay in the presence of 50 μM scrambled peptide (lane 1) or 50 μM AKAPis PKA-anchoring disruptor peptide (lane 2). (B) Co-purification of AKAP220 (top) and AKAP150 (mid) with PKA-RII (bottom) from INS-1(832/13) lysate by cAMP-agarose affinity chromatography. Ethanolamine agarose was used as a control. (C) Immunoblot demonstrating gene silencing of AKAP150. Cells were co-transfected with human growth hormone and pSilencer or p150i, and used in secretion studies; see Supplementary Figure S1C for quantification. (D) Measurement of Glucose-, GLP-1- and (E) forskolin-induced insulin secretion from INS-1(832/13) cells co-transfected with pSilencer or p150i. (F) Schematic of AKAP150KO mice generation. Exon2 of the AKAP5 locus was flanked with loxP sites and mice were crossed onto a EIIa-Cre-deleter line to excise the AKAP5 coding region. (G) Immunoblot detection of AKAP150, PKA-RII and GAPDH loading control from brain and islets of Langerhans lysates from matched WT and AKAP150KO mice. (H) Immunofluorescence detection of AKAP150 (green), E-cadherin (red) and insulin (blue) from paraffin-embedded sections of wild-type and AKAP150 null mouse pancreas, and enlarged composite image (I). (J) Plasma insulin concentrations measured from fasted wild-type and AKAP150KO mice and following IP glucose (1.5 g/kg) injection. (K) Insulin content of acid extracted pancreata from WT (grey) and AKAP150KO (green) mice. (L, M) Depict representative 10 × images from WT and AKAP150KO pancreata (respectively) used for determination of islet area and β-cell mass (Table I) as per Materials and methods; sections are stained for insulin (blue), glucagon (green) and E-Cadherin (red). Data represent mean±s.e.m. *P⩽0.05, **P⩽0.01. Immunoblots are representative of 3–6 independent experiments. Age-matched male mice were used for all experiments (WT: 18.5±0.5 weeks, 29.1±0.5 g; AKAP150KO: 18.6±0.6 weeks, 28.3±0.5 g). Figure source data can be found with the Supplementary data.
More definitive investigation of AKAP150-dependent insulin regulation was performed in AKAP150KO mice (Figure 1F–M). LoxP sites flanking exon 2 of the AKAP5 locus were introduced to permit deletion of the entire open reading frame (Tunquist et al, 2008). Loss of AKAP150 protein from brain and islet extracts was initially confirmed by immunoblot (Figure 1G; Supplementary Figure S1K–M). Subsequent immunofluorescent analyses of paraffin-embedded pancreatic sections revealed that AKAP150 is present in wild-type islets but is not detected in equivalent samples from knockout animals (Figure 1H). At higher magnification, the AKAP150 signal was less prevalent in the non-insulin-positive islet periphery (Figure 1I). Moreover, only trace levels of AKAP150 were present in the α-cell-derived line (αTC1–6; Supplementary Figure S1N). Metabolic studies performed on matched male mice revealed that fasted serum insulin levels were reduced 27.4±8.1% in AKAP150KO mice compared to WT. After IP glucose injection, circulating serum insulin was 27.0±5.6% lower in AKAP150KO animals (Figure 1H; n=21, P<0.05). A similar trend was evident at early time points (Supplementary Figure S1O). Since total pancreatic insulin content (Figure 1K; n=20) and β-cell mass and area (Figure 1L and M, and Table I; n=3) were similar in both genotypes, our findings are consistent with a secretory defect when the AKAP150 gene is deleted.
Table 1. Islet area and β-cell mass of AKAP150 transgenic mice.
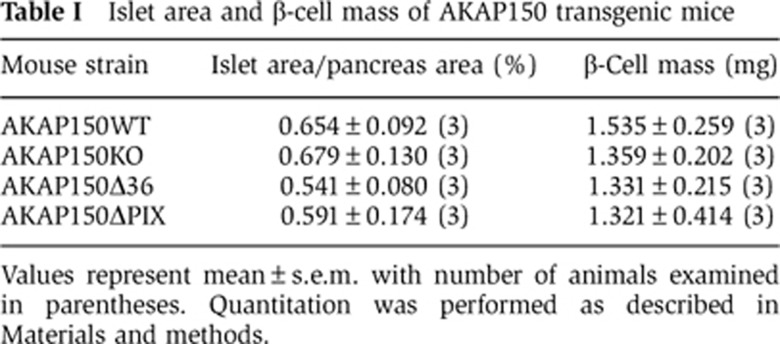
Values represent mean±s.e.m. with number of animals examined in parentheses. Quantitation was performed as described in Materials and methods.
AKAP150 coordinates Ca2+ and cAMP-stimulated insulin secretion from β-cells
Next, we monitored a range of cellular and molecular events associated with insulin secretion in primary islets from AKAP150KO mice. Static insulin release measurements confirmed that islets from both genotypes responded to glucose. However, less insulin was discharged from AKAP150 null islets (Figure 2A). Dynamic insulin release, evaluated by islet perifusion, also detected a modest reduction in the rate of insulin secretion from AKAP150KO islets. The peak secretory response to 11 mM glucose was 31.5±10.9% lower from KO islets, and reached statistical significance upon delivery of the potent insulin secretagogue forskolin (Figure 2B; n=5). The viability of islet preparations was determined by 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay (Supplementary Figure S2A).
Figure 2.

Cellular analysis of insulin release from isolated β-cells. (A) Measurement of glucose-dependent insulin release from isolated islets from WT and AKAP150KO mice. Data are normalized to total cell content (%TCC) of insulin. (B) Dynamic insulin secretion from perifused WT and AKAP150KO islets in response to different concentrations of glucose and forskolin. Integrated responses are shown in Supplementary Figure S2B and C. (C) Representative nifedipine-sensitive, whole-cell Ca2+ current traces from dissociated WT (grey) or AKAP150KO (green) β-cells. (D) Current–voltage relationship of ICa from WT (grey) or AKAP150KO (green) β-cells. (E) ICa voltage dependence of activation and steady-state inactivation from WT (grey) and AKAP150KO (green) β-cells. (F) Montage of images showing the time course of Ca2+ transients in WT and AKAP150KO β-cells in response to 11 mM glucose. Cells were loaded with Fluo-4 AM dye. (G) Fluo-4 AM imaging of glucose-induced intracellular [Ca2+] transients in WT (grey) and AKAP150KO (green) β-cells. (H) Amalgamated data of [Ca2+]i transients in WT and AKAP150KO β-cells after stimulation with 11 or 20 mM glucose. (I) Schematic representation of method used for measuring submembrane cAMP concentration (J, K). cAMP levels were measured using TIRF to detect PKA-catalytic subunit-YFP dissociation from membrane bound PKA-RII-CFP in dispersed β-cells. (J) Representative TIRF traces of the membrane cAMP oscillations in WT (grey) and AKAP150KO (green) islet cells in response to elevated glucose or forskolin. (K) Amalgamated TIRF detection of cAMP production at the membrane of WT (grey) and AKAP150KO (green) β-cells. Data represent mean±s.e.m. *P⩽0.05.
