

Abstract
In specialized capillary beds such as the kidney glomerulus, the sheet-like structure of the basement membrane in conjunction with opposing monolayers of endothelium and epithelium form the functioning filtration unit of the kidney. Using a novel cross-linking method on a collagen substrate, we have created a novel hydrogel scaffold to substitute for the basement membrane. Using a simple casting method to create thin films of the hydrogel scaffold (1–5μm), the scaffolds were suitable for long-term static culture, and supported cell attachment and long term cell viability similar to a standard type I collagen substrate. Bulk diffusion and protein permeability of the hydrogel scaffold were evaluated, in addition to its use in a perfusion chamber where it withstood hydraulic pressures typical for glomerular capillaries. This system thus provided a suitable cell substrate for the co-culture of renal epithelial podocytes and endothelial cells in a device that replicates the geometry of the in vivo juxtaposition of the two cell types in relation to their basement membrane.
Key terms: kidney, podocyte, endothelium, extracellular matrix, collagen, glomerular filtration barrier
INTRODUCTION
The basement membrane (BM) is a specialized extracellular matrix that underlies most epithelial cells, and is unique in both structure and composition (1). In specialized capillary beds, such as the kidney filtration barrier, the BM is a functioning component of the tissue being an integral part of the physical barrier that separates the vascular (blood) space from the urinary space (2). In the kidney, the glomerular capillary wall is essentially a laminated structure, composed of opposing monolayers of fenestrated endothelium in the vascular space (3), a highly specialized epithelial cell (podocyte) in the urinary space (4), separated by a double thickness BM produced by both cell types (5). Other organs with fenestrated epithelia also have similar laminated structures with central BMs such as the respiratory membrane separating the blood space from the air space in the lung (6), and endocrine and glandular structures that involve secretion of hormones from an excretory cell into the blood.
In modeling these epithelium-BM-endothelium tri-laminated structures, the commonly used three dimensional culturing methods that rely on sponges of native or non-native matrices in which the cells are embedded, are inadequate since they result in matrix contact on all cell surfaces (7,8). Although useful for studying tumor or stem cell biology or for tissue engineering applications, they do not accurately recreate polarized epithelial cell growth on a basement membrane where matrix contact occurs only on the basal surface. In addition, the currently used methods to mimic cell growth on BMs typically include a solid support, such as transwells coated with thick layers of reconstituted matrix (9,10,11). These systems can achieve epithelial cell polarization of single cell monolayers, but do not accurately recreate the in vivo dimensions of the tri-laminate structure of the capillary wall. Importantly, the existing systems do not replicate the thin, sheet-like properties of BMs. BMs thickness varies depending on the tissue ranging from the very thin BM of the neuromuscular junction at 10nm to the very thick lens Descemet’s membrane at 10μm. In addition, the use of solid supports makes it difficult to accurately model the mechanical properties of native extracellular matrices, as the physical properties of the matrix such as stiffness and compliance are now known to impart significant environmental cues that dictate cell phenotype (12,13).
To recreate in vitro these tri-laminate cell and matrix structures such as the kidney filtration barrier would require a compound system that closely juxtaposes the specialized epithelial cells with the vascular endothelial cells, separated by a hydrogel that approximates the glomerular BM. A critical component to the design of this system is the fabrication of a cell scaffold that can resemble the glomerular BM in both biochemical and biophysical parameters. We have previously developed a method to covalently cross-link hyaluronan to form a hydrogel, resulting in a biomaterial that is a stable gel under physiological conditions and is biocompatible and immunotolerant (14). We report here an adaption of this cross-linking method for use with gelatin, and describe a method to fabricate an ultrathin hydrogel film that can be used as a BM-like cell scaffold. This system provides for the co-culture of podocytes and endothelial cells on a hydrogel scaffold allowing close opposition of the cell monolayers. In addition, our system does not use rigid supports beneath the hydrogel scaffold, allowing the cells to experience a more typical biomechanical environment of a matrix hydrogel. This system may benefit the in vitro modeling of specialized capillary walls by providing a system in which cell-to-cell paracrine signaling, cell-matrix interactions, and biomechanical forces could be simultaneously investigated.
MATERIALS AND METHODS
Synthesis of tyramine-substituted gelatin (T-gelatin)
Chemicals were purchased form Sigma-Aldrich (St. Louis, MO) and ultrapure water was used for all synthesis and dialysis steps. T-gelatin was synthesized based on a proprietary process previously developed for tyramine-substituted hyaluronan (14). Type A porcine gelatin 300 bloom was hydrated in ultrapure water at 80% of the water volume for a final concentration of 5g/l, and allowed to fully dissolve at 37°C for 24h. The solution was buffered to pH 6.5 by adding 250mM 2-(N-morpholino)ethanesulfonic acid, 75mM sodium hydroxide, 150mM sodium chloride, and the water volume adjusted to the final concentration. Tyramine hydrochloride (868mg per gram of gelatin) was added and dissolved by mixing at room temperature. Carbodiimide-mediated substitution of tyramine onto the carboxyl groups of aspartate and glutamate residues in gelatin was initiated by the addition of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (“EDC”, 960mg per gram of gelatin), and N-hydroxysuccinimide (57.6mg per gram of gelatin), followed by mixing at room temperature for 24h. The T-gelatin was purified from low molecular weight reagents, by-products, and buffer molecules through exhaustive dialysis versus 150mM sodium chloride for 3 days followed by water for 3 days, and then lyophilized.
