

Abstract
A workshop1 to share, consider and discuss the latest developments in understanding xeroderma pigmentosum and other human diseases caused by defects in nucleotide excision repair (NER) of DNA damage was held on September 21–24, 2010 in Virginia. It was attended by approximately 100 researchers and clinicians, as well as several patients and representatives of patient support groups. This was the third in a series of workshops with similar design and goals: to emphasize discussion and interaction among participants as well as open exchange of information and ideas. The participation of patients, their parents and physicians was an important feature of this and the preceding two workshops. Topics discussed included the natural history and clinical features of the diseases, clinical and laboratory diagnosis of these rare diseases, therapeutic strategies, mouse models of neurodegeneration, molecular analysis of accelerated aging, impact of transcriptional defects and mitochondrial dysfunction on neurodegeneration, and biochemical insights into mechanisms of NER and base excision repair.
Keywords: DNA repair, Neurodegeneration, Cancer, Aging, Mouse models
1. Background and workshop overview
Xeroderma pigmentosum (XP), Cockayne syndrome (CS) and trichothiodystrophy (TTD) are caused by inherited mutations in genes encoding proteins that play critical roles in nucleotide excision repair (NER) of DNA damage and transcription of mRNA. Three of these proteins (XPB, XPD and TTDA) are essential subunits of TFIIH, a basal transcription factor required for initiation of mRNA transcription. To date, inherited mutations have been identified in 13 NER genes associated with 11 clinically distinct syndromes, including XP, CS, TTD, and syndromes that combine features of these three diseases (Table 1). The phenotypes of these diseases are heterogeneous, consistent with their genetic complexity and the existence of multiple complementation groups for each disease. The heterogeneous pathology of XP, CS and TTD may also be attributed to different amounts of oxidative stress (and oxidative stress-induced DNA damage) in different tissues and cell types, as well as tissue-specific variation in DNA repair capacity, DNA damage response networks, and patterns of gene expression.
Table 1.
Diseases related to impaired UV DNA damage repair or tolerance and their molecular basis.
| Complementation group | Mutated gene | Clinical disorders
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Xeroderma pigmentosum (XP)b | XP with neurological abnormalities | XP/TTD | Tricho- thiodystrophy (TTD) | XP/CS | COFSc syndrome | COFS/TTD | CS/TTD | XFE progeroid syndromed | Cockayne syndrome (CS) | UV-sensitive syndrome | ||
| XP-A | XPAa | ✓ | ✓ | |||||||||
| XP-B | XPB/ERCC3a | ✓ | ✓ | ✓ | ||||||||
| XP-C | XPCa | ✓ | ||||||||||
| XP-D | XPD/ERCC2a | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | |||
| XP-E | XPE/DDB2a | ✓ | ||||||||||
| XP-F | XPF/ERCC4a | ✓ | ✓ | ✓ | ||||||||
| XP-G | XPG/ERCC5a | ✓ | ✓ | ✓ | ||||||||
| TTD-A | TTDA/GTF2H5a | ✓ | ||||||||||
| ERCC1 | ERCC1a | ✓ | ||||||||||
| CS-A | CSA/ERCC8a | ✓ | ✓ | |||||||||
| CS-B | CSB/ERCC6a | ✓ | ✓ | ✓ | ||||||||
| XP VARIANT | XPV/POLη | ✓ | ||||||||||
| TTD-N1 | TTD-N1/C7orf11 | ✓ | ||||||||||
Mutations cause a nucleotide excision repair defect.
XP without neurological involvement.
Cerebro-ocular-facial-skeletal syndrome.
Accelerated aging of multiple organ systems.
XP is characterized by defects in sunlight-induced skin hyperpigmentation and a >10,000-fold increased risk of cancer in sun-exposed areas of the skin. There is also prominent involvement of the sun-exposed tissues of the eyes, lips and tip of the tongue. Approximately 25% of XP patients seen in the US develop progressive neurodegeneration affecting the sensorineural, peripheral and central nervous systems. CS and TTD patients also may present with extreme photosensitivity, but curiously, lack skin cancer susceptibility. CS patients experience progressive neurological decline. In addition TTD patients have brittle hair with “tiger-tail” banding of hair shafts under polarized microscopy, a reduced number of cysteine cross-links in hair proteins, and mild to severe ichthyosis (scaling) of the skin. The phenotype of cells from XP, CS and TTD patients includes reduced survival after UV exposure, impaired repair of UV-induced DNA damage, and impaired ability to resume RNA transcription after UV exposure, although the extent to which each of these three functions is impaired differs for XP, CS and TTD.
The conceptual framework for understanding the pathophysiology of XP, CS and TTD continues to develop (Fig. 1). There is an ongoing focus on oxidative DNA damage in the nuclear genome, defective transcription and/or promoter transactivation and variable responses of proliferating and post-mitotic cells to DNA damage. More recently, it has been recognized that mitochondrial dysfunction and loss of functional stem cells may also contribute to disease progression.
Fig. 1.

Conceptual framework to explain pathological features of XP, CS, TTD and consequences of failing to repair DNA damage. XP, CS and TTD are linked to mutations in 13 genes whose gene products play well-established roles in global NER or transcription-coupled (TC)-NER. Phenotypic effects of defective global NER include accumulated nuclear DNA mutations, dysregulated cell growth and cancer. Defects in TC-NER result in defective or insufficient transcription, senescence, and cell death, leading to tissue-specific defects, degenerative changes, and segmental accelerated aging. Recent studies indicate that the CSB gene product, which plays a critical role in TC-NER, also plays a putative role in maintaining mitochondrial function and/or mitochondrial BER. Mutations in mtDNA and/or loss of mtDNA lead to altered energy homeostasis and increased oxidative stress especially in tissues with high energy demand. This process could exacerbate the phenotypic effects of co-existing NER and/or TC-NER defects by causing increase oxidative nuclear DNA damage.
