

Abstract
A new approach toward the asymmetric synthesis of optically active trifluromethylated amines was enabled by an unprecedented, highly enantioselective catalytic isomerization of trifluoromethyl imines with a new chiral organic catalyst. Not only aryl but also alkyl trifluoromethylated amines could be obtained in high enantioselecitivities.
The beneficial yet unique impact resulted from the presence of fluorine on organic molecules of pharmaceutical and agrochemical importance has led to a rapidly increasing demand of synthetic methodology providing ready and practical access to organofluorines.1 Presented in many biologically interesting compounds,2 trifluoromethylated amines are recognized to afford improved lipophilicity and metabolic stability over the corresponding methyl amines. In addition trifluoromethylated amines have found applications in medicinal chemistry as non-basic amide bonds surrogates3 and as reagents for ee analysis4.
Accordingly, significant strides have been made in catalytic asymmetric synthesis of trifluoromethylated amines. A variety of asymmetric nucleophilic addition to trifluoromethyl imines have been reported.5 Uneyama6a, Zhou6b and Akiyama6c disclosed highly enantioselective hydrogenations of trifluoromethyl imines with either chiral metal or organic catalysts. Applying phase transfer catalysis, Shibata7 and coworkers achieved highly enantioselective addition of trifluoromethylsilane to designed, conformation-constrained azomethine imines.
In spite of these notable advances, significant challenges remain in catalytic asymmetric synthesis of chiral trifluoromethylated amines. Among them the lack of a general access towards chiral aliphatic trifluoromethylated amines stands as an especially important one. To our knowledge, only two examples of catalytic asymmetric generation of aliphatic trifluoromethylated amines from achiral starting materials in high optical purity have been documented.5a,6b Specifically, Zhou6b reported the Pd-catalyzed asymmetric hydrogenations of N-PMP trifluoromethyl n-butyl and homobenzyl imines in 89% and 92% ee, respectively. Herein, we wish to describe a new approach based on organocatalysis that provides highly enantioselective access toward a broad range of aromatic and, more importantly, aliphatic chiral trifluoromethylated amines.
The catalytic enantioselective isomerization of N-benzyl trifluoromethyl imines8 to the corresponding trifluoromethylated amines via 1,3-proton shift represents an attractive strategy for the asymmetric synthesis of chiral trifluoromethylated amines. Early attempts to realize this strategy by synthetic catalysts were met with limited success, although proton transfer catalysis is a commonly occurring pathway in enzymatic transformations. Particularly relevant to the current study is the report by Soloshonok and Yasumoto,8d in which a room temperature isomerization of N-benzyl trifluoromethyl phenyl imines catalyzed by a cinchonidine derived organic catalyst was documented in up to 35% ee. The extremely long reaction time (52 days, 79% conv.) illustrated the need to improve catalytic activity. Later, Plaquevent8e and co-workers reported a cinchona alkaloid-catalyzed isomerization of an aliphatic trifluoromethyl imine. The aliphatic imine was found to be much less active, as a significantly elevated temperature (50-110 °C) is required for the cinchona alkaloid-catalyzed isomerization to proceed.
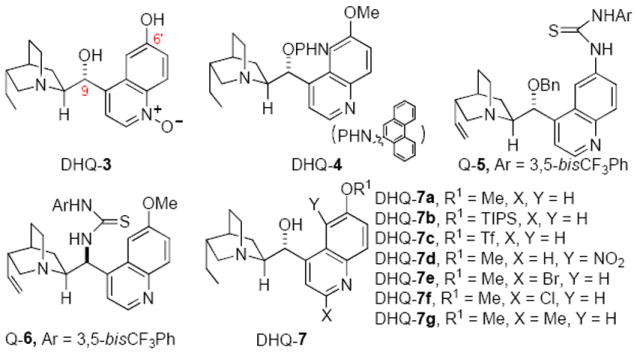
Results from these previous studies indicated that the most daunting challenge in the development of a highly enantioselective isomerization that is applicable to both aryl and alkyl trifluoromethyl imines lies in the discovery of a catalyst of not only high enantioselectivity but also enhanced activity. Recently we9 and the Shi group10 reported cinchona alkaloid-catalyzed enantioselective proton transfer catalysis for the isomerization of butenolides and isomerization of iminoesters, respectively. In both reports the 6’-OH cinchona alkaloids11 were shown to be effective catalysts. These developments prompted us to explore cinchona alkaloid derivatives bearing a hydrogen bond donor moiety as bifunctional catalysts for enantioselective isomerization of N-benzyl protected trifluoromethyl imines.