Evaluation of tyramine substitution by HPLC
The degree of tyramine substitution on T-gelatin was determined by amino acid analysis using cation exchange chromatography by the Molecular Biology-Proteomics Facility, University of Oklahoma based on Moore et al. (15). Lyophilized T-gelatin and stock gelatin used in its synthesis were first heated in evacuated tubes containing 6N hydrochloric acid at 110°C for 20–24h to hydrolyze all peptide (amide) bonds generating free amino acids. This treatment also hydrolyzes the amide bond between tyramine adducts and aspartate and glutamate residues in T-gelatin releasing these amino acid residues and free tyramine for analysis. Hydrolyzed samples were vacuum-dried, dissolved in 0.01N hydrochloric acid and filtered through a 0.45μm nylon filter.
For the common amino acids, elution was accomplished by a two solvent-step elution system: first 0.2N sodium citrate, pH 3.28 was used to elute the first nine amino acids followed by 1.0N sodium chloride (pH 7.4) to elute the remaining amino acids. Amino acids were detected by on-line post column reaction with ninhydrin. Derivatized amino acids were quantified by their absorption at 570nm compared to standards, except for glutamate and proline, which were detected at 440nm. Elution of tyramine required a more aggressive two solvent-step elution system: first 0.3M sodium chloride, 0.2N sodium citrate, pH 5.55 with 5.5% ethanol at a column temperature of 66°C with this buffer eluting most amino acids, including arginine, followed by 2.6M sodium chloride, 0.2N sodium citrate, pH 5.73 with 2.5% ethanol and 9.0% isopropanol at a column temperature of 73°C with this buffer eluting tyramine and internal polyamine standards. All procedures were performed on an automated Beckman Gold HPLC amino acid analyzer system. Each sample was run under both chromatography conditions with the data from each chromatographic run normalized using the recovery of arginine under both conditions.
Hydrogel scaffold fabrication
Lyophilized T-gelatin was hydrated at 70mg/ml in water overnight at 37°C, and then 10 U/ml horseradish peroxidase (HRP) was added, mixed by vortexing, and centrifuged briefly to remove air bubbles. Minusheet o-rings (13mm) were purchased from Minucells and Minutissue GmbH (Bad Abbach, Germany) and the black and white o-rings were separated (Fig. 2A). A 200μl drop of the T-gelatin-HRP mixture was placed on a 1×3 inch glass slide (Fig. 2B). The white oring was dipped onto the T-gelatin drop, slowly lifted such that a film formed by surface tension (Fig. 2C), and briefly (~5 seconds) held vertically to drain excess T-gelatin off the film and back into the drop (Fig. 2D). The extent of drainage of excess liquid T-gelatin from the o-ring dictated the final thickness of the hydrogel scaffold. The white o-ring was rested onto the black o-ring in a humidified chamber, pre-chilled to 4°C (Fig. 2E). The o-rings with the T-gelatin film were allowed to fully gel overnight at 4°C in a humidified chamber. The next day, the white and black o-rings were gently pressed together to fully reassemble the carrier and the T-gelatin film was cross-linked into a stable hydrogel by floating on ice cold 0.03% hydrogen peroxide (H2O2) in phosphate buffered saline (PBS) for 3 minutes (Fig. 2F). The cross-linked T-gelatin carriers were removed from the H2O2, returned to the humidified chamber, and allowed to cure overnight at 4°C. The next day, the fully assembled and cross-linked T-gelatin carriers with hydrogel scaffolds were placed in PBS or culture media and inspected for integrity of the T-gelatin hydrogel scaffolds by light microscopy (10× magnification). Scaffold thickness was estimated with confocal microscopy.
Figure 2. Hydrogel scaffold casting and assembly of the cell culture device.

A–F. The T-gelatin hydrogel scaffold fabrication steps using 13mm Minusheet o-ring carriers from Minucell and Minutissue (described in detail in the Materials and Methods section).
Diffusion and bulk flow testing
The ability of molecules to diffuse through T-gelatin was tested on acellular scaffolds and compared to dialysis tubing of 6–8 or 50kDa molecular weight cutoffs with molecules of various molecular weights, including phenol red (0.4 kDa), albumin (66 kDa), and gamma globulins (150–300 kDa). In a 12 well dish, sufficient PBS (750 μl) was added to the dish (bottom well) such that the fluid level was level with the top of the o-rings, leaving the inner chamber formed by the o-rings (top well) empty (Fig. 3A). The o-ring system does not rest directly on the tissue culture dish. The bottom o-ring is manufactured with four static culture supports to permit exposure of the culture media to the lower surface of the T-gelatin scaffold, thereby permitting fluid diffusion between the top and bottom wells. The top well was filled with PBS (40 μl) containing 0.05% phenol red, 50mg/ml bovine serum albumin (BSA), and 1mg/ml goat gamma globulins. At the beginning (0h) and at the end (24h) of the experiment, both the top and bottom well solutions were sampled proportionally to assure assays reflected the volume difference in the top and bottom wells. Diffused proteins were analyzed for molecular weight using denaturing polyacrylamide gel electrophoresis (4–20% gradient gels, Coomassie staining) and for protein concentration using a standard colorimetric assay (Bradford method) using a commercial kit (BioRad Protein Assay, Hercules, CA). This study was repeated three times and protein assay data are presented as percent of total input protein (mean±standard deviation).