At the two previous workshops in this series (Bohr et al., 2005; Kraemer et al., 2007), patient support groups, clinicians and basic researchers agreed that the highest priority for future research should be to improve the understanding of neurodegenerative processes in patients with XP, CS and TTD. At that time, patients and their families felt uncomfortable with the fact that clinicians could not predict which patients would develop neurological symptoms, when they would manifest, and how debilitating they would be. Although the molecular basis of neurodegeneration in XP, CS and TTD has not yet been clarified, recent studies suggest that neuronal death due to cumulative oxidative DNA damage plays an important role. This may be caused, at least in part, by mitochondrial dysfunction leading to high levels of cellular reactive oxygen species (ROS). Unfortunately, there are still no treatments available to prevent or mitigate neurodegeneration in XP, CS or TTD patients.
At the current workshop, clinicians emphasized the ongoing need to document the neurological decline and cancer incidence in affected patients, and to explore as rapidly as possible novel therapeutic approaches that might protect against disease progression. Mouse models are increasingly facilitating this goal, as strains that better model human clinical features are characterized and appropriate endpoints for rapidly scoring therapeutic efficacy are identified. The lack of a diagnostic center for XP and TTD in the US remains the single largest barrier to instituting early sun protection strategies that delay many sequelae of these diseases, and to establishing clinical trials aimed at disease prevention.
The research scientists and clinicians who work on XP, CS and TTD are strongly aware of the potential impact of their work on the quality of the lives of affected patients and their families. This inspires and motivates their diligence and dedication, which has driven rapid progress in this research area. As this field has matured, patient support and advocacy groups have begun to have more extensive contacts and impact, reaching out to families and communities world-wide who are deeply in need of help. An example of this outreach was described at the workshop by Mrs. Michelle Milota (Executive Director, XP Family Support Group (XPFSG)) (Box 1) who established a unique relationship with a rural community in Guatemala, where the concept of inherited disease was entirely unknown and medical resources for managing the needs of an XP-affected family were completely lacking. XPFSG brought medical assistance to multiple XP-affected children in this remote village. Workshop participants learned about the medical team in the documentary movie Hidden from Light (http://www.hiddenfromlight.com), which was screened during an evening session.
Box 1. Resources for XP, CS and TTD information and support.
Xeroderma Pigmentosum Family Support Group
8495 Folsom Blvd #1
Sacramento, CA 95826, USA
Xeroderma Pigmentosum Support Group
Instron House, Coronation Road, Hugh Wycombe
Bucks, HP12 3SY, United Kingdom
Multidisciplinary Xeroderma Pigmentosum Clinic
Guy’s and St Thomas’ NHS Foundation Trust
Westminister Bridge Road
London SE1 7EH, United Kingdom
http://www.guysandstthomas.nhs.uk/services/dash/dermatology/specialties/photobiology.aspx
Xeroderma Pigmentosum Society
437 Syndertown Road
Craryville, NY 12521, USA
The Share and Care Cockayne Syndrome Network
Box 282, Waterford, VA 20197, USA
Trichothiodystrophy – The Friends of Sammy-Joe
Sammy-Joe Liistro Fund
13 Hursley Court, Craigieburn
Victoria 3064, Australia
Clinical trials
Examination of clinical and laboratory abnormalities in patients with xeroderma pigmentosum, Cockayne syndrome or trichothiodystrophy
National Cancer Institute Protocol 99-C-0099
http://clinicalstudies.info.nih.gov/cgi/detail.cgi?A_99-C-0099.html
Genotype–phenotype correlations in Cockayne syndrome and ultraviolet sensitive syndrome
Contact: Edward Neilan MD, PhD, Dept of Pediatrics, Children’s Hospital Boston and Harvard Medical School, Boston, MA, Edward.Neilan@childrens.harvard.edu
Since the last workshop in this series, researchers and clinicians have made significant progress in several areas of key concern to this community. Aiding this effort, the scientists have had access to larger pools of clinical data and older surviving patients (due to improved sun-protection), and the opportunity to perform autopsies on deceased patients, and to conduct retrospective analyses of several decades of clinical research. These efforts have led to improved understanding of the diseases and their progression, as reported below. The workshop also provided the opportunity for young scientists to participate in a poster session, and five outstanding posters were presented orally during the final plenary session of the workshop. This report highlights selected presentations and key outcomes of the workshop (Box 2).
Box 2. Workshop highlights.
Highlights of clinical studies
The defects in DNA repair or DNA damage tolerance in XP patients dramatically increases the risk of early death from skin cancer and neurodegeneration. The former can be prevented by early diagnosis of XP.
Mothers carrying TTD-affected fetuses are at high risk of complications during pregnancy.
TTD patients are at significantly increased risk of severe infections. Thus early diagnosis is essential for preventing unnecessary death.
Multiple temperature sensitive XPD alleles have been identified leading to transient and reversible exacerbation of symptoms in TTD patients during febrile episodes.
Highlights of genetic and biochemical studies
Structural studies on XPD reveal that mutations that lead to XP, XP/CS and TTD differentially affect protein activity, stability and flexibility, providing a precedent for the critical contribution of structural studies to understanding disease pathogenesis.
CSB helps coordinate mitochondrial DNA repair, and therefore appears to play a critical role in maintaining mitochondrial function and limiting cellular levels of oxidative stress.
XPD forms a novel TFIIH-independent protein complex that appears to be important for chromosome segregation, a function that could contribute to genomic instability in XP and XP/CS patients.
Novel mouse models of premature aging and neurodegeneration
Systemic Ercc1 or neuron-specific knockout mouse models display premature aging and neurodegeneration that can be mitigated by pharmacological inhibition of cell stress responses.
Xpc−/−;Csb−/− double mutant mice have defects in both GG-NER and TC-NER and show severe neurodegeneration, premature aging and short life span, phenotypic symptoms that can be mitigated by exposure to nutraceuticals that reduce oxidative stress.