Previous studies by Soloshonok12 and Plaquevent8e illustrated the significant impact of the N-substituent on the activity of the trifluoromethyl imines toward isomerization. Prior to our catalyst development studies, we first examined a series of trifluoromethyl imines 1 bearing various N-substituents with the goal of identifying the optimal N-substituent in activating the imine toward an Et3N-catalyzed isomerization. In the presence of 10 mol% Et3N in dichloromethane, virtually no isomerization was detected with N-benzyl imine 1Aa after 30 min at room temperature (entry 1, Table 1). Replacing the N-benzyl with either 4-trifluoromethylbenzyl or 4-carboxylatebenzyl resulted in dramatically improved conversion (entries 2-3 vs. 1, Table 1). With the even stronger electron-withdrawing N-4-NO2 or N-2-NO2 benzyl, trifluoromethyl imine 1Da was converted into the corresponding trifluoromethylated amine 2Da in nearly complete conversion after 30 min. Thus, the enantioselective isomerization of 1Da was selected as the model reaction for the screening of chiral catalysts.
Table 1.
Identification of the Optimal N-Substituenta
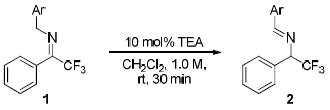
Unless noted, the reaction was carried out with 1 (0.1 mmol) in CH2Cl2 (0.1 mL) in the presence of TEA (0.01 mmol) at rt for 30 min.
Determined by 19F NMR analysis.
Since the 6’-OH cinchona alkaloid DHQ-3 was identified by us as an effective catalyst for enantioselective isomerization of butenolide,9 we first examined its ability for the promotion of the enantioselective isomerization of imine 1Da. As a control, we also investigated the same reaction with a cinchona alkaloid bearing no hydrogen bond donor (DHQ-4). Cinchona alkaloid DHQ-4 afforded much lower conversion in comparison to that by DHQ-3 (entry 2 vs. entry 1, Table 2). To our disappointment, despite affording high enantioselectivity for isomerization of butenolide,9 DHQ-3 was found to furnish practically no enantioselectivity for the isomerization of imine 1Da. Similarly non-enantioselective isomerization was also observed with a cinchona alkaloid (Q-5) bearing a 6’-thiourea (entry 3, Table 2). The catalyst screening was next extended to DHQ-7a, a 9-OH cinchona alkaloid. Although the aliphatic 9-OH should be a weaker hydrogen bond or proton donor than the phenolic 6’-OH, DHQ-7a was found to afford significantly higher activity and enantioselectivity (entry 5 vs. entries 1-3, table 2). Unexpectedly, the corresponding 9’-thiourea cinchona alkaloid 6 was found to be inactive. These results suggested that the overall spatial arrangement of the quinuclidine and the 9-OH group is critical to both the reactivity and enantioselectivity of DHQ-7a. Thus, our catalyst screening study proceeded further with a focus on modified 9-OH cinchona alkaloids.
Table 2.
Reaction optimizationa
| |||||
|---|---|---|---|---|---|
| Entry | cat. | T(°C) | t(h) | conv. (%)b | ee (%)b |
| 1 | DHQ-3 | rt | 0.5 | 47 | 5 |
| 2 | DHQ-3 | rt | 0.5 | 16 | 0 |
| 3 | Q-5 | rt | 0.5 | 38 | 8 |
| 4 | Q-6 | rt | 0.5 | < 5 | - |
| 5 | DHQ-7a | rt | 0.5 | 97 | 26 |
| 6 | DHQ-7b | rt | 0.5 | 80 | 17 |
| 7 | DHQ-7c | rt | 0.5 | 71 | 31 |
| 8 | DHQ-7d | rt | 0.5 | 62 | 38 |
| 9 | DHQ-7e | rt | 0.5 | 90 | 58 |
| c10 | DHQ-7f | rt | 0.5 | 95 | 68 |
| 11 | DHQ-7g | rt | 0.5 | 98 | 28 |
| 12 | DHQ-7f | -20 | 12 | 87 | 78 |
| d13 | DHQ-7f | -20 | 72 | 80 | 82 |
| e14 | DHQ-7f | -20 | 24 | 71 | 88 |
| f15 | DHQ-7f | -20 | 24 | 84 | 88 |
| f16 | DHQ-7f | -30 | 48 | 81 | 90 |
Unless specified, the reaction was carried out with 1Da (0.05 mmol) in CH2Cl2 (0.05 mL) in the presence of catalyst (0.005 mmol).