Figure 3. Diffusion through acellular T-gelatin hydrogel scaffolds.

Diffusion, driven by a concentration gradient, was observed in static culture using PBS containing phenol red, serum albumin (BSA) and gamma globulins and placed in the top chamber reservoir versus PBS in the bottom reservoir. A. Visualization of small molecule (phenol red) diffusion comparing T-gelatin with dialysis tubing of 6–8 kDa or 50kDa molecular weight cutoff. Images were taken at the time points noted. B. Analysis of large molecule (albumin and γ-globulins) diffusion with denaturing polyacrylamide gel electrophoresis and a standard protein assay. Samples from the top (T) and bottom (B) reservoirs before (time point: 0 hour) and after diffusion (time point: 24 hours) and sampling was proportional to the total volume of the top and bottom reservoirs. Note this was a denaturing gel, resulting in dissociation of immunoglobulins into their composite heavy and light chains which then migrated at their individual molecular weights. However, in the experiment these proteins would have diffused as intact high molecular weight macromolecules. Results of independent colorimetric protein assays of the same samples run on the gel are shown below and are an average of three experiments (mean±SD).
A gradient perfusion chamber was purchased from Minucells and Minutissue and used for bulk flow testing (Fig. 4A, C). The perfusion chamber was modified from the manufacturer’s specification as follows: The four static culture supports on the base of the black o-rings were removed with a sharp knife (Fig. 4B). After placing the fully assembled carrier with the hydrogel scaffold into the perfusion chamber, a second white o-ring was placed on top to compensate for the difference in height (Fig. 4C). A syringe barrel was suspended above the perfusion chamber, connected to the upper chamber afferent port, and hydraulic head was changed by altering solution height (Fig. 4A). The lower chamber afferent port was connected to a syringe pump (Harvard Apparatus, Holliston, MA) only to fill the lower chamber and then held static for the experiment. The efferent flow port on the upper perfusion chamber was blocked to force fluid flow through the hydrogel scaffold. Eluate from the perfusion chamber was collected from the lower chamber efferent port at 1 minute intervals, weighed to determine flow volume (assumed 1gm=1ml), and flow rates were calculated. Control materials included an impermeable material (parafilm) to confirm the pressure seal, and materials of various permeability or pore structure. These included dialysis tubing of various molecular weight cutoffs (Spectra/Por, Spectrum Labs, Rancho Dominguez, CA), and 0.4μm isopore polycarbonate and 0.45μm mixed cellulose ester membranes (both from Millipore, Bellerica, MA). Data are presented as mean±SD of three experiments.
Figure 4. Water bulk flow through acellular T-gelatin hydrogel scaffolds.

A–C. Diffusion chamber configuration and modifications used for bulk flow testing. See text for detailed descriptions; white arrow indicates path of fluid flow. D. Flow rates were calculated over a range of pressures reaching a maximal pressure of ~10 mmHg which is the estimated net pressure exerted on the vascular wall of the glomerular capillaries (19). Various other membranes of different pore size and structure were used as references (“DT”, dialysis tubing; molecular weight cut off as indicated on graph). Although the pore size of the isopore polycarbonate and cellulose ester membranes is similar, the pore structure is very different. The isopore polycarbonate membrane is a solid sheet with holes of a defined diameter, whereas the cellulose ester is a fibrillar macromolecular meshwork and is more similar to the pore structure of collagen hydrogels.
Cells and microscopy
Mouse conditionally immortalized glomerular endothelial and podocyte cell lines have been previously described (16,17,18). Both cell lines were propagated as previously described and used for experiments under non-permissive growth conditions. The cell plating area is 1cm in diameter (0.79cm2), and plating density depended on the size of the cells used. For the podocyte cell line, plating at 2.5×104cell gave a confluent monolayer and for the endothelial cell line, 5×104 cells were needed to achieve confluence. Each side of the hydrogel scaffold was seeded with cells sequentially. The assembled device was placed in 1 well of a 12 well cluster dish such that the medium was level with height of the o-rings, but did not fill the top chamber (~300μl). The cells were added to the top chamber (in ~50μl) and allowed to attach. The length of time needed for attachment depended on the cell line, and was monitored by light microscopy (~4–6hrs). After the cells on one side attached, the assembly was inverted and the second cell type was seeded in the same manner.
Cell viability was determined using a Live/Dead Double Staining kit (Oncogene Research Products, San Diego, CA) as recommended by the manufacturer. Podocytes were plated at 2.5×104 cells/well of a 24 well dish that contained a 12mm round coverslip that was either uncoated, coated with rat tail collagen (type I collagen, BDBiosciences, Bedford, MA), or coated with T-gelatin. Cells were cultured for times indicated followed by the Live/Dead assay and total number of live and dead cells were manually counted from six random high power fields per coverslip (using 3 coverslips per time point). Significance was determined by t test.