2. Long-term studies and aging patient populations: insights from a maturing clinical community
2.1. Understanding skin cancer susceptibility in XP patient subgroups
Drs. Kenneth Kraemer and Porcia Bradford (National Cancer Institute, Bethesda, MD) reported a retrospective analysis of clinical data on 106 XP patients seen at the National Institutes of Health (NIH) over 39 years, in which differential sensitivity to skin cancer and neurological degeneration were analyzed (Box 1). The disease-affected population had wide race-ethnicity distribution, broad age distribution (1–73 years of age) and balanced gender representation (48 males, 58 females). The study produced several striking conclusions. For XP patients under the age of 20 years, the non-melanoma skin cancer (NMSC) incidence was 10,000-fold higher than in the US general population, and melanoma incidence was 2000-fold higher. The median age at onset for NMSC was 9 years, 58 years younger than in the general population, while the median age of onset for melanoma was 22 years, 33 years younger than in the general population; this younger age of onset of NMSC compared to melanoma is a relative age reversal from the general population, suggesting a different mechanism of carcinogenesis for NMSC than for melanoma. Approximately 60% of the XP patients had marked sun sensitivity. These XP patients developed skin cancer at a later age than non-photosensitive patients, probably reflecting more extensive use of sun protection at an early age. The median age at death of XP patients was 32 years, 38 years younger than for the general population and metastatic skin cancer and neurological degeneration were the two most frequent causes of death (34.5% and 31%, respectively). XP patients with neurodegeneration had poorer survival than XP patients without neurodegeneration. The majority of patients with neurodegeneration (16/26) were in the XP-D complementation group. These data emphasize the major role of DNA repair genes in the etiology of skin cancer and neurologic degeneration (Table 1).
Dr. Brian Brooks (National Eye Institute, Bethesda, MD) described the ocular manifestations of XP and TTD. XP patients have signs of degeneration and premature aging of the eyes with dry eye, limbal stem cell deficiency, inflammatory lesions, scarring leading to deformities and neoplasms of the lids and conjunctiva. He reported results of a pilot study of conjunctival cytology for early detection of neoplasms, which might reduce the need for surgical biopsies. In contrast, the ocular changes in TTD appear to represent a developmental defect with some features of degeneration with neonatal cataracts that may progress, small corneas, abnormal eye movements and dry eyes.
2.2. Perspectives on TTD
TTD has historically been diagnosed based on a cluster of symptoms known by the mnemonic PIBIDS, an acronym for photosensitivity, ichthyosis, brittle hair, intellectual impairment, DNA repair defect and short stature. However, recent studies by Dr. John DiGiovanna (National Cancer Institute, Bethesda, MD) and colleagues provided evidence that many TTD patients present with clinical features that do not fit this pattern. DiGiovanna reviewed the literature and reported on TTD patients seen at the NIH. He identified the most prevalent and some novel clinical features of TTD. These included intellectual impairment (100%), bone abnormalities (92%), pregnancy complications in the mothers of the patients (88%), ocular abnormalities (80%), recurrent infections (76%) and gastrointestinal dysfunction, all of which were observed often as, or more frequently, than PIBIDS. Susceptibility to infection was noteworthy; indeed a literature review of case reports of 112 TTD patients showed a 20-fold higher than normal rate of infection-related deaths in TTD individuals <10 years of age. This emphasizes the need for early diagnosis of TTD so that precautionary measures can be taken to reduce the risk of infection and infection-related complications.
DiGiovanna also reported that two new syndromes were recently identified that overlap with TTD. Two sisters were identified who have clinical features of both TTD and CS. An infant was also identified with combined features of TTD and cerebro-oculo-facial-skeletal syndrome (COFS) who unfortunately died at 13 months of age. All three of these patients carry germline mutations in XPD. Thus, XPD mutations have now been linked with XP (with or without neurological disease), XP/CS, TTD, TTD/CS, TTD/XP and TTD/COFS (Table 1).
Dr. Melissa Merideth (National Human Genome Research Institute, Bethesda, MD) reviewed a detailed study of pregnancy complications in 24 mothers carrying TTD-affected offspring. There was up to 36-fold higher than expected rate of small for gestational age, pre-term birth, gestational hypertension and proteinuria, hemolysis/elevated liver enzymes/low platelets (HELLP syndrome), and frequent abnormal results for a triple marker screen. These studies suggest that altered gene expression in the fetus during gestation adversely affects fetal development. Genetic testing and counseling during pregnancy may also be of value, where there is reasonable suspicion of high TTD risk (Box 1).
2.3. Brain pathology in XP and TTD patients
Drs. Nicholas Patronas (Clinical Center, NIH) and Tyler Pierson (National Institute of Neurological Disorders and Stroke, Bethesda, MD) reported on neuroimaging studies of 36 XP, 18 TTD and 7 XP/ TTD patients. The results confirmed earlier findings based on approximately half as many patients. Brains from XP patients typically demonstrated cerebral and cerebellar atrophy but no white matter disease. Brains from TTD patients showed cerebral atrophy and white matter disease but no cerebellar atrophy. Brains from XP/TTD patients showed significant white matter disease, thus resembling brains from TTD patients more than XP patients. Among XP-affected individuals, those from XP-A and XP-D complementation groups were more severely affected by brain pathology (as detected by MRI/CT studies) than other complementation groups. Clinically, the XP patients with neurological abnormalities displayed progressive symptoms. The TTD patients could be divided into two groups: a normal-mild or moderate-severe neurophenotype. Both patient groups have significant defects of attention.