Determined by HPLC analysis.
After 48h, the ee value was reduced to 46%.
The reaction was performed in CH2Cl2 (0.50 mL).
The reaction was performed in PhMe (0.50 mL).
The reaction was performed with 1Da (0.05 mmol) and 4 Ǻ MS (5.0 mg) in PhMe (0.50 mL) in the presence of DHQ-7f (0.005 mmol).
Subsequently, we prepared a series of 9-OH cinchona alkaloids via the modifications of the quinoline ring. The introduction of an electron-donating silyloxy group at 6’ position resulted in a decreased enantioselectivity (entry 6, Table 2) while the electron-withdrawing triflate slightly improved the enantioselectivity (entry 7, Table 2). Overall, these 6’-substituted 9-OH cinchona alkaloids provided very low enantioselectivity. A cinchona alkaloid bearing a nitro substituent on the 5’ position (DHQ-7d) afforded slightly better but still low enantioselectivity and diminished activity (entry 8, Table 2). The introduction of substituents at the 2’ position turned out to have the biggest influence on enantioselectivity. With a 2’-Br the cinchona alkaloid DHQ-7e furnished a significantly improved enantioselectivity while providing excellent activity (entry 9, Table 2). Switching the 2’-Br into 2’-Cl, the resulting catalyst (DHQ-7f) afforded further enhanced activity and higher enantioselectivity (entry 10, Table 2), and the enantioselective isomerization produced the chiral trifluoromethylated amine in 68% ee. The electronic nature of the 2’-substituent was found to have a dramatic effect on enantioselectivity, as catalyst DHQ-7g with a 2’-methyl quinoline gave significantly lower enantioselectivity (entry 11 vs. entries 9-10, Table 2).
During the investigation of the isomerization of 1Da, we found that the product 2Da underwent racemization at room temperature if the reaction mixture was left in stirring for more than 24h. On the other hand, virtually no racemization was detected at -20 °C even after three days. At that temperature, the enantioselectivity is also improved. We also found that, by decreasing the reaction concentration from 1.0 M to 0.1 M, the enantioselectivity could be further increased to 82% ee. Nonetheless, a longer reaction time is required to reach a good conversion (entry 13, Table 2). We next found that solvent effect provided us with an additional handle for reaction optimization. The ee of 2Da reached 88% in toluene vs. 82% in dichloromethane (entries 13-14, Table 2) and the reaction rate was also faster. For reactions in toluene, the addition of molecular sieves is beneficial to reaction conversion without compromising enantioselectivity (entry 15, Table 2). Finally, at -30°C a highly enantioselective isomerization of 1Da in high conversion could be attained in toluene (entry 16, Table 2).
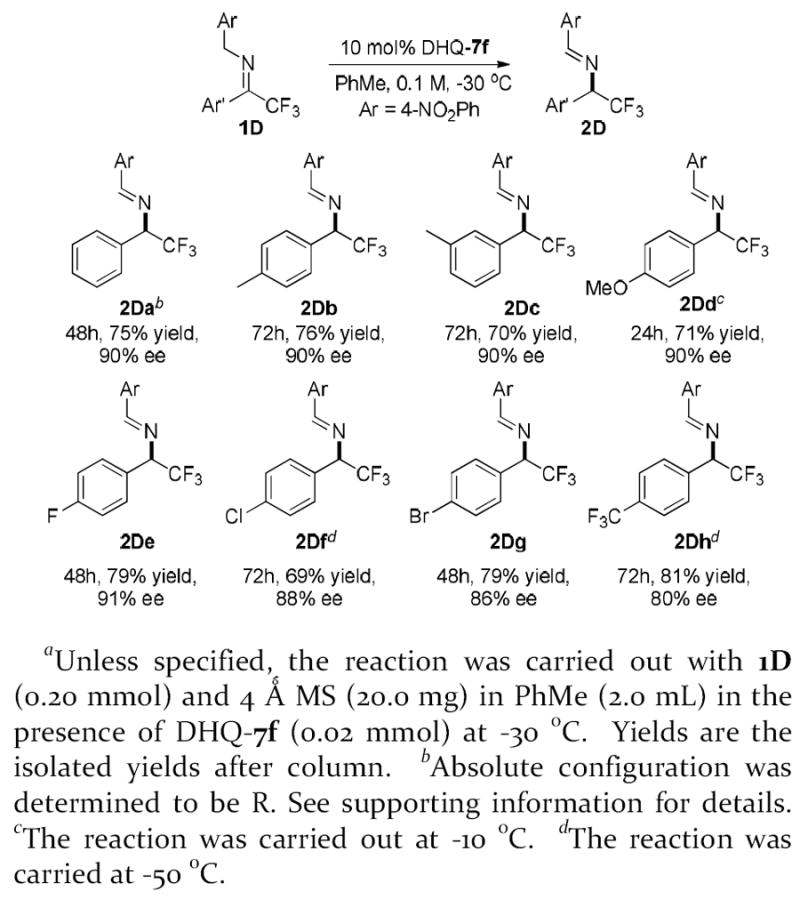
A variety of aryl trifluoromethyl ketimines 1Da-1Dh were subjected to the DHQ-7f-catalyzed isomerization (Table 3). The imine bearing a strongly electron-donating substituent, such as 1Dd, is less active. An elevated temperature was required for the reaction to proceed to high conversion and, fortunately, an excellent enantioselectivity could still be attained. In contrary imines bearing strongly electron withdrawing substitutents were more reactive, thereby optimal enantioselectivity could be obtained at lower temperature. Overall, aryl trifluoromethyl imines bearing a range of electronically differing aryl substituents could be converted into the corresponding chiral tirfluoromethylated amines in synthetically useful yield and high enantioselectivity.