For immunohistochemistry analysis of cell monolayers, the entire cell culture device was processed through fixation and staining steps using a previously published method for cells grown on coverslips (16). Rhodamine-conjugated Phalloidin was used at 1:1000 dilution and nuclei were stained with 1μM TOPRO3 (both from Invitrogen, Carlsbad, CA). Following staining, the device was placed on a drop of mounting media (Vectashield, Vector Laboratories, Burlingame, CA) on a 1×3 inch slide and the T-gelatin scaffolds were released from the o-rings by scoring around the inner edge of the o-ring with a 30g needle. A drop of mounting media was placed on top of the released scaffold and covered with a standard coverslip for confocal microscopy.
RESULTS
In the development of this cell culture device, an important design consideration was incorporating a cell scaffold that resembled the native glomerular BM in both its microscale dimensions and its hydrogel properties. All extracellular matrices are hydrogels (7) and, as the name implies, are typically 80–90% water. Native BMs are primarily composed of type IV collagen, laminins, heparan sulfate proteoglycans, and nidogen, with collagen being the main structural component (5). Because of the structural contributions of collagen in native BMs, we chose to develop a collagen-based hydrogel scaffold for our cell culture device. Gelatin, predominantly composed of type I collagen, was selected as a readily-available collagen source, however, this cross-linking method has the capacity to be adapted to other collagens or other proteins (19). Since gelatin is a liquid at 37°C and gels only at lower temperatures, the gelatin was modified to permit stable gel formation at physiologic temperatures through the introduction of new intermolecular cross-links using a two-step process (14) (Fig. 1). The first step involved the substitution of gelatin with tyramine, a naturally occurring octopamine that resemble the side chain of tyrosine, using conventional carbodiimide chemistry (20,21). The second step involves the enzymatic cross-linking of tyramine-substituted gelatin (T-gelatin) using the hydroxyphenyl groups on tyramine adducts of adjacent gelatin molecules. The cross-linking is mediated by peroxidase in the presence of dilute H2O2 which is able to preferentially extract the phenolic hydroxyl hydrogen atom from tyramine leaving the phenolic hydroxyl oxygen with a single unshared electron; an extremely reactive free radical. The free radical isomerizes to one of the two equivalent ortho-position carbons, and then two such structures dimerize to form a covalent bond, which after enolizing, generates a dityramine cross-link. This chemistry is the same reaction that occurs through the action of endogenous peroxidases on tyrosine residues of native proteins (22). The creation of multiple dityramine cross-links between adjacent gelatin molecules results in the formation of a thermostable hydrogel.
Figure 1. Schematic diagram of the two-step process in generating cross-linked T-gelatin.

Step 1: Tyramine addition to gelatin is initiated with EDC modification (to form a reactive O-acylisourea intermediate) of the carboxyl groups on aspartate (Asp) or glutamate (Glu) residues of the individual collagen molecules, followed by substitution of tyramine (nucleophilic attack of the tyramine amine on the carbonyl carbon of the acylisourea intermediate) and displacement of the acylurea form of EDC (EDU). Step 2: Cross-linking reaction is initiated by the addition of peroxidase and hydrogen peroxide that causes an oxidation of tyramine and results in a dihydroxyphenyl link between two collagen chains forming the stable intermolecular cross-link.
The tyramine substitution takes place on the carboxylic acid side chains of aspartate and glutamate, and gelatin has a relatively high mole percent of aspartate and glutamate residues. Table 1 shows an example of the amino acid composition by residue and mole percent for one T-gelatin preparation and the stock gelatin used in its synthesis. The results showed that the calculated mole percentages for the typical amino acids detected in gelatin were not significantly altered by the tyramine-substitution reaction. Tyramine substitution on gelatin, calculated as the moles of tyramine per total moles of aspartate and glutamate, on several synthesis reactions consistently achieved between 11 and 16% substitution. The acid hydrolysis conditions used not only cleaved peptide bonds, but all amide bonds including those between tyramine and aspartate/glutamate residues in gelatin allowing for detection and quantification of moles of aspartate, glutamate and tyramine in T-gelatin. However, this acid hydrolysis also converts asparagine residues to aspartate and glutamine residues to glutamate. Thus, the degree of tyramine substitution in T-gelatin as determined by this method will be an underestimation, as the mole percent of aspartate and glutamate reported for gelatin upon its hydrolysis also contains the mole percents of asparagine and glutamine, respectively. In the intact gelatin molecule, however, these latter two amino acids are not available for tyramine substitution.
Table 1.
Amino acid composition as mole percent for gelatin before and after tyramine substitution.
| Amino Acid1 | Gelatin Mole % | T-gelatin Mole % |
|---|---|---|
| Aspartic Acid | 5.23 | 5.21 |
| Threonine | 2.00 | 1.78 |
| Serine | 3.72 | 3.71 |
| Glutamic Acid | 8.24 | 8.43 |
| Proline | 14.10 | 14.38 |
| Glycine | 37.24 | 36.46 |
| Alanine | 12.39 | 12.61 |
| Cysteine | 0.00 | 0.18 |
| Valine | 2.55 | 2.50 |
| Methionine | 0.02 | 0.41 |
| Isoleucine | 0.96 | 0.97 |
| Leucine | 2.61 | 2.62 |
| Tyrosine | 0.15 | 0.22 |
| Phenylalanine | 1.52 | 1.47 |
| Lysine | 3.11 | 2.94 |
| Histidine | 0.51 | 0.47 |
| Arginine | 5.65 | 5.63 |
| Total | 100.00 | 100.00 |
| Tyramine2 | 0.00 | 16.07 |
Mole % = moles single amino acid/moles total amino acids × 100.