2.4. Molecular neuropathology in XP brains: insights from autopsies of XP and CS patients
With improved clinical care, XP patients live longer and experience higher quality of life. The close interactions between patient families, patient advocacy groups and medical professionals facilitated opportunities to do autopsies on two XP-afflicted patients. This is critical for characterizing the pathophysiology caused by NER defects. The autopsies of a 44 year-old XP-A patient and a 45 year-old XP-D patient were reported by Drs. Jinping Lai and Yen-Chun Liu (National Cancer Institute, Bethesda, MD). Both patients showed reduced brain weight (approximately half of normal for their age) and generalized brain atrophy. The XP-A patient showed thinning of the corpus callosum, neuronal loss and myelin pallor. This patient also showed denervation of skeletal muscle as a consequence of peripheral neuropathy. The XP-D patient showed evidence of a thick calvarium as well as thinning of the corpus callosum, patchy loss of Purkinje cells and myelin pallor in the cerebellum, and neuronal loss. The XP-A autopsy data was very similar to previous autopsy data on XP-A patients from Japan. The XP-D patient autopsy differed from the XP-A patient primarily in calvarium thickening and Purkinje cell loss.
Dr. Karen Weidenheim (Albert Einstein College of Medicine, New York) described the results of previously reported autopsies of four CS patients (ages 6, 14, 17 and 31 years of age). Their brains uniformly displayed markedly reduced weight, patchy demyelination, basal ganglia calcification, optic and auditory system defects and cerebellar atrophy with relative preservation of the neocortex. Atherosclerotic and arteriosclerotic changes, typical of aging normal brains, were present. The neuronal changes suggest possible defects in neuronal connectivity. The mechanism by which defective DNA repair and/or RNA transcription cause the observed brain pathology is unclear.
3. Diagnosis and treatment of XP, TTD and CS patients
XP is more common in Japan (about 1 per 22,000) than in the US and Europe (about 1 per million). Drs. Chikako Nishigori (Kobe University, Kobe, Japan) and Shinichi Moriwaki (Osaka Medical College, Osaka, Japan) described their experience with Japanese XP patients. Of more than 300 patients studied, about 50% were XP-A and about 25% were XP variant (XP-V). Complementation groups C and D represented only about 11% of the Japanese patients. Of interest, about 10% of the 300 patients could not be classified into the known complementation groups (A–G and V) (Table 1) suggesting that there are still unrecognized genes that when mutated can result in a clinical diagnosis of XP. In contrast, Kraemer reported that in the US, among 106 XP patients studied at NIH, 9% were XP-A, 6% were XP-V, 43% were XP-C and 28% were XP-D. There were 5% unclassified XP patients in the US cohort. The experience in Europe for diagnosis of 80 XP patients was similar to that at NIH as reported by Dr. Alan Lehmann (University of Sussex, Brighton, UK) with 10% XPA, 10% XP-V, 20% XP-C, 14% XP-D and 32% unclassified patients. An XP-A founder mutation was present in about 90% of the Japanese XP-A patients and was estimated to be present in 1% of the Japanese general population. Three mild XP-V Japanese patients were diagnosed as teenagers with symptoms limited to increased freckling. They were found to have reduced or absent POLη protein by immunoblotting and each have different mutations in POLη. An adult onset Japanese CSB patient has thyroid and ovarian cancer and carries a mutation previously only found in a younger UV sensitive patient. This raises the possibility that young UV sensitive syndrome patients, who generally have few clinical symptoms (Table 1), may develop progressively worsening symptoms as they age.
Dr. Edward Neilan (Harvard Medical School, Boston, MA) explained that the clinical diagnosis of CS is based on two major symptomatic criteria: post-natal growth failure and developmental delay with neurological dysfunction/deterioration caused by dysmyelination/demyelination. Less penetrant symptoms may also be present including skin photosensitivity, pigmentary retinopathy, sensorineural hearing loss, dwarfism, dental caries and intracranial calcifications. Laboratory diagnosis of about 300 samples over 5 years revealed mutations in 91 affected children in 85 families. Mutation screening was performed with multiplex ligation-dependent PCR. CSB mutations were found in 80% of the families representing 65 novel mutations. CSA mutations were found in 17 families with 19 novel mutations (Table 1). However, the majority of the samples analyzed did not have mutations in the known CS genes. This suggests either that the clinical diagnosis of CS was not correct or that there are still unrecognized genes that when mutated can result in clinical CS. CS patients are being enrolled in a natural history study (Box 1). A phase I/II treatment study of Prodarsan, an oral anti-oxidant, was closed when the sponsoring company withdrew its support.
Dr. Nicolaas G.J. Jaspers (Erasmus Medical Center, Rotterdam, the Netherlands) described a new, non-radioactive method for measuring unscheduled DNA synthesis in response to UV radiation, a test necessary for diagnosis of XP, CS, and TTD. The new method is based on fluorescence detection of ethynyl-deoxyuridine (EdU) in nuclear DNA to measure DNA repair synthesis.
Publicly accessible resources have been established to provide information and support to patients and their families, and links to research about XP, CS and TTD (Box 1). XP support groups are active in the US and UK, a CS support group exists in the US, and a website has been established to support families of TTD-affected children. Dr. Robert Sarkany (St. Thomas NHS Foundation Trust, London, UK) reported that a multidisciplinary clinic for diagnosis and treatment of XP has been established in the UK. XP, TTD and CS patients can also be enrolled in a multidisciplinary protocol at NIH that is documenting the natural history of these diseases.
4. Spotlight on XPD
4.1. Structural and biochemical studies provide insight into complex pathology
XPD presents an interesting research puzzle because it is linked to eight clinically distinct diseases, and mutations associated with each disease are distributed throughout the XPD gene (Table 1). Two presentations at this workshop highlighted progress towards solving this puzzle. Drs. Jill Fuss (Lawrence Berkeley National Laboratory, Berkeley, CA) and Miria Stefanini (Istituto di Genetica Molecolare Consiglio Nazionale delle Ricerche, Pavia, Italy), provided insight into allele-specific biological effects of XPD mutants.