Insulin stimulus-secretion coupling involves membrane depolarization and Ca2+ influx through voltage-gated Ca2+ channels (Hiriart and Aguilar-Bryan, 2008; Seino et al, 2011). AKAP150 directs phosphorylation-dependent modulation of these channels (Oliveria et al, 2007). Dissociated β-cells, identified on the basis of capacitance (>5pF, Supplementary Figure S2D), were voltage clamped in whole-cell patch mode. Loss of AKAP150 reduced peak current densities by 26.7±6.8% in β-cells (Figure 2C and D; green, n=11, P<0.05). Control experiments confirmed that mRNA levels for the pore-forming α1C and accessory β3 channel subunits, voltage dependence of channel opening, and steady-state inactivation parameters were similar in both genotypes (Figure 2E; Supplementary Figure S2E).
Since Ca2+ currents were altered in AKAP150KO β-cells we next monitored real-time changes in cytoplasmic [Ca2+] (Figure 2F–H). Fluo-4 fluorescence peak intensities showed 29.2±8.3% (n=9, P<0.05) and 20.3±5.8% (n=9, P=0.07) lower [Ca2+]i when AKAP150 null cells were challenged with 11 and 20 mM glucose, respectively (Figure 2G and H, green). Hence, loss of AKAP150 or mislocalization of its binding partners in β-cells not only attenuates L-type Ca2+ currents but also perturbs glucose-induced Ca2+ flux.
Glucose also stimulates local oscillations in cAMP just below the β-cell plasma membrane (Dyachok et al, 2008). These fluctuations of cAMP synthesis augment insulin stimulus secretion via PKA and Epac2-dependent mechanisms (Hatakeyama et al, 2006; Holz et al, 2006; Idevall-Hagren et al, 2010). Moreover, AKAP150 is localized in membrane-associated macromolecular complexes that include calcium-sensitive adenylyl cyclases (Efendiev et al, 2010; Willoughby et al, 2010). Therefore, dynamic cAMP production was measured by total internal reflection fluorescence (TIRF) microscopy detection of a plasmalemmal localized ratiometric biosensor (Figure 2I–K). Glucose induced a pulsatile cAMP response in WT β-cells that was essentially absent in cells derived from AKAP150KO mice (Figure 2J). Amalgamated data from fluorescence ratio traces showed a 72.3±13.1% reduction in glucose-stimulated cAMP production in KO islets (Figure 2K; n=17; P<0.05), but no significant difference in forskolin-induced changes. Collectively, the data in Figure 2 show that loss of AKAP150 impairs insulin secretion from β-cells. The mechanism involves reduced influx of Ca2+ through L-type channels and impaired oscillatory production of cAMP.
AKAP150 null mice exhibit enhanced insulin sensitivity
Deficient insulin secretion often results in glucose intolerance. However, in some cases reduced availability of insulin paradoxically improves glucose homeostasis by heightening sensitivity to this hormone (Elchebly et al, 1999). Consistent with this latter concept, intraperitoneal glucose tolerance tests revealed that the glycemic excursion was 35.7±6.5% lower in AKAP150KO than in WT mice (Figure 3A; Supplementary Figure S3A; n=30, P<0.01). Since pyruvate and glucagon tolerance was similar in both genotypes (Figure 3B and C), it seemed that compensatory mechanisms such as reduced glycogen mobilization or gluconeogenesis do not contribute to the improved glucose tolerance of AKAP150KO mice. Consistent with this postulate, no alterations in mRNA abundance of G6Pase or PEPCK were observed in gastrocnemius muscle or liver samples (Supplementary Figure S3G and H). Hence, we reasoned that AKAP150 null mice may have adapted to less circulating insulin by enhancing peripheral insulin sensitivity (Ahren and Pacini, 2004). To test this hypothesis, insulin (0.5 U/kg) was injected into the IP cavity of fed mice and clearance of blood glucose was monitored over time. In AKAP150 null mice, blood glucose declined to 63.0±4.7 mg/dl at 60 min post insulin (Figure 3D; green), whereas it only reached 82.6±8.7 mg/dl in control mice (Figure 3D; grey), indicating significantly enhanced insulin action in the peripheral tissues of AKAP150KO animals.
Figure 3.

Glucose tolerance and peripheral insulin sensitivity in AKAP150KO mice. (A) Blood glucose profiles of WT and AKAP150KO mice during IP glucose (1.5 g/kg) tolerance test (IPGTT). Integrated glycemic responses are found in Supplementary Figure S3A. (B) Blood glucose profiles of WT (grey) and AKAP150KO (green) mice following IP injection of pyruvate (1.5 g/kg) into fasted animals as an index of gluconeogenesis. (C) Changes in blood glucose in response to IP injection of glucagon (25 nmol/kg) in fed WT (grey) and AKAP150KO (green) mice as an index of glycogenolysis. (D) Glycemic profiles of WT (grey) and AKAP150KO (green) mice following IP injection of recombinant insulin (0.5 U/kg). (E) Immunoblot detection of AKAP150, PKA-RII and GAPDH in gastrocnemius muscle, liver and epididymal fat pad homogenates from WT and AKAP150KO mice. (F) Immunoblot detection of active (P)Ser473-Akt, total Akt and GAPDH from gastrocnemius muscle homogenates from WT and AKAP150KO mice 15 min after IP saline or insulin (1.0 U/kg) injection. Two biological replicates are shown for each genotype and condition. Quantification of compiled results was performed by densitometry analysis of active (P)Ser473-Akt normalized to total respective protein level (G). (H) Immunoblot detection and (I) densitometry analysis of active (P)Ser473-Akt, total Akt and GAPDH from liver homogenates from WT and AKAP150KO mice as per (F, G). (J) Immunoblot analysis of active (P)Thr172-AMPK, total AMPK and GAPDH from skeletal muscle homogenates from WT and AKAP150KO mice. Two biological replicates are shown for each genotype and condition. Quantification of compiled results was performed by densitometry analysis of active (P)Thr172-AMPK normalized to total respective protein level (K). (L) Immunoblot detection and (M) densitometry analysis of (P)Thr172-AMPK, total AMPK and GAPDH from liver homogenates from WT and AKAP150KO mice as per (J, K). Data represent mean±s.e.m. *P⩽0.05, **P⩽0.01. Immunoblots are representative of three independent experiments; densitometry represents results from six individual animals of each genotype. Figure source data can be found with the Supplementary data.
A survey of insulin target tissues revealed that AKAP150 is expressed in skeletal muscle and liver but is not present in adipose tissue (Figure 3E, top panels; lanes 1, 3 and 5). Therefore, we monitored the activity status of two metabolically relevant protein kinases in gastrocnemius muscle and liver. Akt/protein kinase B is a principle intracellular mediator of insulin action including control of glucose transport (Whiteman et al, 2002). Endogenous Akt activity can be assessed with antibodies that recognize phosphorylated Ser473. Under basal conditions the skeletal muscle (P)Ser473-Akt signal was minimal in both genotypes (Figure 3F, top panel, lanes 1–4, n=6). However, 15 min after insulin injection, the (P)Ser473-Akt signal was augmented 1.9±0.3-fold in samples from AKAP150KO mice as compared to WT (Figure 3F, top panel, lanes 5–8 and 3G; P<0.05, n=6). Interestingly, Akt activation was not altered when experiments were performed in liver extracts from AKAP150 null animals (Figure 3H and I). IRS-1 is an upstream effector of insulin receptor activation, and phosphorylation on Ser612 is a marker for insulin action (Mothe and Van Obberghen, 1996). We detected enhanced phosphorylation of IRS-1 in skeletal muscle extracts from AKAP150KO mice (Supplementary Figure S3I and J).