Mole % = moles tyramine/(moles aspartic + glutamic acid) × 100.
T-gelatin can be suspended in most common physiologic solutions, and when hydrated before cross-linking is a liquid at room temperature. For this application, T-gelatin was hydrated and cross-linked at a concentration of 70mg/ml (7%), creating a scaffold that would approximate the native glomerular BM water content of 93% (23). To form the thin sheet of T-gelatin to replicate the characteristic dimensions of BMs, a thin film of liquid T-gelatin was cast directly within o-ring carriers (Fig. 2A–F), followed by cross-linking of the thin film into the stabile hydrogel scaffold (described in detail in the Materials and Methods section). This casting method routinely produced hydrogel scaffolds in the range of 1–5μm, but individual films were uniform in thickness with the exception of a meniscus near the edge of the o-ring.
Because our application of the cell culture device is directed at modeling the kidney filtration barrier, the T-gelatin hydrogel scaffolds were tested for simple hydrodynamic properties related to movement of water and proteins. Diffusion of molecules of various molecular weights was tested in static conditions driven by a concentration gradient on acellular scaffolds (Fig. 3A,B). In comparison to dialysis tubing of defined molecular weight cutoffs, small molecules (phenol red, 0.4kDa) similarly diffused through the both the dialysis tubing and hydrogel scaffold at early (4h) and later (24h) time points (Fig. 3A). However, larger proteins such as serum albumin and gamma globulins (66kDa and ~150kDa respectively, both used at physiologically relevant concentrations), did not appreciably diffuse through the dialysis tubing as expected with the respective molecular weight restrictions, however, there was diffusion of these larger molecules through the T-gelatin as determined by gel electrophoresis and a colorimetric protein assay (Fig. 3B).
Water bulk flow (the movement of water under pressure) was tested on acellular hydrogel scaffolds using a perfusion chamber modified as described in Fig. 4A–C. A range of pressures was used up to ~10 mmHg pressure, which is the estimated net pressure exerted on the vascular wall of kidney glomerular capillaries (24). Various other porous materials were used as comparison and flow rates were determined at various pressures (Fig. 4D). The acellular T-gelatin scaffolds were able to withstand ~10mmHg pressure, and flow rates most closely approximated the flow rate observed for dialysis tubing of 50 kDa molecular weight cutoff.
The ability of the hydrogel scaffolds to support cell growth was tested in static culture using conditionally immortalized murine glomerular podocyte and glomerular vascular endothelial cell lines developed by the Mundel and Madaio labs, respectively (25,16). Both cell types rapidly attached and formed monolayers on T-gelatin, and the fluid compartments formed by the sides of the o-rings permitted easy seeding of the respective sides. The complete cell culture device consisting of the suspended hydrogel scaffold seeded with opposing cell monolayers is visually transparent by light microscopy (Fig. 5A). In addition, the complete cell culture device was easily adapted to standard cell fixation and staining protocols, and was handled similar to cells grown on coverslips. For confocal microscopy, however, the scaffolds were released from the orings and mounted under a coverslip. An example of a confocal imaging study is shown in Fig. 5B showing F-actin (phalloidin) and nuclei (TOPRO3) staining of cells grown for 7 days in static culture. Interestingly, the endothelial cell F-actin staining demonstrated a trans-cellular alignment of cytoskeletal elements suggesting the monolayer was developing a higher order of cellular organization characteristic of a tissue. The thickness of the hydrogel scaffold was estimated from the confocal XZY images (Fig. 5C), but may represent an over estimation because when released from the o-rings, the scaffolds are no longer in tension, contract slightly, thereby altering the scaffold dimensions resulting in a thicker hydrogel.
Figure 5. Use of ultrathin T-gelatin hydrogel scaffolds in static cell culture.
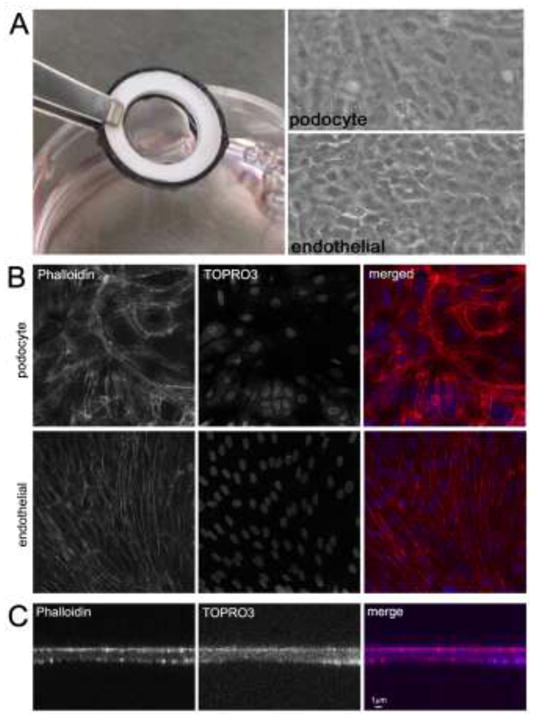
A. Left: The fully assembled cell culture system seeded with opposing cell monolayers which is optically transparent. Note that the T-gelatin scaffold is taut and does not sag when hydrated and seeded with cells. Right: Light micrograph images of both monolayers (podocyte and endothelial); both images are the same field but in different focal planes. These images appear slightly unfocused due to the optical diffraction caused by the close proximity of the two cell monolayers and the use of transmitted light at low power (10×) magnification. B. Confocal images (XYZ plane) of cell monolayers; all images are the same field, but different focal planes (63× magnification). C. Confocal images (XZY plane) through the height (z plane) of a cell culture system showing scaffold thickness.