Table 3.
Asymmetric isomerization of aryl trifluoromethyl iminesa
|
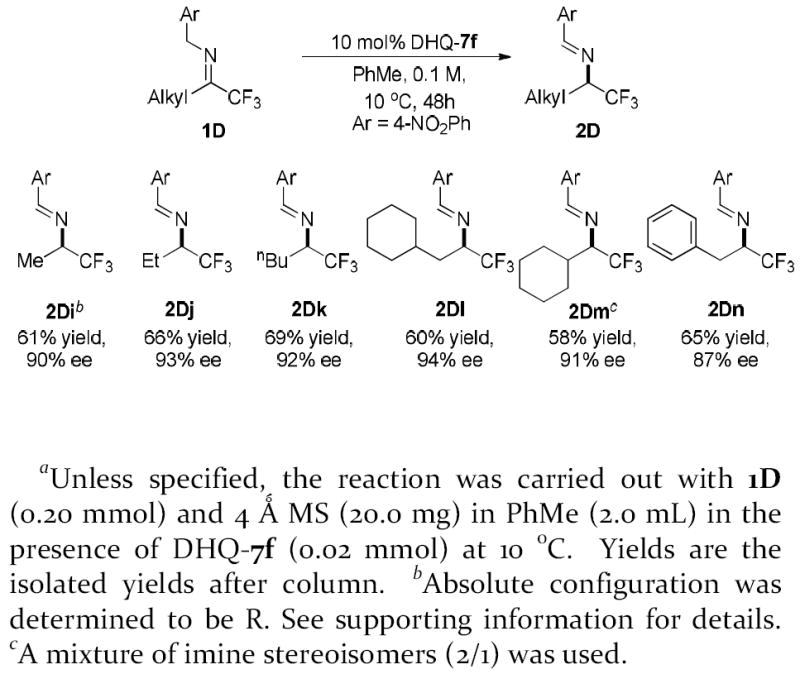
As expected the alkyl trifluoromethyl imines were much less reactive than the aryl trifluoromethyl imines. When applying condition optimized for aryl trifluoromethyl imines to methyl trifluoromethyl imine 1Di, the reaction proceeded to less than 5% conversion after 48 hour. We were pleasantly surprised to find that isomerization of methyl trifluoromethyl imine 1Di with DHQ-7f proceeded smoothly at 10 °C to form the desired trifluoromethylated chiral amine 2Di in 90% ee and 72% conv. (61% yield). Considering that a high temperature of 80 °C was required as reported in the previous studies of cinchona alkaloid-catalyzed isomerization of alkyl trifluoromethyl imines,8b,8e the activity of DHQ-7f demonstrated for the isomerization of 1Di is remarkable. Gratifyingly, similarly high enantioselectivity was obtained with isomerizations of a broad range of alkyl trifluoromethyl imines, which include linear (2Dj, 2Dk), α-, β-branched alkyl (2Dl, 2Dm) and a benzyl (2Dn) substituents. These results render the current reaction the most general catalytic enantioselective approach toward chiral aliphatic trifluoromethylated amines. Subjected to literature protocols with minor modifications8d, the imine groups in both the aryl and alkyl product readily underwent hydrolysis to give the chiral desired trifluoromethylated amines in good yields (Figure 1).