AMP-activated protein kinase (AMPK) is a major cellular regulator of lipid and glucose metabolism. In humans, exercise-induced stimulation of insulin sensitivity correlates with elevated AMPK activity. Likewise, the glucose-lowering effect of the anti-diabetes drug metformin has been attributed to the tonic stimulation of AMPK (Kahn et al, 2005; Hinke et al, 2007). Phosphorylation of Thr172 on AMPK indicates the activity status of this energy sensor kinase. Immunoblot analysis of skeletal muscle extracts revealed that the (P)Thr172-AMPK signal was enhanced 1.9±0.4-fold in AKAP150KO over WT in the fasted state (Figure 3J, top panel, and Figure 3K; P<0.05, n=6). In contrast, liver AMPK activity was not statistically different in either genotype (Figure 3L and M; n=6). Hence, the data in Figure 3 argue that heightened insulin sensitivity in skeletal muscle is an adaptive response to reduced serum hormone levels in AKAP150 knockout mice.
Conditional Ins2-Cre deletion of AKAP150 affects insulin secretion
In order to delineate between the effects of AKAP150 in the pancreas and peripheral tissues mice bearing LoxP sites flanking the coding exon of AKAP150 (AKAP150fl/fl) were backcrossed onto Ins2-Cre mice (Postic et al, 1999) to conditionally delete the anchoring protein in insulin expressing cells (Figure 4A). Immunoblot analysis detected equivalent levels of AKAP150 in brain extracts from both Ins2-Cre+/− and −/− animals (Figure 4B, top panel, lanes 1 and 2). In islet extracts, the anchoring protein was detected in AKAP150fl/fl tissue, but only trace amounts from AKAP150βKO (AKAP150fl/fl/Ins2-Cre+/−) islets (Figure 4B, top panel, lanes 3 and 4). Metabolic studies performed on matched male mice revealed that fasted serum insulin levels were equivalent in AKAP150βKO mice compared to control mice. After IP glucose injection, circulating serum insulin was 26.1±4.6% lower in AKAP150βKO animals (Figure 4C; n=20, P<0.05), despite significantly elevated pancreatic insulin content (Figure 4D). AKAP150βKO mice displayed impaired glucose tolerance (Figure 4E; Supplementary Figure S4A), implying the enhanced insulin content (Figure 4D) was a physiological response to oppose this phenotype. In contrast to global AKAP150KO mice, insulin sensitivity was similar in both AKAP150βKO and controls (Figure 4F). This suggests that conditional ablation of AKAP150 from insulin expressing cells attenuates insulin secretion in a specific manner but importantly does not alter insulin sensitivity in peripheral tissues.
Figure 4.

Metabolic profiling of Ins2-Cre+/−.AKAP5fl/fl (AKAP150βKO) conditional deletion mice. (A) Schematic depicting the deletion of the floxed AKAP5 coding region in insulin expressing tissues by Ins2 promoter-driven Cre-recombinase. (B) Immunoblot detection of AKAP150 from brain and isolated islets of Langerhans lysates of AKAP150fl/fl (control) and AKAP150βKO mice. (C) Plasma insulin concentrations from matched control and AKAP150βKO mice following overnight fasting and IP glucose (1.5 g/kg) injection. (D) Insulin content of acid extracted pancreata from control and AKAP150βKO mice. (E) Blood glucose profiles of control (grey) and AKAP150βKO (orange) mice during IP glucose tolerance testing (1.5 g/kg). (F) Blood glucose profiles of control (grey) and AKAP150βKO (orange) mice following IP injection of insulin (0.5 U/kg). Data represent mean±s.e.m. *P⩽0.05, **P⩽0.01, ***P⩽0.001. Immunoblots are representative of three independent experiments. Age-matched male mice were used in all experiments (control: 20.4±0.4 weeks, 29.5±0.6 g; AKAP150βKO: 20.4±0.4 weeks, 28.1±0.5 g). Figure source data can be found with the Supplementary data.
PKA anchoring to AKAP150 has little effect on glucose homeostasis
Since each AKAP150-anchored enzyme influences distinct metabolic events, we investigated glucose homeostasis in mouse models where discrete elements of the AKAP150 signalling complex were disrupted. An amphipathic helix that binds the regulatory (R) subunits of PKA is a defining characteristic of AKAPs. This structural element is located between residues 705 and 724 of AKAP150. Mice expressing a form of the anchoring protein lacking this region (AKAP150Δ36; Weisenhaus et al, 2010) are unable to anchor PKA but retain the ability to tether PP2B (Figure 5A–C; Supplementary Figure S5A and B). Metabolic profiling of matched male AKAP150Δ36 mice selectively evaluated the contribution of anchored PKA to glucose homeostasis.
Figure 5.

Metabolic profiling of knock-in mice lacking the PKA binding domain of AKAP150. (A) Schematic depicting the insertion of a premature stop codon into the coding region of the AKAP5 locus to generate a truncated AKAP150 protein (AKAP150Δ36) unable to anchor PKA. (B) Immunoblot detection AKAP150 and AKAP150Δ36 from brain and isolated islets of Langerhans lysates. (C) Immunoprecipitation of AKAP150 and AKAP150Δ36 complexes, and immunoblot detection of AKAP150 and co-immunoprecipitating PKA-RII and PP2BB. (D) Plasma insulin concentrations from matched WT and AKAP150Δ36 mice following overnight fasting and IP glucose (1.5 g/kg) injection. (E) Insulin content of acid extracted pancreata from WT and AKAP150Δ36 mice. (F) Measurement of glucose-stimulated insulin release (%TCC) from isolated WT and AKAP150Δ36 mouse islets of Langerhans. (G) Dynamic insulin secretion from perifused WT and AKAP150Δ36 islets in response to different concentrations of glucose and forskolin. Integrated responses are shown in Supplementary Figure S5C and D. (H) Blood glucose profiles of WT (grey) and AKAP150Δ36 (blue) mice during IP glucose tolerance testing (1.5 g/kg). (I) Blood glucose profiles of WT (grey) and AKAP150Δ36 (blue) mice following IP injection of insulin (0.5 U/kg). (J) Immunoblot detection of active (P)Ser473-Akt, total Akt and GAPDH from gastrocnemius muscle homogenates from WT and AKAP150Δ36 mice 15 min after IP saline or insulin (1.0 U/kg) injection. Two biological replicates are shown for each genotype and condition. Quantification of compiled results was performed by densitometry analysis of active (P)Ser473-Akt normalized to total respective protein level (K). Data represent mean±s.e.m. Immunoblots are representative of three independent experiments; densitometry represents results from three individual animals of each genotype. Age-matched male mice were used in all experiments (WT: 18.4±0.7 weeks, 26.9±0.6 g; AKAP150Δ36: 18.9±0.6 weeks, 25.8±0.5 g). Figure source data can be found with the Supplementary data.