To confirm biocompatibility of the T-gelatin, initial cell attachment and long term viability were assessed in comparison to uncoated and type I collagen-coated substrates (Fig. 6). Coverslips with a molecular coating of Type I collagen were used as a control since unmodified gelatin solublizes at 37°C and would not remain on the coverslip once placed in culture, and because gelatin is primarily composed of type I collagen. The total numbers of live and dead cells were quantified using a Live/Dead assay from 1h to 7d after plating podocytes on either T-gelatin-coated, type I collagen-coated, or uncoated coverslips. At the initial 1h time point, the number of live cells attached to T-gelatin-coated coverslips was significantly higher compared to type I collagen-coated or uncoated, however, by the 2h time point, the type I collagen-coated coverslips match the degree of attachment of the T-gelatin. Thus, both the T-gelatin and type I collagen coatings accelerated cell attachment compared to uncoated, both achieving maximal attachment by 2hs. At 24h, all three had reach similar and not significantly different degrees of cell attachment, and since the initial plating was at 75% confluence, the 48h and 7d cells counts increased similarly reflecting cell proliferation until reaching confluence. Following a week of culture and despite the initial differences in attachment kinetics, there were no significant differences in total attached cells or occurrence of cell death between the three groups. There was no significant difference in the number of dead cells at any time point with the exception of the 1h comparison between T-gelatin and uncoated. However, at this time point, the ratio of live to dead cells on the T-gelatin and uncoated coverslips was similar (12.6% vs 11.8% dead cells respectively), suggesting this apparent difference may only reflect the considerably higher total number of cells at the first time point for T-gelatin. Taken together, these studies demonstrate T-gelatin scaffolds were easily adaptable to standard immunostaining and imaging techniques, and there were no significant issues with biocompatibility with regard to supporting cell attachment and long term viability.
Figure 6. Cell attachment and viability on T-gelatin.

Live/Dead assay of podocytes plated on coverslips coated with T-gelatin or control coverslips (uncoated and type I collagen coated) at a cell density to achieve ~75% confluence on the initial plating. Coverslips were fixed at given time points and live and dead cells were counted. Data are mean±SD; significant differences in live cell or dead cell numbers among the three groups are noted with asterisks (*P<0.01, **P<0.001).
DISCUSSION
Recapitulating complex tissue physiology with in vitro culturing systems has expanded in recent years resulting in many advances in tumor and stem cell biology and tissue bioengineering (26). In addition, investigating the complex interrelationship between the biochemical (cell-cell and cell-matrix signaling) and mechanical (membrane stiffness, fluid flow and pressure) events that influence cell and tissue function necessitates a system that recreates the properties of the BM and the unique juxtaposition of cell monolayers. This cell culture system addresses some of these current issues by replicating the thin, linear structure of the BM that is both free of solid supports and provides dual apical fluid exposure, and has the benefit of being able to accommodate the accurate juxtaposition of two cell types similar to the in vivo tissue structure. Although these studies focused on an application to modeling the kidney filtration barrier, other cell types could be substituted for the renal cells used here, for example, co-culturing alveolar epithelium with microvascular epithelial cells to replicate the respiratory membrane.
The application of tyramine cross-linking technology to gelatin has produced a versatile hydrogel scaffold, however, native BMs are composed of type IV collagen and other characteristic glycoproteins, not solely type I collagen. Although type 1 collagen is frequently used as a tissue culture coating to promote the growth and differentiation of cells including podocytes (27), both type IV collagen and the other prototypic BM proteins are known to provide distinct and important signaling information to both the adherent podocytes and endothelial cells (28,29,30). In this regard, one advantage of T-gelatin is that the cross-linking chemistry can be applied to intact proteins, protein fragments or peptides that contain tyrosine residues. The phenolic side chain of tyrosine is identical to tyramine, and thus, coating or impregnating the T-gelatin with tyrosine-containing peptides before cross-linking would permit covalent attachment of these peptides into the final hydrogel scaffold. For example, to better approximate the unique composition of the glomerular basement membrane, peptides containing important cell adhesion domains of laminin 521 and type IV collagen α3α4α5 could be integrated into the T-gelatin matrix. Similarly, incorporating larger molecules, such as sulfated glycosaminoglycans, into the hydrogel scaffolds to replicate contributions by the BM heparan sulfate proteoglycan will be another important consideration, as these charged moieties appear to be important in the filtration barrier function (31), and like hyaluronan, are also amendable to inclusion via the tyramine cross-linking chemistry (14).