Figure 1.
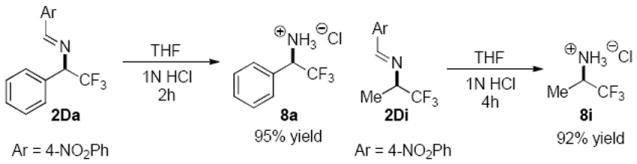
Hydrolysis of the N-protecting group.
In conclusion, we have developed a new catalytic asymmetric synthesis of trifluoromethylated amines enabled by the realization of an unprecedented highly enantioselective catalytic isomerization that is applicable to both aryl and alkyl trifluoromethyl imines. Critically to the successful development of this asymmetric isomerization is the discovery of DHQ-7f, a 9-OH cinchona alkaloid, as an effective catalyst for imine isomerization via proton transfer catalysis. This discovery is noteworthy as both of the two disclosed organic catalysts for highly enantioselective proton transfer catalysis for double bond isomerizations are 6’-OH cinchona alkaloids.9,10 To our knowledge, the current study also provides the first use of DHQ-7f as an effective chiral catalyst.
Supplementary Material
Scheme 1.
Cinchona Alkaloids derivatives
Table 4.
Asymmetric isomerization of alkyl trifluoromethyl iminesa
|
Acknowledgments
We are grateful for financial support from the National Institute of Health (GM-61591). We thank Professor Christine Thomas’ group for the use of IR spectrometer.
Footnotes
Supporting Information
Experimental procedures and characterization of the products. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.For selected reviews, see: Ma J-A, Cahard D. Chem Rev. 2004;104:6119. doi: 10.1021/cr030143e.. Shimizu M, Hiyama T. Angew Chem Int Ed. 2005;44:214. doi: 10.1002/anie.200460441.. Müller K, Faeh C, Diederich F. Science. 2007;317:1881. doi: 10.1126/science.1131943.. Nie J, Guo H-C, Cahard D, Ma J-A. Chem Rev. 2010;111:455. doi: 10.1021/cr100166a.. Zimmer LE, Sparr C, Gilmour R. Angew Chem Int Ed. 2011;50:11860. doi: 10.1002/anie.201102027.
- 2.For selected examples, see: Lim J, Taoka B, Otte RD, Spencer K, Dinsmore CJ, Altman MD, Chan G, Rosenstein C, Sharma S, Su H-P, Szewczak AA, Xu L, Yin H, Zugay-Murphy J, Marshall CG, Young JR. J Med Chem. 2011;54:7334. doi: 10.1021/jm200909u.. Zhang N, Ayral-Kaloustian S, Nguyen T, Afragola J, Hernandez R, Lucas J, Gibbons J, Beyer C. J Med Chem. 2007;50:319. doi: 10.1021/jm060717i.. Grunewald GL, Caldwell TM, Li Q, Criscione KR. J Med Chem. 1999;42:3315. doi: 10.1021/jm980734a.. Grunewald GL, Lu J, Criscione KR, Okoro CO. Bioorg Med Chem Lett. 2005;15:5319. doi: 10.1016/j.bmcl.2005.08.033.. O’Shea PD, Chen C-Y, Gauvreau D, Gosselin F, Hughes G, Nadeau C, Volante RP. J Org Chem. 2009;74:1605. doi: 10.1021/jo8020314.
- 3.(a) Volonterio A, Bravo P, Zanda M. Org Lett. 2000;2:1827. doi: 10.1021/ol005876p. [DOI] [PubMed] [Google Scholar]; (b) Molteni M, Volonterio A, Zanda M. Org Lett. 2003;5:3887. doi: 10.1021/ol0354730. [DOI] [PubMed] [Google Scholar]
- 4.Wang Y, Mosher HS. Tetrahedron Lett. 1991;32:987. [Google Scholar]
- 5.For examples of catalytic asymmetric nucleophilic addition to trifluoromethyl imines, see: Funabiki K, Nagamori M, Goushi S, Matsui M. Chem Commun. 2004:1928. doi: 10.1039/b406000h.. Gosselin F, O’Shea PD, Roy S, Reamer RA, Chen C-Y, Volante RP. Org Lett. 2005;7:355. doi: 10.1021/ol047431x.. Lauzon C, Charette AB. Org Lett. 2006;8:2743. doi: 10.1021/ol0607847.. Fu P, Snapper ML, Hoveyda AH. J Am Chem Soc. 2008;130:5530. doi: 10.1021/ja8001343.. Sukach VA, Golovach NM, Pirozhenko VV, Rusanov EB, Vovk MV. Tetrahedron: Asymmetry. 2008;19:761.. Enders D, Gottfried K, Raabe G. Adv Synth Catal. 2010;352:3147.. Kim SY, Kim SJ, Jang DO. Chem Eur J. 2010;16:13046. doi: 10.1002/chem.201002071.. Hara N, Tamura R, Funahashi Y, Nakamura S. Org Lett. 2011;13:1662. doi: 10.1021/ol2001039.. Huang G, Yang J, Zhang X. Chem Commun. 2011;47:5587. doi: 10.1039/c1cc10403a.. Husmann R, Sugiono E, Mersmann S, Raabe G, Rueping M, Bolm C. Org Lett. 2011;13:1044. doi: 10.1021/ol103093r.. Liu Y-L, Shi T-D, Zhou F, Zhao X-L, Wang X, Zhou J. Org Lett. 2011;13:3826. doi: 10.1021/ol201316z.