An unanticipated outcome of these studies was that most aspects of glucose homeostasis are comparable between AKAP150Δ36 and WT mice (Figure 5D–K). Circulating insulin levels were minimally lower upon glucose challenge in AKAP150Δ36 mice than in controls (Figure 5D; n=14, P>0.05) and the pancreatic insulin content was similar in both genotypes (Figure 5E; n=18). Glucose-stimulated insulin release from islets was the same for both genotypes (Figure 5F and G) and superimposable glycemic profiles were observed during IP glucose tolerance tests (Figure 5H; Supplementary Figure 5E; n=16). In keeping with this trend, peripheral insulin sensitivity was similar in both genotypes (Figure 5I; n=12). Likewise, minimal changes in the activity status of Akt or AMPK were detected in skeletal muscle extracts from AKAP150Δ36 mice (Figure 5J and K; Supplementary Figure S5F and G, n=3). Collectively, this battery of metabolic and molecular tests shows that AKAP150Δ36 mice exhibit normal glucose handling.
Loss of PP2B anchoring recapitulates the AKAP150 null phenotype
Biochemical approaches have identified a ‘PIxIxIT’ motif located between residues 655 and 661 of AKAP150 that forms a binding surface for the Ca2+/calmodulin-dependent phosphatase PP2B. Deletion of this motif (PIAIIIT) from AKAP79/150 (ΔPIX) eliminates PP2B binding in vitro (Dell'Acqua et al, 2002). Gene targeting deleted the 21-bp sequence that encodes these residues to generate AKAP150ΔPIX knock-in mice (Figure 6A). Removal of these seven residues did not alter expression of this AKAP150 derivative (Figure 6B) or its ability to anchor PKA, but prevented association with PP2B (Figure 6C, bottom and mid panels, lane 6; Supplementary Figure S6A and B).
Figure 6.

Metabolic profiling of AKAP150 knock-in mice unable to anchor PP2B. (A) Schematic depicting deletion of 21 bases in the coding region of the AKAP5 locus to generate an internally truncated AKAP150 protein (AKAP150ΔPIX) unable to bind PP2B. (B) Immunoblot detection AKAP150 and AKAP150ΔPIX from brain and islets of Langerhans lysates. (C) Immunoprecipitation of AKAP150 and AKAP150ΔPIX complexes, and immunoblot detection of AKAP150 and co-immunoprecipitating PKA-RII and PP2BB. (D) Plasma insulin concentrations from matched WT and AKAP150ΔPIX mice following overnight fasting and IP glucose (1.5 g/kg) injection. (E) Insulin content of acid extracted pancreata from WT and AKAP150ΔPIX mice. (F) Measurement of glucose-stimulated insulin release (%TCC) from isolated WT and AKAP150ΔPIX mouse islets of Langerhans. (G) Dynamic insulin secretion from perifused WT and AKAP150ΔPIX islets in response to different concentrations of glucose and forskolin. Integrated responses are shown in Supplementary Figure S5C and D. (H) Blood glucose profiles of WT (grey) and AKAP150ΔPIX (purple) mice during IP glucose tolerance testing (1.5 g/kg). (I) Blood glucose profiles of WT (grey) AKAP150ΔPIX (purple) mice following IP injection of insulin (0.5 U/kg). (J) Immunoblot detection of active (P)Ser473-Akt, total Akt and GAPDH from gastrocnemius muscle homogenates from WT and AKAP150ΔPIX mice 15 min after IP saline or insulin (1.0 U/kg) injection. Two biological replicates are shown for each genotype and condition. Quantification of compiled results was performed by densitometry analysis of active (P)Ser473-Akt normalized to total respective protein level (K). Data represent mean±s.e.m. *P⩽0.05. Immunoblots are representative of three independent experiments; densitometry represents results from three individual animals of each genotype. Age-matched male mice were used in all experiments (WT: 18.3±0.7 weeks, 26.1±0.6 g; AKAP150ΔPIX: 19.1±0.6 weeks, 26.6±0.6 g). Figure source data can be found with the Supplementary data.
Metabolic profiling of matched male mice revealed that the pancreatic insulin content and fasting serum insulin levels were similar for each genotype (Figure 6D and E). However, AKAP150ΔPIX mice exhibited 21.6±5.1% lower plasma insulin following a glucose challenge (Figure 6D; n=16, P<0.05). This provided the first hint that the disruption of PP2B–AKAP150 interaction in vivo has similar phenotypic consequences as the complete removal of the anchoring protein. In keeping with this observation, static glucose-induced insulin release was reduced by as much as 35% in islets from AKAP150ΔPIX mice when compared to controls (Figure 6F; n=5, P<0.05). A comparable trend was observed during islet perifusion (Figure 6G). This indicates that AKAP150 tethering of PP2B influences insulin secretion from β-cells.
By analogy to the AKAP150KO mouse, we reasoned that perturbing PP2B tethering might improve glucose tolerance. Accordingly, AKAP150ΔPIX mice more efficiently cleared blood glucose as reflected by a 19.6±9.4% reduction in the integrated glycemic excursion (Figure 6H; Supplementary Figure S6E; n=15, P<0.05). In keeping with this trend, AKAP150ΔPIX mice also displayed heightened insulin sensitivity as blood glucose more rapidly declined following exogenous insulin injection in AKAP150ΔPIX mice than in WT (Figure 6I; n=12).
Next, we monitored the activity status of Akt and AMPK from AKAP150ΔPIX mice. In the basal state, the skeletal muscle (P)Ser473-Akt signal was equivalent in both genotypes, but was augmented 1.8±0.2-fold by insulin in AKAP150ΔPIX samples relative to WT controls (Figure 6J and K; n=3, P<0.05). In fasted skeletal muscle from AKAP150ΔPIX mice, the (P)Thr172-AMPK signal was enhanced 2.1-fold compared to WT (Supplementary Figure S6F and G; n=3). Remarkably, the results in Figure 6 infer that the targeting of PP2B is the predominant molecular determinant in AKAP150-mediated control of glucose homeostasis.
Discussion
Glucose homeostasis involves the intake, production, utilization and storage of this essential nutrient. Serum glucose levels decrease in response to insulin whereas counterregulatory hormones such as glucagon, adrenaline, cortisol and growth hormone reverse this response. Clinical interest in this process is heightened because chronic changes in the availability or sensitivity to insulin underlie the pathophysiology of metabolic syndrome and diabetes (Lee and Cox, 2011). Gene silencing and genetic manipulation of mice has allowed us to pinpoint AKAP150 as a positive intermediary of nutrient-mediated insulin secretion from pancreatic islets. AKAP150 impacts these essential metabolic processes by directing Ca2+ and cAMP responsive enzymes towards elements of the insulin release machinery. The cAMP responsive component of this process proceeds through two distinct effector pathways. The Epac2 guanine nucleotide exchange factor modulates SUR1 KATP channel subunits and activates the small GTPase Rap-1 whereas PKA phosphorylates ion channels and proteins of the exocytotic machinery to facilitate insulin release (Seino and Shibasaki, 2005; Holz et al, 2006; Hinke, 2009). Perhaps surprisingly, a body of data in Figure 5 suggests that PKA anchoring to AKAP150 is not essential for insulin release, as AKAP150Δ36 mice experience near normal glucose homeostasis. However, Epac2 may be an AKAP150 binding partner (Nijholt et al, 2008), suggesting that submembrane targeting of this guanine nucleotide exchange factor might convey the cAMP component of AKAP150-mediated insulin release. Consequently, anchored PKA regulation of insulin exocytosis must proceed through other β-cell anchoring proteins such as MyRIP and AKAP15/18 that target this kinase to different subcellular microenvironments (Fraser et al, 1998; Goehring et al, 2007).