Another advantage of this cell culture system is that it uses a hydrogel without rigid supports, providing a unique substrate for culturing cells that permits analysis of cellular responses to important physical forces such as stretch, tension, and compression. Information on how these mechanical forces signal to cells and alter their behavior is just beginning to emerge (12,13), and are clearly an integral component of the in vivo environment, as the capillary wall is constantly under shear and hydraulic forces. In addition, little is known about the combined structural architecture of the tissue with regard to the integration of cellular cytoskeleton with the extracellular matrix proteins that networks the structural components of the cells into the structural components of the matrix (32,33). Thus, the complete cell culture device may provide an integrated system that can evaluate not only the individual components of the filtration barrier, but also their integrated structure and physiological responses to environmental changes or disease.
In the specific application of the T-gelatin hydrogel scaffolds to the study of the kidney capillaries and the blood filtration barrier, the diffusion and bulk flow results of the T-gelatin hydrogel appeared to be consistent with gel sieving properties typical of cross-linked macromolecules (34). In addition, our observations were similar to the known restrictions on macromolecular diffusion as the native glomerular BM, which is a barrier to leukocytes and larger proteins with approximate molecular weight restriction near the size of albumin (35). Our T-gelatin hydrogel system exhibited some diffusion of larger molecules, and the bulk flow of water most closely approximated dialysis tubing of 50kDa molecular weight cut off. For future studies using the cell culture device in the perfusion chamber, the thin hydrogel scaffolds was also found to be strong enough to withstand physiological forces typical of the in vivo pressures acting in the kidney glomerular capillaries. Thus, the application of the T-gelatin hydrogel scaffold here has the potential to provide a new cell culture platform to investigate unique aspects in kidney cell responses to physical forces involved in blood filtration.
In summary, we have developed a method to form stable hydrogels using gelatin and a cell culture system that supports seeding and long term culture of opposing monolayers of cells. It is simple to fabricate, is amenable to standard imagining techniques, and has potential applications to a variety of specialized capillary structures. The novel aspects of the system will provide for unique opportunities in evaluating cell-cell and cell-matrix responses, and also the in vivo physiological forces that will permit the investigation of not just individual cellular responses, but the integrated dynamics of a multi-cellular tissue.
Acknowledgments
We thank Dr. Michael Madaio for providing the endothelial cell line, Zhenzhen Wu for technical assistance, and Sydney Calabro for assistance with the images. We also acknowledge the helpful input from Drs. Jeffrey Simske, John Sedor, Tyler Miller, and William Fissell for discussions and review of the manuscript. This study was supported by NIH grant DK077668 and MetroHealth Medical Center institutional funds.
Abbreviations
- BM
basement membrane
- BSA
bovine serum albumin
- HRP
horseradish peroxidase
- T-gelatin
tyramine-substituted gelatin
Footnotes
CONFLICT OF INTEREST
The authors (AC and AD) have licensed the tyramine substitution and cross-linking technology for commercialization to Lifecore Biomedical, Inc., Chaska, MN and have received benefits for personal or professional use indirectly related to the subject matter of this manuscript. Benefits have been used for research funding at The Cleveland Clinic, a nonprofit organization, with which these authors are associated.
References
- 1.Martin GR, Timpl R, Kuhn K. Basement membrane proteins: molecular structure and function. Adv Protein Chem. 1988;39:1–50. doi: 10.1016/s0065-3233(08)60374-5. [DOI] [PubMed] [Google Scholar]
- 2.Patrakka J, Tryggvason K. Molecular make-up of the glomerular filtration barrier. Biochem Biophys Res Commun. 2010;396:164–169. doi: 10.1016/j.bbrc.2010.04.069. [DOI] [PubMed] [Google Scholar]
- 3.Ballermann BJ. Contribution of the endothelium to the glomerular permselectivity barrier in health and disease. Nephron Physiol. 2007;106:19–25. doi: 10.1159/000101796. [DOI] [PubMed] [Google Scholar]
- 4.Faul C, Asanuma K, Yanagida-Asanuma E, Kim K, Mundel P. Actin up: regulation of podocyte structure and function by components of the actin cytoskeleton. Trends Cell Biol. 2007;17:428–437. doi: 10.1016/j.tcb.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 5.Miner JH. Glomerular basement membrane composition and the filtration barrier. Pediatr Nephrol. 2011;26:1413–1417. doi: 10.1007/s00467-011-1785-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maina JN, West JB. Thin and strong! The bioengineering dilemma in the structural and functional design of the blood-gas barrier. Physiol Rev. 2005;85:811–844. doi: 10.1152/physrev.00022.2004. [DOI] [PubMed] [Google Scholar]
- 7.Cushing MC, Anseth KS. Materials science. Hydrogel cell cultures. Science. 2007;316:1133–1134. doi: 10.1126/science.1140171. [DOI] [PubMed] [Google Scholar]