Structural biochemistry of XPD has advanced significantly, at least in part because of the availability of a low resolution electron microscopy structure of TFIIH and a high resolution crystal structure of archael XPD protein. Fuss mapped known XPD mutations onto the Sulfolobus acidocaldarius XPD (SaXPD) crystal structure and observed that the mutations associated with XP, XP/ CS and TTD localize to different domains of the protein. XP-causing mutations map to the DNA binding and ATPase domains; XP/CS-causing mutations, associated with more severe pathology, map to one of the two helicase domains of SaXPD and reduce protein flexibility; and TTD-causing mutations are scattered throughout the protein, but share the attribute that they all reduce protein stability. Although K446 and G447 are adjacent amino acids in SaXPD, structural analyses predict that XP-associated mutation K446L (R601L in human XPD) would have a less severe impact on XPD function than the XP/CS-associated mutation G447D (G602D in human XPD), because only the latter is expected to strongly reduce protein flexibility. Biochemical studies also demonstrated that G447D-XPD has much lower residual ATPase activity than K446L SaXPD. Fuss also characterized SaXPD equivalents of two human arch domain mutants from patients, showing that both mutations inactivate XPD helicase, although by different mechanisms. Despite their structural proximity, one mutation disrupts ATPase activity, while the other mutation does not. These data exemplify how structural studies of disease-associated protein variants coupled with biochemical analyses provide critical insight into the pathophysiology of disease.
Stefanini described four Italian TTD patients from three families who exhibited transient, reversible worsening of symptoms including hair loss when febrile. All of these patients are compound heterozygotes for novel mutations in XPD, one of the genes responsible for TTD (Table 1). Studies on cultured patient cells support the conclusion that these mutations cause a decrease in the efficiency of NER and in the amount of XPD, and thereby TFIIH, which is more pronounced at elevated temperature. These alterations are associated with a temperature-dependent reduction in the rate of basal transcription. At the organismal level, decreased TFIIH and decreased capacity for mRNA synthesis could have profound effects on differentiated cells, such as those involved in hair structure, neuromyelination and epidermal differentiation, thereby contributing to the reported aggravation of clinical features in patients when febrile.
Dr. Jean-Marc Egly (Institut de Génétique et de Biologie Moléculaire et Cellulaire, Center National de la Recherche Scientifique, INSERM, Université de Strasbourg, Illkirch cedex, France) described studies on the critical role played by XPD in TFIIH-dependent activation of global and tissue-specific gene expression in XPD mutant cells. Egly presented studies on a mouse model of TTD, in which animals homozygous for the common XPD R722W mutation display dysmyelination in the brain, mimicking the phenotype of TTD patients. These mice demonstrate selective deregulation of thyroid hormone-responsive genes, including myelin basic protein (MBP) and myelin proteolipid (PLP), in the central nervous system. Egly speculates that the low level of TFIIH containing mutant XPD destabilizes complexes between thyroid hormone nuclear receptors and responsive elements of receptor target genes, including MBP. This implies that TFIIH functions as a co-activator of gene expression. Egly also presented evidence that NER proteins, including XPG, XPC, XPF, ERCC1, XPA and CSB, are recruited to the promoters of activated genes, suggesting that mutations in these genes may also interfere with proper activation of gene expression in some differentiated cells. Therefore, cells from XP patients, as well as cells from CS and TTD patients, may have defects in transcription and/or promoter activation.
Adding a new functional attribute to the already complex XPD landscape, Dr. Kyoji Tanaka (Osaka University, Osaka, Japan) reported the discovery of a novel, TFIIH-independent role for XPD. Tanaka characterized an XPD-containing protein complex, composed of MMS19, MIP18/FAM96B, Ciao1, ANT2 and XPD. MMS19, MIP18 and XPD localize to the mitotic spindle during mitosis and siRNA-mediated knockdown of MMS19, MIP18, or XPD caused defects in chromosome segregation. Tanaka speculated that defects in chromosome segregation could contribute to disease pathology in some cases of XP and XP/CS.
5. Therapeutics: new territory for mouse models of neurological dysfunction
Mouse models of XP, CS and TTD have historically played an important role in research on the physiological consequences of defective DNA repair. Recent work has extended their application to testing therapeutic interventions for neurodegeneration. Such studies enable testing of focused hypotheses about the molecular mechanism of pathogenesis. They also have the potential to identify drugs that could delay or attenuate neurodegeneration in XP, CS and TTD. Importantly, such therapeutic agents, once developed and tested, might also be useful for preventing or delaying neurodegenerative processes associated with normal aging.
Dr. Laura Niedernhofer (University of Pittsburgh, Pittsburgh, PA) presented studies based on mouse models she developed while working in Dr. Jan Hoeijmakers’ laboratory (Erasmus Medical Center, Rotterdam, the Netherlands). Niedernhofer developed several models involving mutations in Ercc1, which encodes one subunit of the ERCC1-XPF endonuclease required for NER. The previously characterized Ercc1 knockout mouse (Ercc1−/−) displays signs of accelerated aging in the musculoskeletal, neurologic, hematopoietic, epidermal, hepatobiliary systems and premature death at 4 weeks of age. These mice have a dramatically more severe phenotype than other mouse strains defective in NER. This is attributed to the fact that the ERCC1-XPF endonuclease participates in other DNA repair pathways in addition to NER, including interstrand crosslink repair and double-strand break repair. Hence, it is thought that the ERCC1-deficient mice accumulate a greater number and broader spectrum of endogenous DNA lesions more rapidly than other NER-deficient mice.