Experiments in Figure 2 use AKAP150 null pancreatic islets to assess the contribution of this enzyme complex in the modulation of molecular events that trigger insulin secretion. Three elements of this pathway appear to be under AKAP150 control. First, AKAP150 tethered PKA and PP2B govern the phosphorylation-dependent modulation of voltage-gated L-type Ca2+ currents, in part because this anchoring protein binds directly to the cytoplasmic tail of the channel (Oliveria et al, 2007). Electrophysiological recordings show that loss of AKAP150 suppresses these currents to reduce the influx of calcium that is a prelude to insulin release. Second, Fluo-4 imaging experiments show that ablation of AKAP150 perturbs glucose-induced intracellular calcium flux. Third, this interruption in local Ca2+ influx was accompanied by perturbation of submembrane oscillations of cAMP levels that are crucial for insulin release (Dyachok et al, 2008; Idevall-Hagren et al, 2010; Tian et al, 2011). This latter effect may be indicative of compromised modulation of Ca2+-sensitive adenylyl cyclases that are normally under the control of AKAP150 anchored enzymes (Efendiev et al, 2010; Willoughby et al, 2010). The observation that deletion of AKAP150 impairs glucose-stimulated cAMP oscillations but not forskolin-induced cAMP production, yet defective insulin secretion was observed in response to forskolin, infers that the scaffold affects both proximal and distal steps. The important new concept that emerges from these findings is that reduced insulin release from AKAP150KO islets is not a consequence of interrupted signalling at a single intracellular locus, but rather an amalgamation of changes in discrete molecular events at sites throughout the secretory cascade.
Metabolic profiling of AKAP150 null mice revealed that these animals have reduced circulating insulin yet they display improved glucose tolerance. This confounding but metabolically favourable phenotype can be explained by compensatory changes at the endocrine and molecular level. Data presented in Supplementary Figure S3D–F show that plasma levels of incretin hormones and glucagon are altered in fed AKAP150KO animals. Since the net effect would lower blood glucose we propose that these compensatory endocrine changes represent an attempt to re-establish normoglycemia upon nutrient uptake. Another adaptive mechanism that could counteract reduced insulin secretion is our evidence that sensitivity to insulin is boosted in peripheral tissues in AKAP150 null mice. Skeletal muscle accounts for >75% of insulin-mediated glucose uptake (Baron et al, 1988). Furthermore, enhanced activation of Akt is a recognized hallmark of increased insulin action (Whiteman et al, 2002). Taken together, these latter observations provide context for our evidence in Figure 3G that insulin-sensitive Akt activity is elevated ∼2-fold in the skeletal muscle of AKAP150KO mice. AMPK is considered to be the metabolic master-switch controlling target tissue responsiveness to insulin (Kahn et al, 2005). Data suggest that the activity of AMPK is elevated in the skeletal muscle of AKAP150KO mice. Multiple signals converge on the α-subunit of this kinase. For example, the upstream kinases CaMKK and LKB1 can phosphorylate Thr172 to stimulate phosphotransfer (Kahn et al, 2005; Hinke et al, 2007). In contrast, PKA phosphorylation of the adjacent residue Thr173 inhibits AMPK activity (Djouder et al, 2010). We postulate that the involvement of AKAP150 in the control of AMPK activity may be two-fold: in response to Ca2+, anchored PP2B could preferentially dephosphorylate both sites on AMPK, yet upon elevation of cAMP, anchored PKA might phosphorylate Thr173. Thus, AKAP150 binding partners may somehow influence the enzymes that set insulin sensitivity. Additional albeit indirect support for this concept is provided by examination of Ins2-Cre conditional deletion of AKAP150. Although there are recognized caveats associated with conditional deletion from insulin expressing cells (Wicksteed et al, 2010), this mouse model duplicated the circulating insulin phenotype of the global knockout, but importantly lacked enhanced peripheral insulin sensitivity.
Perhaps, the most intriguing conclusion comes from the metabolic profiling of AKAP150ΔPIX mice where mislocalization of PP2B phenocopies the changes in glucose homeostasis that occur in the complete absence of AKAP150. Remarkably, the data in Figure 6 show that deletion of seven amino acids from AKAP150 not only disrupts PP2B anchoring in vivo, but also creates a favourable metabolic state in terms of glucose handling. This is contrary to the tissue-specific ablation of this phosphatase from β-cells, which is deleterious and generates a diabetic state that includes defects in NFAT transcriptional signalling, diminished insulin biosynthesis and a loss of islet mass (Heit et al, 2006; Bernal-Mizrachi et al, 2010). Since none of these adverse effects were detected in AKAP150KO or AKAP150ΔPIX mice, one can deduce that targeting this phosphatase close to the plasma membrane through its association with AKAP150 is a little recognized but important determinant of β-cell function. A recent study on the architecture of the AKAP79/150 signalling complex may shed further mechanistic light on why the anchored phosphatase is prominent at this location (Figure 7). Native mass spectrometry analyses indicate that the anchoring protein is a dimer that coordinates two PKA holoenzymes and four heterodimers of PP2B (Gold et al, 2011; Figure 7A). Furthermore, the conformation of the PIAIIIT motif is a β strand with protruding hydrophobic side chains that simultaneously contact two A subunits of PP2B (Figure 7B and C). Thus, exploiting the AKAP150–PP2B interface, and the PIAIIIT sequence in particular, as a viable target for therapeutic intervention could enhance insulin sensitivity in selected peripheral tissues.
Figure 7.

The AKAP150–PP2B interface. (A) Model of the molecular architecture of a membrane-associated AKAP150/(PP2B)2/PKA dimer. An AKAP79/150 homodimer coordinates four PP2B holoenzymes and two PKA holoenzymes localized to the plasma membrane. (B) The structure of the PP2B–PIAIIIT peptide complex and (C) magnified cartoon diagram of the PP2B-PIAIIIT-PP2B interface. Structural information and coordinates obtained from Gold et al (2011) and Li et al (2012) [PDB ID: 3LL8].
Materials and methods
Experimental animals
Animals were housed at the University of Washington, under a 12-h light/dark cycle with free access to food (LabDiet 5001) and water. All procedures were approved by the institutional IACUC review process. Animals were maintained on a C57Bl/6 background (Jackson Laboratories); experiments were performed on littermate or age-matched mice from het X het breeding pairs, or age-matched mice from F1 homozygous crosses. Generation of AKAP150KO (Tunquist et al, 2008) and AKAP150Δ36 (Weisenhaus et al, 2010) mice have been previously described. The AKAP150ΔPIX mutation, removing the PIAIIIT motif (a.a. 655–661), was introduced into the mouse genome by homologous recombination (see Supplementary data). Ins2-Cre mice (Postic et al, 1999) on a pure C57Bl/6 background were obtained from the Jackson Laboratories.
Cell culture and islet isolation
INS-1(832/13) insulinoma cells were cultured as described (Hohmeier et al, 2000). For transfection, 3 × 106 cells were seeded into 10 cm dishes (Falcon) and cultured for 2 days; cells were transfected with Lipofectamine2000 as per the supplied protocol (Invitrogen). Constructs used were pSilencer (Ambion) with a rat-specific AKAP150 targeted shRNA insert (p150i) (Hoshi et al, 2005), siGENOME RISC-Free siRNA control (Dharmacon D-001220-01) or rat AKAP220 siRNA duplex 2 (Dharmacon D-090987-02), hGH (pFox.hGH.CMV) (Goldfine et al, 1997), and pEGFP (Invitrogen). In all, 10 μg of DNA/RNA was transfected per 10 cm dish, consisting of 5 μg hGH plasmid, 0.5 μg pEGFP, and siRNA, p150i or controls. Cells recovered overnight were subcultured into 24-well plates (Falcon) at 5 × 105 cells/well and grown for another 48 h before use in secretion experiments.