- 8.Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 2005;23:47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 9.Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15:378–386. doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 10.Hunt JL, Pollak MR, Denker BM. Cultured podocytes establish a size-selective barrier regulated by specific signaling pathways and demonstrate synchronized barrier assembly in a calcium switch model of junction formation. J Am Soc Nephrol. 2005;16:1593–1602. doi: 10.1681/ASN.2004080679. [DOI] [PubMed] [Google Scholar]
- 11.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science. 2010;328:1662–1668. doi: 10.1126/science.1188302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Janmey PA, Miller RT. Mechanisms of mechanical signaling in development and disease. J Cell Sci. 2011;124:9–18. doi: 10.1242/jcs.071001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Discher DE, Janmey P, Wang YL. Tissue cells feel and respond to the stiffness of their substrate. Science. 2005;310:1139–1143. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 14.Darr A, Calabro A. Synthesis and characterization of tyramine-based hyaluronan hydrogels. J Mater Sci Mater Med. 2009;20:33–44. doi: 10.1007/s10856-008-3540-0. [DOI] [PubMed] [Google Scholar]
- 15.Moore S, Spackman DH, Stein WH. Automatic recording apparatus for use in the chromatography of amino acids. Fed Proc. 1958;17:1107–1115. [PubMed] [Google Scholar]
- 16.Mundel P, Reiser J, Zuniga Mejia BA, Pavenstadt H, Davidson GR, Kriz W, Zeller R. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp Cell Res. 1997;236:248–258. doi: 10.1006/excr.1997.3739. [DOI] [PubMed] [Google Scholar]
- 17.Schwartz EJ, Cara A, Snoeck H, Ross MD, Sunamoto M, Reiser J, Mundel P, Klotman PE. Human immunodeficiency virus-1 induces loss of contact inhibition in podocytes. J Am Soc Nephrol. 2001;12:1677–1684. doi: 10.1681/ASN.V1281677. [DOI] [PubMed] [Google Scholar]
- 18.Akis N, Madaio MP. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int. 2004;65:2223–2227. doi: 10.1111/j.1523-1755.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- 19.Moreira Teixeira LS, Feijen J, van Blitterswijk CA, Dijkstra PJ, Karperien M. Enzyme-catalyzed crosslinkable hydrogels: Emerging strategies for tissue engineering. Biomaterials. 2012;33:1281–1290. doi: 10.1016/j.biomaterials.2011.10.067. [DOI] [PubMed] [Google Scholar]
- 20.Sehgal D, I, Vijay K. A method for the high efficiency of water-soluble carbodiimide-mediated amidation. Anal Biochem. 1994;218:87–91. doi: 10.1006/abio.1994.1144. [DOI] [PubMed] [Google Scholar]
- 21.Aslam M, Dent A. Bioconjugation: protein coupling techniques for the biomedical sciences. 1998;188:369–372. [Google Scholar]
- 22.Furtmuller PG, Zederbauer M, Jantschko W, Helm J, Bogner M, Jakopitsch C, Obinger C. Active site structure and catalytic mechanisms of human peroxidases. Arch Biochem Biophys. 2006;445:199–213. doi: 10.1016/j.abb.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 23.Comper WD, Lee AS, Tay M, Adal Y. Anionic charge concentration of rat kidney glomeruli and glomerular basement membrane. Biochem J. 1993;289:647–652. doi: 10.1042/bj2890647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maddox DA, Brenner BM. The Kidney. 6. 2000. Glomerular Ultrafiltration; pp. 319–328. [Google Scholar]
- 25.Akis N, Madaio MP. Isolation, culture, and characterization of endothelial cells from mouse glomeruli. Kidney Int. 2004;65:2223–2227. doi: 10.1111/j.1523-1755.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- 26.Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol. 2006;7:211–224. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 27.Shankland SJ, Pippin JW, Reiser J, Mundel P. Podocytes in culture: past, present, and future. Kidney Int. 2007;72:26–36. doi: 10.1038/sj.ki.5002291. [DOI] [PubMed] [Google Scholar]
- 28.Pozzi A, Zent R. Integrins: sensors of extracellular matrix and modulators of cell function. Nephron Exp Nephrol. 2003;94:e77–e84. doi: 10.1159/000072025. [DOI] [PubMed] [Google Scholar]
- 29.Kretzler M. Regulation of adhesive interaction between podocytes and glomerular basement membrane. Microsc Res Tech. 2002;57:247–253. doi: 10.1002/jemt.10083. [DOI] [PubMed] [Google Scholar]
- 30.Schnaper HW, Kleinman HK, Grant DS. Role of laminin in endothelial cell recognition and differentiation. Kidney Int. 1993;43:20–25. doi: 10.1038/ki.1993.5. [DOI] [PubMed] [Google Scholar]
- 31.Deen WM, Lazzara MJ, Myers BD. Structural determinants of glomerular permeability. Am J Physiol Renal Physiol. 2001;281:F579–F596. doi: 10.1152/ajprenal.2001.281.4.F579. [DOI] [PubMed] [Google Scholar]
- 32.Wang N, Ingber DE. Control of cytoskeletal mechanics by extracellular matrix, cell shape, and mechanical tension. Biophys J. 1994;66:2181–2189. doi: 10.1016/S0006-3495(94)81014-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Janmey PA, Weitz DA. Dealing with mechanics: mechanisms of force transduction in cells. Trends Biochem Sci. 2004;29:364–370. doi: 10.1016/j.tibs.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 34.Gehrke SH, Fisher JP, Palasis M, Lund ME. Factors determining hydrogel permeability. Ann N Y Acad Sci. 1997;831:179–207. doi: 10.1111/j.1749-6632.1997.tb52194.x. [DOI] [PubMed] [Google Scholar]
- 35.Haraldsson B, Nystrom J, Deen WM. Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev. 2008;88:451–487. doi: 10.1152/physrev.00055.2006. [DOI] [PubMed] [Google Scholar]