Mice carrying one null and one hypomorphic Ercc1 allele with a C-terminal deletion (Ercc1−/Δ) have a milder phenotype than the Ercc1 knockout mice and longer lifespan of approximately 7 months. These mice display a nearly identical spectrum of premature aging symptoms as the Ercc1−/− mice and patients with XFE progeroid syndrome, which is caused by mutations in XPF (Table 1). Importantly, unlike mice defective only in NER, symptoms of neurodegeneration appear spontaneously in ERCC1-deficient mice, mimicking the neurologic features of XP. A third Ercc1 model (Ercc1−/flox) carries one null allele and one floxed allele that can be conditionally inactivated by tissue- or cell type-specific expression of Cre recombinase (e.g., crossing the Ercc1−/flox mice with a nestin-Cre transgenic strain to knockout expression of ERCC1 only in neurons). Because Ercc1−/Δ are relatively unaffected at a young age, but display rapidly progressing neurodegeneration over a period of several months, these mice, or the tissue-specific ERCC1-knockout mice, provide an excellent experimental system for testing therapeutics aimed at delaying the onset, or attenuating the severity of neurodegeneration caused by NER defects.
Dr. Ingrid van der Pluijm reported on experiments performed at DNage, Inc. and the Departments of Genetics and Neurosciences, Erasmus University, Rotterdam, The Netherlands, that illustrate the role of oxidative DNA damage in neurodegenerative processes in CS. Initial studies identified characteristic pathological features in the brains of mouse models of CS, including expression of glial acidic filament protein (GFAP, a marker of neurodegeneration), increased numbers of activated phagocytosing microglia (i.e., Mac2-positive microglia), and expression of caspase 3 and p53 (markers of apoptosis and the DNA damage response, respectively). These features were most pronounced in mice with a transcription coupled NER (TC-NER) defect, including Csa−/−, Csb−/−, XpdXP/CS, Xpa−/−Csb−/− mice. They proposed to test libraries of small molecules as single agent or combination therapeutics in Ercc1−/Δ and Csb−/−Xpa−/flox mice;CAMKII-Cre and Csb−/− mice. A proprietary combination of two small molecules called Prodarsan® showed promising results in mice. The profound neurodegeneration associated with XP and CS and mouse models of these diseases support the conclusion that DNA damage promotes neuronal loss, and that this process could potentially be delayed by appropriate interventions.
The molecular mechanisms by which neurodegeneration and neurological dysfunction could be delayed is under investigation. Recent studies by Dr. Paul Robbins (University of Pittsburgh, Pittsburgh, PA) and colleagues suggest that activation of cellular stress responses contributes to cell loss and dysfunction in the absence of NER. Robbins showed that NF-κB, a transcription factor activated by inflammatory, oxidative and genotoxic stress, is more active in mouse embryo fibroblasts from Ercc1−/− mice than in wild-type control cells. Because NF-κB regulates cell survival in response to stress, this transcription factor may offer a novel target for delaying or attenuating degenerative symptoms in XP, CS and TTD. Significantly, inhibitors of NF-κB are currently being tested in human clinical trials.
The Xpc−/− mouse has a global genome NER (GG-NER) defect, but shows few spontaneous symptoms. Similarly, Csa−/− and Csb−/ − mice, which are defective in TC-NER but not GG-NER, have a much milder phenotype than most human CS patients carrying an equivalent genetic defect. However, double mutant Xpc−/−;Csb−/− mice, which lack GG- and TC-NER, which display a severe post-natal growth defect and very short life span, provide a useful mouse model for the more severe forms of CS. Dr. James Cleaver (University of California, San Francisco) and colleagues reported extensive characterization of these animals. These mice are normal at birth but exhibit post-natal whole-body wasting, ataxia and neural loss and die by postnatal day 21 with a 50% reduction in body weight. Granule cells and Purkinje cells are depleted in the mouse cerebellum and demyelination (loss of staining for myelin basic protein) is observed in the corpus callosum, hippocampus, and anterior commissure. Although oligodendrocytes were not severely depleted in Xpc−/−;Csb−/− mice, they appear to lose their proliferative capacity prematurely. Cleaver’s interpretation of these data is that oligodendrocytes in Xpc−/−;Csb−/− mice enter premature senescence and fail to differentiate normally, leading to defective CNS myelination. This provides novel insight into the pathophysiology of CS neurodevelopmental defects.
6. ROS, mitochondria, oxidative DNA damage and aging: focus on CSB
Transcriptional-coupled NER (TC-NER) is a well-established NER subpathway, which requires CSA and CSB proteins. Recent studies from Dr. Vilhelm Bohr’s laboratory (National Institute of Aging, NIH, Baltimore, MD) suggest that CSB may also play a direct role in BER, as well as a role in maintaining mitochondrial function. Several lines of evidence support the idea that CSB plays a role in BER. For example, CSB interacts with multiple essential BER proteins and stimulates their activity, including apurinic endonuclease 1 (APE1), NEIL-1, PARP-1, and 8-oxoguanine glycosylase (OGG1). CSB, APE1, OGG1, FEN1 and NEIL-1 localize to both the nuclear and mitochondrial compartments, and are thought to contribute to BER in both organelles (Fig. 2). Several other proteins that contribute to the DNA damage response have also been identified in mitochondria, raising the question of which DNA repair pathways exist in this organelle. Although PARP-1 is predominantly nuclear, a subfraction of PARP-1 may also localize to mitochondria, and indirect evidence suggests that NEIL-1 is required for optimal mitochondrial BER. These proposed roles of CSB are consistent with the hypothesis that mitochondrial dysfunction leads to increased production of damaging ROS, the free radical theory of aging, and the accelerated aging associated with CS.
Fig. 2.