Collagenase P (Roche) digested pancreatic islets were isolated as previously described (Delmeire et al, 2003) by handpicking under a stereo microscope. Islets were cultured in suspension overnight in RPMI supplemented with 10% FBS, 10 mM HEPES, 1 mM sodium pyruvate and antibiotics.
Immunoprecipitation, western blotting and tissue analysis
Cultured cells and mouse tissues were homogenized in lysis buffers according to the experiments performed: RIPA buffer for immunodetection, immunoprecipitation buffer (0.5% NP-40, 100 mM NaCl, 50 mM Tris–HCl, pH7.4), or phosphatase inhibitor buffer (1% Triton X-100, 60 mM β-glycerophosphate, 20 mM MOPS, 5 mM EDTA, 5 mM EGTA, 1 mM Na3VO4, 20 mM NaF, pH 7.2), each with protease inhibitors added (1 mM benzamidine, 10 μM AEBSF, 25 μg/ml leupeptin). Immunoprecipitation was performed for 2 h at 4°C in the presence of 25 μl protein A-agarose beads (Upstate), 2 μg antibody, and 1 mg of lysate. Immune complexes were washed thrice in IP buffer and bound proteins eluted by boiling in 2 × NuPage sample buffer with 0.5 M DTT (Invitrogen), and resolved by SDS–PAGE (Invitrogen NuPage). This method was also adapted to cAMP-agarose (BioLog) and calmodulin-agarose (Sigma). Conventional western blotting was performed by loading 50 μg/lane, electrophoretic separation, transfer onto nitrocellulose, and blocking in 1 × Blotto. Antibodies used in this report are described in Supplementary Methods.
Total pancreatic insulin content was measured as previously described in Supplementary Experimental procedures. Clarified extract was analysed for protein content (BCA assay, Pierce) and insulin content (ELISA, Millipore). Immunofluorescent imaging of paraffin-embedded pancreatic sections, and quantitation of islet area and β-cell mass are described in Supplementary data.
For examination of phospho-AMPK and phospho-Akt in skeletal muscle and liver samples, fasted mice were injected with saline or 1.0 U/kg recombinant human insulin (HumulinR; Eli Lilly). At 15 min, animals were sacrificed, the liver and gastrocnemius muscles were briefly rinsed in ice-cold PBS and snap frozen. Tissues were homogenized in phosphatase inhibitor containing solubilization buffer, before protein assay and immunoblot analysis. Densitometry was performed using NIH ImageJ software (v10.2).
Hormone secretion studies
INS-1(832/13) cells transfected with hGH and gene silencing vectors or siRNA were washed twice with 37°C HEPES-buffered Kreb’s ringer bicarbonate buffer (KRBH; mM: 25 HEPES, 5 NaHCO3, 1.2 MgSO4, 1.2 KH2PO4, 4.74 KCl, 125 NaCl, 1 CaCl2) with 2 mM glucose, and pre-incubated 1 h at 37°C/5%CO2. Media was replaced with 1 ml of KRBH with 2 or 20 mM glucose, with or without GLP-1 (Bachem) or forskolin (Sigma), and incubated for 1 h. Supernatants were collected, centrifuged, and stored at −20°C until hGH or Insulin was measured by ELISA (Roche or Millipore, respectively) according to manufacturer’s instructions.
For static insulin release experiments from overnight cultured primary islets, 15 size matched (∼70–300 μm in diameter) islets per tube were tested in triplicate, pre-incubated (45 min/37°C) in 2 mM glucose RPMI with 10 mM HEPES and 0.1% BSA (Sigma), followed by 45 min stimulation with 2, 8, 15 mM glucose, with or without 10 μM forskolin. Following centrifugation (1200, r.p.m./4°C/5 min), a sample of supernatant was removed for insulin determination, and remaining media and islet pellet were acidified and sonicated for normalization purposes. Islet perifusion was performed as described (Delmeire et al, 2003) on re-picked overnight-cultured islets (100 per chamber) sandwiched in Biogel P2 (Bio-Rad). Islets were pre-perifused with 37°C 2 mM RPMI/10 mM HEPES/0.1% BSA continuously bubbled with carbogen, at a flow rate of 0.5 ml/min for 20 min; fractions were collected at 1 min intervals following stimulation. Subsequently, total cell insulin content was acid extracted by sonication for normalization. Under these conditions, mouse islets do not display distinct first and second phase insulin responses (Hansotia et al, 2004; Zawalich et al, 2008). Insulin was measured using mouse insulin-specific ELISA kits from Millipore or Alpco according to included instructions.
In vivo metabolism studies
Glucose tolerance tests were performed on mice following overnight food deprivation. Conscious mice were injected with 1.5 g/kg glucose (10%w/v; Sigma) into the intraperitoneal cavity after basal samples were obtained. Blood glucose was measured from tail vein samples using a handheld glucometer (OneTouch Ultra, LifeScan) at the times indicated in the figures over a 90-min period. For plasma hormone determination, whole blood was collected using heparinized capillary tubes (Fisher), and plasma separated by refrigerated centrifugation. Samples were stored at −20°C until hormone measurement by commercial ELISA kits: ultrasensitive mouse insulin (CrystalChem), mouse C-peptide (Alpco), glucagon (Yanaihara), GLP-1 (Alpco) and GIP (Millipore). The pyruvate tolerance test was performed identically to the IP glucose tolerance test, but substituting sodium pyruvate (10%w/v; Sigma), and tracking the appearance of glucose into the circulation. Insulin tolerance testing was performed on matched fed mice injected with 0.5 U/kg recombinant human insulin (HumulinR, Eli Lilly); the decrease in blood glucose from baseline was monitored by tail vein whole blood samples. The glucagon tolerance test was performed by IP injecting fed mice with 25 nmol/kg synthetic glucagon (Bachem) dissolved in 0.9% saline, and measuring the release of glucose into the blood stream.
Isolated islet studies
The viability and metabolism of overnight-cultured isolated islets were determined by the MTT assay, as described (Janjic and Wollheim, 1992; Hinke et al, 2007). Islets were re-picked into triplicate tubes and washed twice in 2 mM glucose KRBH. After a 30-min pre-incubation, media was changed to KRBH containing 2, 11 or 20 mM glucose and 0.5 mg/ml MTT (Sigma), and incubated for 2 h/37°C. Following centrifugation (3000, r.p.m./4°C/10 min) and removal of the supernatant, formazan crystals were dissolved in DMSO and absorbance read at A562 and A690.