- 6.For examples of catalytic asymmetric hydrogenation of trifluoromethyl imines, see: Abe H, Amii H, Uneyama K. Org Lett. 2001;3:313. doi: 10.1021/ol0002471.. Chen M-W, Duan Y, Chen Q-A, Wang D-S, Yu C-B, Zhou Y-G. Org Lett. 2010;12:5075. doi: 10.1021/ol1020256.. Henseler A, Kato M, Mori K, Akiyama T. Angew Chem Int Ed. 2011;50:8180. doi: 10.1002/anie.201103240.
- 7.Kawai H, Kusuda A, Nakamura S, Shiro M, Shibata N. Angew Chem Int Ed. 2009;48:6324. doi: 10.1002/anie.200902457. [DOI] [PubMed] [Google Scholar]
- 8.For a review on the isomerization of imines, see: Han J, Sorochinsky AE, Ono T, Soloshonok VA. Curr Org Synth. 2011;8:281.. For examples on enantioselective isomerization of N-benzyl trifluoromethyl imines, see: Soloshonok VA, Kirilenko AG, Galushko SV, Kukhar VP. Tetrahedron Lett. 1994;35:5063.. Soloshonok VA, Ono T. J Org Chem. 1997;62:3030. doi: 10.1021/jo970425c.. Soloshonok VA, Yasumoto M. J Fluorine Chem. 2007;128:170.. Michaut V, Metz F, Paris J-M, Plaquevent J-C. J Fluorine Chem. 2007;128:500.. For selected examples on enantioselective isomerization of other imines, see: Guthrie RD, Meister W, Cram DJ. J Am Chem Soc. 1967;89:5288.. Kuzuhara H, Komatsu T, Emoto S. Tetrahedron Lett. 1978;19:3563.. Deschenaux R, Bernauer K. Helv Chim Acta. 1984;67:373.. Zimmerman SC, Breslow R. J Am Chem Soc. 1984;106:1490.. Willems JGH, de Vries JG, Nolte RJM, Zwanenburg B. Tetrahedron Lett. 1995;36:3917.. Hjelmencrantz A, Berg U. J Org Chem. 2002;67:3585. doi: 10.1021/jo0106748.. Knudsen KR, Bachmann S, Jørgensen KA. Chem Commun. 2003:2602. doi: 10.1039/b308395k.. Bachmann S, Knudsen KR, Jørgensen KA. Org Biomol Chem. 2004;2:2044. doi: 10.1039/b404381b.
- 9.Wu Y, Singh RP, Deng L. J Am Chem Soc. 2011;133:12458. doi: 10.1021/ja205674x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao X, Xie Y, Su C, Liu M, Shi Y. J Am Chem Soc. 2011;133:12914. doi: 10.1021/ja203138q. [DOI] [PubMed] [Google Scholar]
- 11.For examples with 6’-OH bifunctional cinchona alkaloids, see: Li H, Wang Y, Tang L, Deng L. J Am Chem Soc. 2004;126:9906. doi: 10.1021/ja047281l.. Li H, Wang Y, Tang L, Wu F, Liu X, Guo C, Foxman BM, Deng L. Angew Chem Int Ed. 2005;44:105. doi: 10.1002/anie.200461923.. Li H, Wang B, Deng L. J Am Chem Soc. 2006;128:732. doi: 10.1021/ja057237l.
- 12.Soloshonok VA, Ono T. Tetrahedron. 1996;52:14701. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.