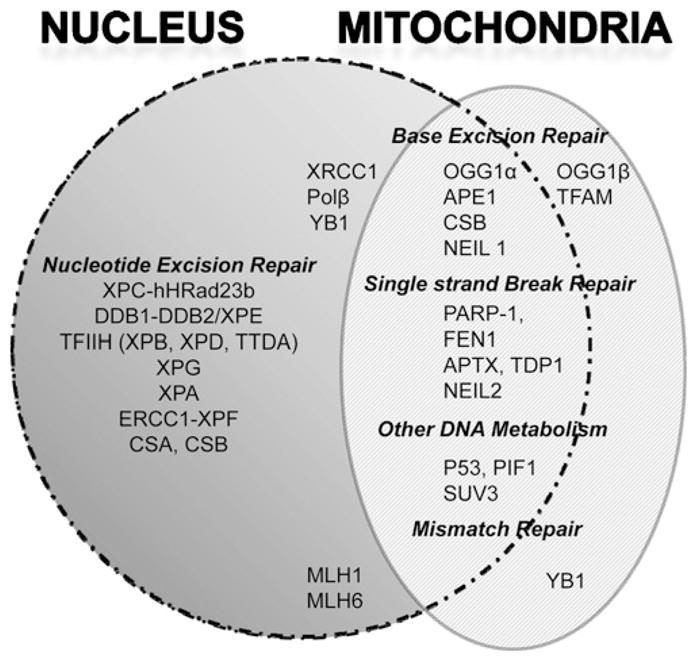
Distribution of DNA repair proteins in the nucleus and mitochondrion. Schematic diagram summarizing subcellular localization of DNA repair proteins in eukaryotic cells. Proteins listed in the overlap region localize to both nuclei and mitochondria.
Recent work by Drs. Bohr, Berquist, and David Wilson (National Institute on Aging, NIH, Baltimore, MD) shows that CSB localizes to the mitochondrial nucleoid and co-localizes with mitochondrial transcription factor A (TFAM) in cells treated with the oxidant menadione. TFAM is a high mobility group (HMG)-like protein that may act as a structural scaffold for the mitochondrial nucleoid, to which it also recruits proteins involved in mitochondrial DNA replication and repair. Using Seahorse technology, Bohr’s laboratory demonstrated that cells from a CS patient have increased respiratory capacity, oxygen consumption, and more ROS than control cells. Consistent with this, Csbm/m mice have a higher metabolic rate than normal mice, most likely as a consequence premature loss of subcutaneous fat. In this regard, the mutant mice phenotypically resemble CS patients.
These data suggest that CSB may play an important role in preventing mitochondrial dysfunction and oxidative stress. Therefore, CS pathology could be caused at least in part by a vicious cycle, in which impaired repair of mitochondrial DNA could lead to mitochondrial dysfunction and increased production of ROS, which in turn causes more oxidative DNA damage, both in the nuclear and mitochondrial genomes (Fig. 3).
Fig. 3.

Proposed contribution of mitochondrial dysfunction to premature aging in Cockayne syndrome and related diseases. Upper panel: defects in CSB or other proteins involved in mtBER cause accumulation of mtDNA mutations or loss of mtDNA. Mutated or depleted mtDNA leads to altered respiration and increased production of ROS/RNS, which in turn causes more cellular damage, including even more mtDNA damage (arrows). This vicious cycle accelerates the rate of accumulating dysfunctional mitochondria. Lower panel: a single young cell contains many functional mitochondria (left). Normal aging is associated with slow accumulation of dysfunctional mitochondria. Phenotypic signs appear at high degree of mitochondrial heteroplasmy (right). Defective mtBER could increase the rate of accumulation of dysfunctional mitochondria (upper panel) in individuals with CS or other diseases characterized by accelerated aging.
Several rare human diseases, including those with profound neurodegenerative symptoms, are caused by defects in proteins that play critical roles in mitochondrial DNA metabolism: Alpers syndrome (caused by mutations in the gene encoding DNA polymerase γ, the mitochondrial DNA polymerase), chronic progressive external ophthalmoplegia (caused by mutations in DNA polymerase γ or twinkle, a mitochondrial DNA helicase), the family of spinocerebellar ataxias, and Ataxia With Oculomotor Apraxia Type 1 (AOA1; caused by mutations in APTX, which encodes aprataxin, critical for single-strand break repair). In related workshop presentations, Drs. Ben van Houten (University of Pittsburgh, Pittsburgh, PA) and Matthew Longley (National Institute of Environmental Health Sciences, NIH, Research Triangle Park, NC) described cell and molecular studies on twinkle and DNA polymeraseγ, and discussed the disease states associated with defects in these two mitochondrial replication proteins.
Ongoing studies by Wilson, Bohr and colleagues have continued to improve our understanding of the biochemical activities of CSB. Although CSB protein has conserved DNA helicase motifs, it has no demonstrable DNA helicase activity in vitro. However, CSB is a DNA-dependent ATPase that anneals homologous single-stranded DNA in vitro. CSB potently stimulates BER in vitro in an ATP-independent manner. TFAM interacts physically and functionally with CSB. CSB displaces pre-bound TFAM and other DNA binding proteins from DNA in an ATP-independent manner. Wilson proposed that CSB may promote chromatin remodeling in an ATP-dependent manner, but that its DNA damage sensing and repair functions are ATP-independent. Dr. P.J. Brooks (National Institute of Alcohol Abuse and Alcoholism, Bethesda, MD) reported that CSB is found in the nuclear matrix fraction of human brain cells. Dr. Leon Mullenders (Leiden University, the Netherlands) provided evidence that CSB ATPase is essential for recruiting XPG to NER complexes in the nuclei of human cells. Taken together, these studies highlight multiple potential functions of CSB in multiple cellular compartments, and offer potential novel therapeutic targets for CS.
7. Perspectives
This workshop on XP, CS, TTD and related diseases presented an opportunity for clinicians and researchers to review and discuss progress in understanding disease pathogenesis and to identify promising areas for future study. As prioritized in the last workshop, much progress has been made in understanding the pathophysiology and molecular mechanisms driving neurodegeneration in these rare diseases. Although there are only a few hundred patients affected by these inherited diseases, the experience and knowledge gained in interacting with and helping them is invaluable because it promotes understanding of normal human biology and the causes of diseases affecting the general population. For example, XP pathology has taught us that both melanoma and non-melanoma skin cancers are caused directly by UV-induced DNA damage from the sun. Similarly, insights into the mechanisms of the premature aging and neurodegenerative features in XP, CS and TTD patients are relevant to the understanding of aging-related neurodegeneration in normal individuals. The development and recent characterization of mouse models for testing the efficacy of anti-aging and neuro-protective therapeutics, some of which may have value for the general population, is an exciting development in this field.