Ca2+ currents in β-cells were examined using the whole-cell configuration of the patch-clamp technique with an Axopatch 200B. Isolated islets were dispersed into single cells by trypsin/EDTA (Invitrogen), and cultured overnight on poly-lysine coated coverslips. During experiments, cells were superfused with a solution composed of (in mM): 138 NaCl, 5.4 KCl, 1 MgCl2, 10 CaCl2, 2 Glucose and 10 HEPES adjusted to pH 7.4. Pipettes were filled with a solution with the following constituents (in mM): 87 Cs-aspartate, 20 CsCl, 1 MgCl2, 5 MgATP, 10 HEPES and 10 EGTA adjusted to pH 7.2 with CsOH. Protocols for measuring the current–voltage relationship of nifedipine-sensitive Ca2+ currents, voltage dependence of activation and steady-state inactivation of Ca2+ currents are described in Supplementary Methods.
For [Ca2+]i imaging experiments, dispersed β-cells were loaded with the Fluo-4 AM (5 μM; 30 min) and perfused with (in mM): 125 NaCl, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 5 NaHCO3, 2 CaCl2, 10 HEPES, and 2, 11 or 20 glucose, pH 7.4. Images were acquired using a Bio-Rad Radiance 2100 confocal system coupled to an inverted Nikon TE300 microscope equipped with a Nikon PlanApo (60 × , NA=1.4) oil-immersion lens, and analysed using NIH-ImageJ. Background-subtracted fluorescence signals were normalized by dividing the fluorescence (F) intensity at each time point by the resting fluorescence (F0).
Sub-membrane [cAMP] was measured by TIRF microscopy and a membrane anchored, truncated form of PKA-RIIβ with a CFP tag and YFP-tagged PKA catalytic Cα subunit. The dissociation of Cα-YFP from the membrane-targeted regulatory subunit permits the ratiometric determination of local cAMP levels (Dyachok et al, 2008). Islets cultured 2–5 days were infected with adenoviral constructs encoding the biosensor subunits. Twenty-four hours after infection, islets were allowed to attach to coverslips and superfused (0.3 ml/min) with media containing (mM) 125 NaCl, 4.8 KCl, 1.3 CaCl2, 1.2 MgCl2, 25 HEPES, pH 7.4. The islets equilibrated for 30 min in basal medium (3 mM glucose) before stimulating with 20 mM glucose or 5 μM forskolin. The chamber was mounted on the stage of a Ti microscope with a 60 × 1.45NA oil-immersion lens (Nikon), acquiring image pairs at 5 s intervals with a back-illuminated EMCCD camera (DU-887; Andor Technology) controlled by MetaFluor software (Molecular Devices). CFP and YFP were excited by 458 and 514 nm light using an argon laser (Creative Laser Production). All imaging experiments were performed at 37°C.
Quantitative PCR was performed on mRNA obtained from islets cultured overnight in suspension. mRNA was isolated from 100 to 150 islets using RNeasy spin columns with on column DNAse I treatment (Qiagen). Messenger RNA from liver or gastrocnemius muscle was isolated from fresh snap frozen tissue samples according to Qiagen protocols for RNeasy and RNeasy Fibrous Tissue kits. cDNA was generated using a TaqMan reverse transcription kit (Applied Biosystems). Real-time PCR was performed using TaqMan expression assays for CaV1.2α1C (Mm00437917_m1), CaVβ3 (Mm00432244_m1), Kir6.2 (Mm00440050_s1), SUR1 (Mm00803450_m1), GAPDH (4352932E) and TaqMan universal PCR master mix (4324018) and islet mRNA; skeletal muscle and liver samples were assayed using TaqMan primer/probe sets for G6Pase (Mm00839363_m1) and PEPCK (Mm01247058_m1) and GAPDH. mRNA abundance was quantified by the ΔΔCt method and GAPDH as the internal housekeeping transcript, and expressed as fold-WT.
Statistical analyses and molecular modelling
All values are reported as mean±standard error (s.e.m.) and the number of times the experiment was repeated (n) shown on the figures. Data were analysed using GraphPad Prism software (v5.0b). Statistical significance was determined by Student’s t-tests, with P=0.05 as the significance level. All molecular representations were rendered using Pymol (Delano Scientific), as described in Supplementary data.
Supplementary Material
Acknowledgments
We thank Professors Chris Newgard for the INS-1(832/13) cells, Mike German for the hGH plasmid, G Stanley McKnight for the AKAP150Δ36 mice. SAH was supported by postdoctoral fellowships from the Canadian Institutes of Health Research and the Canadian Diabetes Association. PJN is supported by NIH grant GM07750. This study was supported in part from NIH grants to JDS (GM48231), MFN (HL98200) and LFS (HL85686 and HL85870). Generation of AKAP150ΔPIX mice was supported by an NIH grant to MLD (NS40701 and NS048154). VC is supported by JDRF grants 43-2009-791 and 17-2011-620 and ADA grant 1-11-BS-28.
Author contributions: SAH, MFN, AU, JLW, PJN, GT, AJ-C, LKL, VC, AT, MD, LFS and JDS designed, performed and analysed experiments. SAH, PJN, LKL and JDS designed figures and wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Ahren B, Pacini G (2004) Importance of quantifying insulin secretion in relation to insulin sensitivity to accurately assess beta cell function in clinical studies. Eur J Endocrinol 150: 97–104 [DOI] [PubMed] [Google Scholar]
- Baron AD, Brechtel G, Wallace P, Edelman SV (1988) Rates and tissue sites of non-insulin- and insulin-mediated glucose uptake in humans. Am J Physiol 255: E769–E774 [DOI] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD (2006) Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell 23: 925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Cras-Meneur C, Ye BR, Johnson JD, Permutt MA (2010) Transgenic overexpression of active calcineurin in beta-cells results in decreased beta-cell mass and hyperglycemia. PLoS ONE 5: e11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratanova-Tochkova TK, Cheng H, Daniel S, Gunawardana S, Liu YJ, Mulvaney-Musa J, Schermerhorn T, Straub SG, Yajima H, Sharp GW (2002) Triggering and augmentation mechanisms, granule pools, and biphasic insulin secretion. Diabetes 51: Suppl 1S83–S90 [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Dodge KL, Tavalin SJ, Scott JD (2002) Mapping the protein phosphatase-2B anchoring site on AKAP79. Binding and inhibition of phosphatase activity are mediated by residues 315-360. J Biol Chem 277: 48796–48802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmeire D, Flamez D, Hinke SA, Cali JJ, Pipeleers D, Schuit F (2003) Type VIII adenylyl cyclase in rat beta cells: coincidence signal detector/generator for glucose and GLP-1. Diabetologia 46: 1383–1393 [DOI] [PubMed] [Google Scholar]
- Djouder N, Tuerk RD, Suter M, Salvioni P, Thali RF, Scholz R, Vaahtomeri K, Auchli Y, Rechsteiner H, Brunisholz RA, Viollet B, Makela TP, Wallimann T, Neumann D, Krek W (2010) PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J 29: 469–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyachok O, Idevall-Hagren O, Sagetorp J, Tian G, Wuttke A, Arrieumerlou C, Akusjarvi G, Gylfe E, Tengholm A (2008) Glucose-induced cyclic AMP oscillations regulate pulsatile insulin secretion. Cell Metab 8: 26–37 [DOI] [PubMed] [Google Scholar]
- Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, Dessauer CW (2010) AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem 285: 14450–14458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elchebly M, Payette P, Michaliszyn E, Cromlish W, Collins S, Loy AL, Normandin D, Cheng A, Himms-Hagen J, Chan CC, Ramachandran C, Gresser MJ, Tremblay ML, Kennedy BP (1999) Increased insulin sensitivity and obesity resistance in mice lacking the protein tyrosine phosphatase-1B gene. Science 283: 1544–1548 [DOI] [PubMed] [Google Scholar]