Acknowledgments
This research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, the Office of Rare Diseases of the NIH, the National Institute of Aging and the National Institute of Environmental Health Sciences as well as the University of Pittsburgh Cancer Institute.
Footnotes
The workshop, held in Chantilly, Virginia from 21 to 24 September 2010, was co-organized by Drs. Vilhelm Bohr (National Institute of Aging), Kenneth Kraemer (National Cancer Institute), and Laura Niedernhofer (University of Pittsburgh) and sponsored by the National Institutes of Health Office of Rare Diseases; Center for Cancer Research, National Cancer Institute; National Institute on Aging, Laboratory of Molecular Gerontology; University of Pittsburgh; The Ellison Medical Foundation; Trevigen; Xeroderma Pigmentosum Family Support Group; Fisher Scientific, Invitrogen and Integrated DNA Technologies, Inc.
Contributor Information
Laura J. Niedernhofer, University of Pittsburgh, Pittsburgh, PA, United States
Vilhelm A. Bohr, Laboratory of Molecular Gerontology, National Institute of Aging, Baltimore, MD, United States
Miriam Sander, Page One Editorial Services, Boulder, CO, United States.
Kenneth H. Kraemer, DNA Repair Section, Dermatology Branch, Center for Cancer Research, National Cancer Institute, Building 37, Room 4002, MSC 4258, Bethesda, MD 20892-4258, United States.
References
- Bohr VA, Sander M, Kraemer KH. Rare diseases provide rare insights into DNA repair pathways, TFIIH, aging and cancer. DNA Repair. 2005;4:293–302. doi: 10.1016/j.dnarep.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Sander M, Bohr VA. New areas of focus at workshop on human diseases involving DNA repair deficiency and premature aging. Mech Ageing Dev. 2007;128:229–235. doi: 10.1016/j.mad.2006.11.028. [DOI] [PubMed] [Google Scholar]
Further reading
- Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu M, Tian J, de Souza-Pinto NC, Scheibye-Knudsen M, Wilson DM, 3rd, Stevnsner T, Bohr VA. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010;24:2334–2346. doi: 10.1096/fj.09-147991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford PT, Goldstein AM, Tamura D, Khan SG, Ueda T, Boyle J, Oh KS, Imoto K, Inui H, Moriwaki S, Emmert S, Pike KM, Raziuddin A, Plona TM, Digiovanna JJ, Tucker MA, Kraemer KH. Cancer and neurologic degeneration in xeroderma pigmentosum: long term follow-up characterises the role of DNA repair. J Med Genet. 2011;48:168–176. doi: 10.1136/jmg.2010.083022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Waard MC, van der Pluijm I, Zuiderveen BN, Comley LH, Haasdijk ED, Rijksen Y, Ridwan Y, Zondag G, Hoeijmakers JH, Elgersma Y, Gillingwater TH, Jaarsma D. Age-related motor neuron degeneration in DNA repair-deficient Ercc1 mice. Acta Neuropathol. 2010;120:461–475. doi: 10.1007/s00401-010-0715-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghri S, Tamura D, Kraemer KH, Digiovanna JJ. Trichothiodystrophy: a systematic review of 112 published cases characterises a wide spectrum of clinical manifestations. J Med Genet. 2008;45:609–621. doi: 10.1136/jmg.2008.058743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Tan LJ, Andoh D, Narita T, Seki M, Hirano Y, Narita K, Kuraoka I, Hiraoka Y, Tanaka K. MMXD, a TFIIH-independent XPD-MMS19 protein complex involved in chromosome segregation. Mol Cell. 2010;39:632–640. doi: 10.1016/j.molcel.2010.07.029. [DOI] [PubMed] [Google Scholar]
- Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype-phenotype relationship. Neuroscience. 2007a;145:1388–1396. doi: 10.1016/j.neuroscience.2006.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laposa RR, Huang EJ, Cleaver JE. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc Natl Acad Sci USA. 2007;104:1389–1394. doi: 10.1073/pnas.0610619104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le May N, Mota-Fernandes D, Vélez-Cruz R, Iltis I, Biard D, Egly JM. NER factors are recruited to active promoters and facilitate chromatin modification for transcription in the absence of exogenous genotoxic attack. Mol Cell. 2010a;38:54–66. doi: 10.1016/j.molcel.2010.03.004. [DOI] [PubMed] [Google Scholar]
- Le May N, Egly JM, Coin F. True lies: the double life of the nucleotide excision repair factors in transcription and DNA repair. J Nucleic Acids. 2010b:10. doi: 10.4061/2010/616342. Article ID 616342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moslehi R, Signore C, Tamura D, Mills JL, Digiovanna JJ, Tucker MA, Troendle J, Ueda T, Boyle J, Khan SG, Oh KS, Goldstein AM, Kraemer KH. Adverse effects of trichothiodystrophy DNA repair and transcription gene disorder on human fetal development. Clin Genet. 2010;77:365–373. doi: 10.1111/j.1399-0004.2009.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenych V, de Jesus BB, Zhovmer A, Egly JM, Coin F. Molecular insights into the recruitment of TFIIH to sites of DNA damage. EMBO J. 2009;28:2971–2980. doi: 10.1038/emboj.2009.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanini M, Botta E, Lanzafame M, Orioli D. Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair. 2010;9:2–10. doi: 10.1016/j.dnarep.2009.10.005. [DOI] [PubMed] [Google Scholar]
- Usuda T, Saijo M, Tanaka K, Sato N, Uchiyama M, Kobayashi T. A Japanese trichothiodystrophy patient with XPD mutations. J Hum Genet. 2011;56:77–79. doi: 10.1038/jhg.2010.123. [DOI] [PubMed] [Google Scholar]