- Fraser ID, Cong M, Kim J, Rollins EN, Daaka Y, Lefkowitz RJ, Scott JD (2000) Assembly of an A kinase-anchoring protein-beta(2)-adrenergic receptor complex facilitates receptor phosphorylation and signaling. Curr Biol 10: 409–412 [DOI] [PubMed] [Google Scholar]
- Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD (1998) A novel lipid-anchored A-kinase Anchoring Protein facilitates cAMP-responsive membrane events. EMBO J 17: 2261–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao T, Yatani A, Dell'Acqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM (1997) cAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron 19: 185–196 [DOI] [PubMed] [Google Scholar]
- Goehring AS, Pedroja BS, Hinke SA, Langeberg LK, Scott JD (2007) MyRIP anchors protein kinase A to the exocyst complex. J Biol Chem 282: 33155–33167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold MG, Stengel F, Nygren PJ, Weisbrod CR, Bruce JE, Robinson CV, Barford D, Scott JD (2011) Architecture and dynamics of an A-kinase anchoring protein 79 (AKAP79) signaling complex. Proc Natl Acad Sci USA 108: 6426–6431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine ID, German MS, Tseng HC, Wang J, Bolaffi JL, Chen JW, Olson DC, Rothman SS (1997) The endocrine secretion of human insulin and growth hormone by exocrine glands of the gastrointestinal tract. Nat Biotechnol 15: 1378–1382 [DOI] [PubMed] [Google Scholar]
- Hansotia T, Baggio LL, Delmeire D, Hinke SA, Yamada Y, Tsukiyama K, Seino Y, Holst JJ, Schuit F, Drucker DJ (2004) Double incretin receptor knockout (DIRKO) mice reveal an essential role for the enteroinsular axis in transducing the glucoregulatory actions of DPP-IV inhibitors. Diabetes 53: 1326–1335 [DOI] [PubMed] [Google Scholar]
- Hatakeyama H, Kishimoto T, Nemoto T, Kasai H, Takahashi N (2006) Rapid glucose sensing by protein kinase A for insulin exocytosis in mouse pancreatic islets. J Physiol 570: 271–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK (2006) Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature 443: 345–349 [DOI] [PubMed] [Google Scholar]
- Hinke SA (2009) Epac2: a molecular target for sulfonylurea-induced insulin release. Sci Signal 2: pe54 [DOI] [PubMed] [Google Scholar]
- Hinke SA, Hellemans K, Schuit FC (2004) Plasticity of the beta cell insulin secretory competence: preparing the pancreatic beta cell for the next meal. J Physiol 558: 369–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinke SA, Martens GA, Cai Y, Finsi J, Heimberg H, Pipeleers D, Van de Casteele M (2007) Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol 150: 1031–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiriart M, Aguilar-Bryan L (2008) Channel regulation of glucose sensing in the pancreatic beta-cell. Am J Physiol Endocrinol Metab 295: E1298–E1306 [DOI] [PubMed] [Google Scholar]
- Hohmeier HE, Mulder H, Chen G, Henkel-Rieger R, Prentki M, Newgard CB (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49: 424–430 [DOI] [PubMed] [Google Scholar]
- Holz GG, Kang G, Harbeck M, Roe MW, Chepurny OG (2006) Cell physiology of cAMP sensor Epac. J Physiol 577: 5–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD (2005) Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol 7: 1066–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idevall-Hagren O, Barg S, Gylfe E, Tengholm A (2010) cAMP mediators of pulsatile insulin secretion from glucose-stimulated single beta-cells. J Biol Chem 285: 23007–23018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janjic D, Wollheim CB (1992) Islet cell metabolism is reflected by the MTT (tetrazolium) colorimetric assay. Diabetologia 35: 482–485 [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG (2005) AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab 1: 15–25 [DOI] [PubMed] [Google Scholar]
- Lee AW, Cox RD (2011) Use of mouse models in studying type 2 diabetes mellitus. Expert Rev Mol Med 13: e1. [DOI] [PubMed] [Google Scholar]
- Lester LB, Langeberg LK, Scott JD (1997) Anchoring of protein kinase A facilitates hormone-mediated insulin secretion. Proc Natl Acad Sci USA 94: 14942–14947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Pink MD, Murphy JG, Stein A, Dell'Acqua ML, Hogan PG (2012) Balanced interactions of calcineurin with AKAP79 regulate Ca2+-calcineurin-NFAT signaling. Nat Struct Mol Biol 19: 337–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mothe I, Van Obberghen E (1996) Phosphorylation of insulin receptor substrate-1 on multiple serine residues, 612, 632, 662, and 731, modulates insulin action. J Biol Chem 271: 11222–11227 [DOI] [PubMed] [Google Scholar]
- Nijholt IM, Dolga AM, Ostroveanu A, Luiten PG, Schmidt M, Eisel UL (2008) Neuronal AKAP150 coordinates PKA and Epac-mediated PKB/Akt phosphorylation. Cell Signal 20: 1715–1724 [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA (2007) AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55: 261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, Shelton KD, Lindner J, Cherrington AD, Magnuson MA (1999) Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem 274: 305–315 [DOI] [PubMed] [Google Scholar]
- Scott JD, Pawson T (2009) Cell signaling in space and time: where proteins come together and when they're apart. Science 326: 1220–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Shibasaki T (2005) PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev 85: 1303–1342 [DOI] [PubMed] [Google Scholar]
- Seino S, Shibasaki T, Minami K (2011) Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest 121: 2118–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim AT, Baldwin ML, Rostas JA, Holst J, Ludowyke RI (2003) The role of serine/threonine protein phosphatases in exocytosis. Biochem J 373: 641–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Sandler S, Gylfe E, Tengholm A (2011) Glucose- and hormone-induced cAMP oscillations in alpha- and beta-cells within intact pancreatic islets. Diabetes 60: 1535–1543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD (2008) Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci USA 105: 12557–12562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, Hell JW, McKnight GS (2010) Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS ONE 5: e10325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiteman EL, Cho H, Birnbaum MJ (2002) Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab 13: 444–451 [DOI] [PubMed] [Google Scholar]
- Wicksteed B, Brissova M, Yan W, Opland DM, Plank JL, Reinert RB, Dickson LM, Tamarina NA, Philipson LH, Shostak A, Bernal-Mizrachi E, Elghazi L, Roe MW, Labosky PA, Myers MG Jr, Gannon M, Powers AC, Dempsey PJ (2010) Conditional gene targeting in mouse pancreatic ss-Cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59: 3090–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D, Masada N, Wachten S, Pagano M, Halls ML, Everett KL, Ciruela A, Cooper DM (2010) AKAP79/150 interacts with AC8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J Biol Chem 285: 20328–20342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Nakayama Y, Hattori N, Liu B, Inagaki C (2008) GABAC-receptor stimulation activates cAMP-dependent protein kinase via A-kinase anchoring protein 220. J Pharmacol Sci 106: 578–584 [DOI] [PubMed] [Google Scholar]
- Zawalich WS, Yamazaki H, Zawalich KC (2008) Biphasic insulin secretion from freshly isolated or cultured, perifused rodent islets: comparative studies with rats and mice. Metabolism 57: 30–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.